

Abstract
Purpose of review
This review will highlight recent advances in developing strategies to accelerate muscle regeneration and to slow muscle degeneration in myositis, focusing primarily on inclusion body myositis (IBM).
Recent findings
Therapies for accelerating muscle regeneration, primarily through inhibition of myostatin, have shown promise in the laboratory and are now entering clinical trials. Recent studies have implicated autophagy, a key cellular process involved in clearance of ubiquitinated aggregates, in the pathogenesis of familial and sporadic inclusion body myositis (sIBM). IBM has joined a growing list of diseases known as TDP-43 proteinopathies, in which this protein becomes mislocalized to the cytoplasm; however, it is unclear whether these protein aggregates or others are pathogenic in this disease.
Summary
New discoveries of biomarkers in sIBM and new insights into the pathogenesis of familial IBM are opening novel therapeutic pathways for these disorders. In particular, drugs that stimulate autophagy, already in development for cancer and neurodegenerative diseases, are candidates for clinical trials. These disease-specific therapies combined with novel therapies to accelerate muscle regeneration hold promise for future therapy for this devastating disease.
Keywords: autophagy, follistatin, inclusion body myositis, myostatin, regeneration, TDP-43, valosin-containing protein
Introduction
For most myositis patients, prognosis depends upon premorbid health and the ability of the disease to respond to immunosuppressive therapy. Skeletal muscle has an incredible ability to regenerate following an acute injury, and patients with immunotherapy-responsive idiopathic inflammatory myopathies have an excellent prognosis. In contrast, patients with immunotherapy-resistant myositis often have a relentlessly progressive course. Although newer immunotherapies may have improved efficacy in refractory cases, for patients with sporadic inclusion body myositis (sIBM), novel treatment strategies are desperately needed.
Muscle strength in disease can be thought of as a balance between regeneration and degeneration, and this balance is strongly influenced by the immune system in myositis. For example, cytokines released from inflammatory cells may stimulate satellite cells to proliferate and thus enhance regeneration. Therefore, a better understanding of the complex interplay between the inflammatory response and muscle regeneration is critical to the development of therapies designed to enhance myogenesis (reviewed in [1]). Furthermore, although the immune system is believed to directly cause muscle degeneration in most forms of myositis, there are clearly nonimmune mechanisms (reviewed in [2]), and these may be the primary culprit in sIBM. Understanding the cellular mechanisms of myofiber degeneration will likely lead to novel treatment strategies for myositis in the future. This review will focus on strategies to suppress myofiber degeneration and to enhance myofiber regeneration, and will not discuss emerging immunotherapies (recently reviewed in [3••]). These emerging therapies may be helpful in conjunction with immunosuppressive medications in many forms of myositis, and may become the primary mode of therapy for sIBM.
Strategies to accelerate muscle growth
In theory, therapies that enhance muscle regeneration could be beneficial in all muscle diseases regardless of cause. For this reason, intense research has been under-taken to develop drugs that stimulate myofiber regeneration in addition to cell-mediated therapies to replace injured myofibers. Androgens to stimulate muscle hypertrophy have long been used by body builders, but their use in patients is limited due to side-effects. Oxandrolone is a synthetic testosterone analog that is well tolerated in patients, and has shown efficacy in a small, double-blinded placebo-controlled trial in IBM [4]. Research over the last decade has implicated the myostatin pathway as a central regulator of myogenesis during development. More recently, inhibition of the myostatin pathway has shown great promise in enhancing muscle regeneration in animal models of muscle diseases.
Myostatin inhibition
During vertebrate development, one of the critical regulators of muscle growth is the myostatin pathway. Myostatin is a member of the transforming growth factor (TGF)-beta family of secreted growth factors and is a potent suppressor of muscle growth. Myostatin knockout mice have a two-fold to three-fold increase in muscle mass, without abnormalities in other organs such as the heart [5]. This pathway is clearly important in humans, as patients with a loss-of-function mutation in myostatin have marked muscle hypertrophy and increased strength [6]. Thus, inhibitors of myostatin may be potent stimulators of muscle growth.
Myostatin is synthesized as a prepropeptide and forms a homodimer via a covalent disulfide bond. After cleavage extracellularly, the amino-terminal proprotein remains noncovalently attached to the carboxy-terminal peptide, keeping it in an inactive state. A second cleavage event by a metalloproteinase activates myostatin, allowing it to bind to its receptor, activin receptor type IIB (ActRIIB). Myostatin can be inactivated by binding to proteins including follistatin, follistatin-related gene (FLRG) and GASP-1. Activation of ActRIIB leads to activation of a Smad complex that enters the nucleus and activates the transcription of myogenic genes, inhibiting both the proliferation and differentiation of myogenic precursors.
Most myostatin inhibitors in development bind to myostatin extracellularly and reduce its bioavailability. For example, an antibody against myostatin, MYO-029, was used in a double-blind, placebo-controlled phase I clinical trial in 116 adult muscular dystrophy patients [7]. Although the study was not powered to evaluate clinical efficacy, this drug was well tolerated, with the major side effect being a cutaneous hypersensitivity reaction at high doses. Other companies are developing a soluble ActRIIB protein that can similarly bind to and inactivate myostatin [8•]. sActRIIB has been shown to be very effective in mouse muscular dystrophy models [9]. Other therapies in development include the propeptide that keeps myostatin inactive [10] and a dominant-negative myostatin analog [11].
One of the most promising myostatin inhibitors in development is follistatin [12•]. Follistatin binds to and inhibits myostatin and other TGF-beta family members such as activin. Interestingly, muscle-specific overexpression of follistatin causes an even greater increase in muscle mass than the myostatin knockout mouse [13], suggesting that in addition to inhibition of myostatin, follistatin also acts to increase muscle growth independently of myostatin. Indeed, follistatin overexpression in myostatin knockout mice causes a quadrupling of muscle mass [14].
Mendell and colleagues utilized the natural propensity of the AAV virus to infect muscle cells to overexpress the follistatin (FSTN) gene in animal models. AAV gene vectors have proven to be safe, and a single injection can lead to lifelong expression of the gene of interest. Injection of AAV-FSTN into muscles of wild-type mice led to a two-fold increase in muscle size, and even had efficacy in uninjected muscles, demonstrating that it is released into the circulation [15]. Furthermore, AAV-FSTN has a long-term benefit in the mouse muscular dystrophy models when injected into the quadriceps muscle [15]. A recent study in nonhuman primates injected with AAV-FSTN into the quadriceps muscle showed a long-term, marked increase in muscle size without adverse effects [16••]. An alternative approach to gene therapy is to upregulate follistatin expression. Indeed, histone deacetylase inhibitors upregulate follistatin and show efficacy in mouse models of muscular dystrophy [17,18].
One concern regarding follistatin therapy is a potential adverse effect on fertility (it was initially discovered as an inhibitor of FSH secretion). However, an alternatively spliced isoform of follistatin containing the carboxy-terminus (named FS-344) inhibits the ability of follistatin to bind activin, and thus has little effect on reproduction in mouse or monkey animal models [15,16••]. A potentially beneficial side-effect of follistatin therapy in myositis is an anti-inflammatory effect [19].
Stem cell therapy
In addition to stimulation of endogenous muscle stem cells with growth factors, enhancing muscle growth with exogenous stem cells is a promising area of therapeutic development [20•]. In particular, the possibility of converting patient’s own cells into muscle progenitors using induced pluripotent stem (iPS) cell technology is a new and exciting field of research [21], although many obstacles to clinical application remain. Satellite cells have long been known to lie quiescent within the myofiber basal lamina, proliferate in response to muscle injury, and replace degenerating myofibers. In recent years, improvements in isolating satellite cells from muscle have been made, but the utility of satellite cells in cell-based therapy is severely limited by their poor expansion in culture and inability to cross the endothelial wall, prohibiting systemic delivery. Embryonic stem cells, hematopoietic stem cells, mesoangioblasts (MABs), and other myoblast progenitors may be able to circumvent these problems and are capable of differentiating into myofibers in vitro or in vivo [22,23]. MABs, isolated from blood vessel walls, have shown particular promise, leading to improved muscle regeneration and strength in animal models of muscular dystrophy when delivered intraarterially [24,25]. An improved understanding of the efficacy and safety of these stem cells will likely lead to clinical trials for cell-based therapy of skeletal muscle diseases in the near future.
Strategies to slow muscle degeneration
Although stimulating muscle regeneration shows promise in improving muscle strength in myositis patients, it does not address the underlying disease pathogenesis. In fact, in some patients, autoantibodies may recognize antigens that are enriched in regenerating fibers [26], making it possible that enhancing regeneration without suppressing inflammation could actually be counter-productive. In patients with immunotherapy-refractory myositis, such as sIBM, addressing the underlying pathogenic process is critical. Even in patients with immune-mediated myositis, there are clearly nonimmune mechanisms that cause muscle degeneration (reviewed in [2]).
Is inclusion body myositis a primary degenerative disease?
The mechanisms of pathogenesis of neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease have been intensely investigated over the last several decades, and more recently, some of these mechanisms have been studied in IBM (Fig. 1). sIBM shares features with Alzheimer’s disease including an association with aging and accumulation of amyloidogenic proteins. These pathologic features, along with the lack of responsiveness to steroids, have led to the hypothesis that sIBM is a primary degenerative disease with a secondary inflammatory response [27•]. However, there is an association of sIBM with specific HLA haplotypes, autoantibodies, and other autoimmune diseases. Furthermore, some patients have an overlap syndrome with polymyositis [28], and some sIBM patients with proximal limb weakness may be responsive to corticosteroids, at least early in the course of the disease.
Figure 1. Model for pathogenesis of sporadic inclusion body myositis – disruption of protein homeostasis.

This model shows an interrelation of pathological hallmarks in sporadic inclusion body myositis (sIBM) including vacuoles, muscle atrophy, degeneration, accumulation of protein aggregates, and inflammation (shown in boxes). The consequences of aging (circled as the ‘initiating’ factor) include reactive oxygen species (ROS) and accumulation of ubiquitinated protein aggregates, degenerating mitochondria, and other macromolecules. In this model, these accumulations are both directly toxic to myofibers and also induce inflammation via induction of NF-κB. Protein aggregates are normally degraded via the ubiquitin–proteasome system (UPS) and autophagy, both of which are induced by the transcription factor FOXO. Growth factors such as IGF-1 stimulate protein synthesis and inhibit autophagy via the PI3K/Akt/TOR pathway to enable muscle growth. Disruption of autophagolysosome degradation may lead to accumulation of these structures as ‘rimmed vacuoles’ in sIBM patients. Thus, autophagy may be tightly regulated in muscle cells such that it is induced during cellular stress to allow degradation of toxic aggregates and inhibited during cell growth to allow synthesis of structural proteins.
Thus, both the autoimmune and the degenerative hypotheses have merit, and both pathologic processes are likely involved in the progression of the disease.
One prominent hypothesis for the etiology of sIBM is that the disease is triggered by an accumulation of amyloid-beta peptide (Ab), analogous to the amyloid hypothesis in Alzheimer’s disease. In fact, a transgenic mouse ‘model’ of sIBM has been generated by over-expressing Ab precursor protein (APP) [29], and these mice are being used to study pathogenesis and new treatments for sIBM. In one such study [30], transgenic mice vaccinated with Ab led to amelioration of the disease phenotype, prompting suggestions for a clinical trial of the vaccine in sIBM patients. However, the Ab hypothesis is controversial [31,32•], and many other proteins have been found to accumulate in sIBM muscle. It is unknown whether Ab plays a role in the pathogenesis of sIBM or if it is merely one of many proteins that accumulate in diseased muscle.
Although the specific toxic molecule(s) that initiate the pathogenic cascade in IBM are unknown, oxidative stress associated with aging may trigger an accumulation of toxic macromolecules and organelles that is characteristic of IBM. As shown in Fig. 1, cells have several protective responses to these insults, such as upregulating chaper-ones to help refold misfolded proteins and inducing degradation of toxic compounds via autophagy or the ubiquitin–proteosome system (UPS) (reviewed in [33]). Furthermore, an accumulation of misfolded proteins within the endoplasmic reticulum (ER) causes an ER stress response that can trigger inflammation via NF-κB, induce autophagy, and suppress protein synthesis. Growth factors such as IGF-1 have the opposite effect via activation of the PI3K/Akt/Tor pathway. Thus, a complex homeostatic mechanism, similar to that seen in neurodegenerative diseases, regulates the cellular response to the deleterious effects of aging. The development of disease may be due to an imbalance in this homeostatic response triggering a feed-forward loop that leads to progressive degeneration.
One of the central questions in neurodegenerative diseases is whether ubiquitinated aggregates are toxic or beneficial; it may be different for each disease, but the leading hypothesis is that small oligomers are toxic and larger aggregates are likely cytoprotective. Primary targets for neurodegenerative disease therapy are agents that prevent protein aggregation. One class of proteins that function to prevent protein misfolding and aggregation are heat shock proteins, chaperones that are induced under conditions of cell stress. Arimoclomol is a drug that induces expression of heat shock proteins. In a mouse model of amyotrophic lateral sclerosis (ALS), arimoclomol slowed disease progression [34], prompting a clinicaltrial in ALS patients [35]. A phase I safety trial of arimoclomol in ALS showed that it is well tolerated and safe [36]. Phase II efficacy trials of arimoclomol are currently underway in sIBM and ALS and are expected to be completed soon.
Is inclusion body myositis caused by defective autophagy?
One of the most intensively investigated fields of neuro-degeneration research in recent years is that of autophagy. As shown in Fig. 2, autophagy is the cellular process whereby macromolecules or organelles are engulfed by a double-membrane ‘phagophore’ to form an ‘autophago-some’ that degrades the engulfed material upon fusion with the lysosome [37,38]. The rimmed vacuoles seen in IBM are believed to be an autophagolysosomal compartment [39,40]. Inhibition of autophagy in neurons is sufficient to cause ubiquitinated cytoplasmic aggregates and neurodegeneration [41,42]. Similarly, muscle-specific disruption of autophagy leads to muscle atrophy and accumulation of ubiquitinated aggregates, demonstrating that autophagy is critical for maintaining muscle mass [43,44•].
Figure 2. Role of autophagy in inclusion body myositis pathogenesis and potential therapeutic avenues.
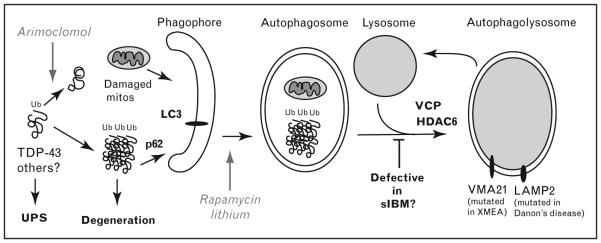
Myofiber degeneration in inclusion body myositis (IBM) is proposed to be due to toxic oligomers of misfolded proteins such as TDP-43. Arimoclomol is a drug being tried in IBM and neurodegenerative diseases due to its ability to induce expression of chaperones that help prevent accumulations of misfolded proteins. Normally, these ubiquitinated protein aggregates and damaged mitochondria (mitos) are degraded via the ubiquitin–proteasome system (UPS) or autophagy. Ubiquitinated aggregates may be recruited to the LC3 receptor on a newly forming double-membrane phagophore via the ubiquitin-binding protein p62/SQSTM1. Rapamycin and lithium both stimulate autophagy and help clear toxic aggregates such as TDP-43. VCP, mutated in familial IBM, functions in maturation of autophagosomes into an autophagolysosome. Overexpression of HDAC6 rescues the VCP phenotype, by stimulating autophagosome maturation. This step may be defective in sporadic inclusion body myositis (sIBM) as well, explaining the accumulation of rimmed vacuoles in addition to the numerous aggregated proteins. Upon fusion of autophagosomes with the lysosome, the ubiquinated aggregates and damaged mitochondria are degraded. A similar vacuolar pathology is seen in two rare inherited vacuolar myopathies in which myofiber degeneration is caused by defective lysosomal function termed XMEA (X-linked myopathy with ‘excessive’ autophagy, caused by loss of VMA21 function, required for lysosomal acidification) and Danon’s disease (caused by mutations in lysosome-associated membrane protein 2 (LAMP2) involved in lysosome fusion).
Recently, Askanas and colleagues have shown that autophagy is defective in sIBM [45••]. They determined that activity of lysosomal enzymes Cathepsin B and D are reduced in IBM, and this defect is specific for IBM, as enzymatic activity is actually increased in polymyositis. Furthermore, the autophagosome marker LC3-II is upregulated specifically in sIBM, consistent with the increase in autophagolysosomes seen in IBM muscle. Similarly, Temiz et al. [28] showed that LC3 is the most frequent protein seen in aggregates in IBM and in polymyositis with mitochondrial disorder. Interestingly, the receptor for LC3 on autophagsomes, p62, a ubiquitin-binding protein, also forms prominent inclusions specifically in sIBM [46•]. Thus, the accumulation of rimmed vacuoles in IBM may be due to a defect in lysosomal degradation of auto-phagosomal material, and this in turn may lead to the accumulation of ubiquitinated aggregates and other toxic macromolecules. Nogalska et al. [45••] propose that the decreased lysosomal enzyme activity in IBM is due to ER stress, as experimentally induced ER stress in cultured muscle fibers leads to a similar reduction in lysosomal enzymatic activity. Similar accumulations of autophago-somes are seen in the vacuolar myopathies XMEA (X-linked myopathy with excessive autophagy) and Danon’s disease, in which the defect is believed to be in lysosomal function [47,48]. Thus, a primary defect in autophagoly-somal function can directly explain many of the pathological hallmarks of IBM, namely rimmed vacuoles, ubiquitinated aggregates, and muscle degeneration.
Further evidence for defective autophagy in IBM has come from the study of valosin-containing protein (VCP), mutated in the familial disease IBMPFD (IBM, Paget disease of bone, and frontotemporal dementia). Two independent studies have shown that VCP is essential for autophagy, and in its absence, rimmed vacuoles accumulate, suggesting that VCP is required for maturation of autophagosomes to autophagolysosomes [49,50••]. Interestingly, overexpression of HDAC6, a neuroprotective protein also involved in autophagosome maturation, rescues the cellular phenotype in this model [51,52]. In addition to ubiquitinated aggregates and rimmed vacuoles, a primary defect in autophagy in IBM can explain the accumulation of abnormal mitochondria in many patients, a characteristic feature of the disease [53]. Mitophagy, a specialized form of autophagy, is important for degrading degenerating mitochondria. Accumulation of damaged mitochondria may amplify oxidative stress associated with aging. A recent study may shed light on one potential link between mitochondrial degeneration and inflammation, whereby mitochondrial proteins released from dying cells activate the innate immune response due to their ancestral bacterial origin [54,55].
Thus, stimulation of autophagy may accelerate the removal of toxic macromolecules and organelles in degenerative diseases. If autophagy is indeed defective in IBM, drugs that stimulate autophagy may be an important therapeutic intervention. Along this line, rapamycin (Sirolimus) may be a reasonable drug for clinical trials in IBM given its potent anti-inflammatory activity in addition to its ability to induce autophagy. Similarly, lithium is being tested in IBM, primarily for its ability to inhibit the kinase GSK-3 that phosphorylates tau and APP (phospho-tau is yet another abnormal inclusion in IBM), but it is also an inducer of autophagy [56].
Although increasing autophagy may help clear toxic molecules, it may also lead to autophagic cell death or atrophy. One of the pathways for inducing muscle atrophy in cachexia and aging is the FOXO-mediated pathway that stimulates autophagy and the UPS [57]. These pathways are believed to be therapeutic targets for preventing age-associated muscle atrophy. Thus, too much or too little autophagy can lead to muscle atrophy.
Is inclusion body myositis a TDP-43 proteinopathy?
The ubiquitinated aggregates and expanded autophagosomal vacuoles that are the pathological hallmarks of IBM suggest the possibility that a primary defect in autophagy leads to the accumulation of toxic macromolecular complexes or organelles. Many such protein aggregates have been identified in sIBM, as previously described (Ab, phospho-tau, LC3, and p62 to name a few), suggesting that IBM may be caused by a generalized defect in protein degradation. Several recent studies have added a new member to this list, the RNA-binding protein TDP-43. TDP-43 is the major ubiquitinated aggregate-forming protein found in many neurodegenerative diseases, referred to as TDP-43 proteinopathies, including ALS and frontotemporal lobar degeneration (FTD) (reviewed in [58,59•]). Recently, several groups have identified TDP-43 protein aggregates in IBM muscle fibers, and these aggregates are not seen in polymyositis or dermatomyositis [60,61,62••]. Greenberg et al. report that sarcoplasmic TDP-43 aggregates are seen in 23% of fibers, much more frequently than amyloid or phospho-tau, seen in less than 3% of fibers in their study [62••].
TDP-43 is normally nuclear, though it shuttles between the cytoplasm and nucleus. TDP-43 was initially discovered as a binding partner of the HIV-1 TAR terminal repeats, suggesting the intriguing possibility that the increased incidence of IBM in HIV patients may be related. In ALS and other TDP-43 proteinopathies, TDP-43 accumulates in the cytoplasm and is depleted from the nucleus. A central role of TDP-43 in the pathogenesis of ALS is suggested by the finding of autosomal dominant mutations in TDP-43 in familial ALS [63,64]. As in sporadic ALS, mutant TDP-43 is ubiquitinated and forms cytoplasmic aggregates, suggesting that cytoplasmic TDP-43 directly mediates toxicity. A central question in ALS and other TDP-43 proteinopathies is the mechanism of TDP-43-mediated toxicity.
A recent study [65•] suggests that TDP-43 is a major source of toxicity in IBMPFD. A genetic screen performed in a Drosophila model of IBMPFD identified the fly homolog of TDP-43 as a potent suppressor of VCP-mediated neurodegeneration. Epistasis experiments determined that TDP-43 toxicity functions downstream of VCP, suggesting that VCP-mediated degeneration is at least partially due to accumulation of TDP-43 in the cytoplasm. Importantly, clearance of TDP-43 accumulation in cell culture is accelerated with Rapamycin, suggesting that TDP-43 aggregates are removed by autophagy [66•].
Conclusion
Novel therapies to accelerate muscle regeneration and to prevent degeneration are on the horizon. Myostatin inhibition to accelerate muscle regeneration is a promising therapeutic approach for many muscle diseases including myositis, and clinical trials are underway. We are just beginning to understand the pathogenesis of IBM, and findings of defects in autophagy and accumulations of TDP-43 suggest that these may be new therapeutic targets. One limitation to clinical trials for IBM is a clear biomarker of disease, and future research should focus on identifying which of these new pathologic features correlate with disease activity.
Acknowledgements
T.E.L. is funded by the NIH (K08-NS062890) and The Robert Packard Center for ALS Research at Johns Hopkins. The author would like to thank Paul Taylor for his comments on this manuscript.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 000–000).
- 1.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1173–R1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henriques-Pons A, Nagaraju K. Nonimmune mechanisms of muscle damage in myositis: role of the endoplasmic reticulum stress response and autophagy in the disease pathogenesis. Curr Opin Rheumatol. 2009;21:581–587. doi: 10.1097/BOR.0b013e3283319265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3••.Dalakas MC. Immunotherapy of myositis: issues, concerns and future prospects. Nat Rev Rheumatol. 2010;6:129–137. doi: 10.1038/nrrheum.2010.2. This excellent review summarizes current and emerging immunotherapies for myositis. One of the key messages is that a common cause of ‘treatment failure’ in myositis is misdiagnosis. [DOI] [PubMed] [Google Scholar]
- 4.Rutkove SB, Parker RA, Nardin RA, et al. A pilot randomized trial of oxan-drolone in inclusion body myositis. Neurology. 2002;58:1081–1087. doi: 10.1212/wnl.58.7.1081. [DOI] [PubMed] [Google Scholar]
- 5.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 6.Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 7.Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 8•.Cadena SM, Tomkinson KN, Monnell TE, et al. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. 2010 doi: 10.1152/japplphysiol.00866.2009. [Epub ahead of print]. Soluble ActRIIB protein is a promising myostatin inhibitor that is in development by several companies for muscle diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SJ, Reed LA, Davies MV, et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci U S A. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogdanovich S, McNally EM, Khurana TS. Myostatin blockade improves function but not histopathology in a murine model of limb-girdle muscular dystrophy 2C. Muscle Nerve. 2008;37:308–316. doi: 10.1002/mus.20920. [DOI] [PubMed] [Google Scholar]
- 11.Bartoli M, Poupiot J, Vulin A, et al. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not alpha-sarcoglycan deficiency. Gene Ther. 2007;14:733–740. doi: 10.1038/sj.gt.3302928. [DOI] [PubMed] [Google Scholar]
- 12•.Rodino-Klapac LR, Haidet AM, Kota J, et al. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve. 2009;39:283–296. doi: 10.1002/mus.21244. This paper reviews the myostatin pathway and discusses the rationale for follistatin treatment in muscle diseases such as IBM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SJ. Quadrupling muscle mass in mice by targeting TGF-beta signaling pathways. PLoS One. 2007;2:e789. doi: 10.1371/journal.pone.0000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haidet AM, Rizo L, Handy C, et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci U S A. 2008;105:4318–4322. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Kota J, Handy CR, Haidet AM, et al. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci Transl Med. 2009;1 doi: 10.1126/scitranslmed.3000112. 6ra15. This important preclinical study demonstrates the potential of AAV-follistatin gene therapy in enhancing muscle regeneration. Cynomolgus macaques injected with this therapy into the quadriceps had significant and long-lasting increases in muscle size and strength without detectable adverse effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iezzi S, Di Padova M, Serra C, et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6:673–684. doi: 10.1016/s1534-5807(04)00107-8. [DOI] [PubMed] [Google Scholar]
- 18.Minetti GC, Colussi C, Adami R, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 19.Jones KL, Mansell A, Patella S, et al. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci U S A. 2007;104:16239–16244. doi: 10.1073/pnas.0705971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Tedesco FS, Dellavalle A, Diaz-Manera J, et al. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest. 2010;120:11–19. doi: 10.1172/JCI40373. This timely review nicely summarizes the field of muscle stem cell research, including recent clinical trials of muscle stem cells in muscular dystrophies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamanaka S. A fresh look at iPS cells. Cell. 2009;137:13–17. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 22.Barberi T, Bradbury M, Dincer Z, et al. Derivation of engraftable skeletal myoblasts from human embryonic stem cells. Nat Med. 2007;13:642–648. doi: 10.1038/nm1533. [DOI] [PubMed] [Google Scholar]
- 23.Darabi R, Gehlbach K, Bachoo RM, et al. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat Med. 2008;14:134–143. doi: 10.1038/nm1705. [DOI] [PubMed] [Google Scholar]
- 24.Sampaolesi M, Torrente Y, Innocenzi A, et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301:487–492. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- 25.Sampaolesi M, Blot S, D’Antona G, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574–579. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- 26.Mammen AL, Casciola-Rosen LA, Hall JC, et al. Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum. 2009;60:3784–3793. doi: 10.1002/art.24977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Karpati G, O’Ferrall EK. Sporadic inclusion body myositis: pathogenic considerations. Ann Neurol. 2009;65:7–11. doi: 10.1002/ana.21622. This outstanding review by the late George Karpati gives an unbiased assessment of what is known about the pathogenesis of sIBM. [DOI] [PubMed] [Google Scholar]
- 28.Temiz P, Weihl CC, Pestronk A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J Neurol Sci. 2009;278:25–29. doi: 10.1016/j.jns.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Sugarman MC, Yamasaki TR, Oddo S, et al. Inclusion body myositis-like phenotype induced by transgenic overexpression of beta APP in skeletal muscle. Proc Natl Acad Sci U S A. 2002;99:6334–6339. doi: 10.1073/pnas.082545599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitazawa M, Vasilevko V, Cribbs DH, et al. Immunization with amyloid-beta attenuates inclusion body myositis-like myopathology and motor impairment in a transgenic mouse model. J Neurosci. 2009;29:6132–6141. doi: 10.1523/JNEUROSCI.1150-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberg SA. Theories of the pathogenesis of inclusion body myositis. Curr Rheumatol Rep. 2010;12:221–228. doi: 10.1007/s11926-010-0102-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32•.Greenberg SA. How citation distortions create unfounded authority: analysis of a citation network. BMJ. 2009:339, b2680. doi: 10.1136/bmj.b2680. This article discusses how citation bias may have led to premature acceptance of the Ab hypothesis for IBM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirkin V, McEwan DG, Novak I, et al. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 34.Kieran D, Kalmar B, Dick JR, et al. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- 35.Lanka V, Wieland S, Barber J, et al. Arimoclomol: a potential therapy under development for ALS. Expert Opin Investig Drugs. 2009;18:1907–1918. doi: 10.1517/13543780903357486. [DOI] [PubMed] [Google Scholar]
- 36.Cudkowicz ME, Shefner JM, Simpson E, et al. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve. 2008;38:837–844. doi: 10.1002/mus.21059. [DOI] [PubMed] [Google Scholar]
- 37.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Arencibia M, Hochfeld WE, Toh PP, et al. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol. 2010 doi: 10.1016/j.semcdb.2010.02.008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumamoto T, Ueyama H, Tsumura H, et al. Expression of lysosome-related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol. 2004;107:59–65. doi: 10.1007/s00401-003-0774-2. [DOI] [PubMed] [Google Scholar]
- 40.Askanas V, Engel WK, Nogalska A. Inclusion body myositis: a degenerative muscle disease associated with intra-muscle fiber multiprotein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol. 2009;19:493–506. doi: 10.1111/j.1750-3639.2009.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 42.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:819–820. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 43.Raben N, Hill V, Shea L, et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17:3897–3908. doi: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. This study shows that disruption of autophagy specifically within myofibers causes muscle atrophy and degeneration. [DOI] [PubMed] [Google Scholar]
- 45••.Nogalska A, D’Agostino C, Terracciano C, et al. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol. 2010:177. doi: 10.2353/ajpath.2010.100050. Epub ahead of print. This elegant study shows that lysosomal enzyme activity is specifically reduced in IBM muscles and attributes this dysfunction to ER stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46•.Nogalska A, Terracciano C, D’Agostino C, et al. p62/SQSTM1 is over-expressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009;118:407–413. doi: 10.1007/s00401-009-0564-6. This study shows that p62, a ubiquitin-binding protein that may link protein aggregates to the autophagosome, is overexpressed and forms inclusions in sIBM muscle. [DOI] [PubMed] [Google Scholar]
- 47.Ramachandran N, Munteanu I, Wang P, et al. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell. 2009;137:235–246. doi: 10.1016/j.cell.2009.01.054. [DOI] [PubMed] [Google Scholar]
- 48.Hirano M, DiMauro S. VMA21 deficiency: a case of myocyte indigestion. Cell. 2009;137:213–215. doi: 10.1016/j.cell.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tresse E, Salomons FA, Vesa J, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6:217–227. doi: 10.4161/auto.6.2.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50••.Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187:875–888. doi: 10.1083/jcb.200908115. This study suggests that mutations in VCP that cause familial IBM do so by disrupting autophagy. These findings directly implicate defective autophagy in the pathogenesis of familial IBM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ju JS, Miller SE, Hanson PI, Weihl CC. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J Biol Chem. 2008;283:30289–30299. doi: 10.1074/jbc.M805517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JY, Koga H, Kawaguchi Y, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oldfors A, Moslemi AR, Jonasson L, et al. Mitochondrial abnormalities in inclusion-body myositis. Neurology. 2006;66:S49–S55. doi: 10.1212/01.wnl.0000192127.63013.8d. [DOI] [PubMed] [Google Scholar]
- 54.Manfredi AA, Rovere-Querini P. The mitochondrion: a Trojan horse that kicks off inflammation? N Engl J Med. 2010;362:2132–2134. doi: 10.1056/NEJMcibr1003521. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Terracciano C, Nogalska A, Engel WK, Askanas V. In AbetaPP-overexpressing cultured human muscle fibers proteasome inhibition enhances phosphorylation of AbetaPP751 and GSK3beta activation: effects mitigated by lithium and apparently relevant to sporadic inclusion-body myositis. J Neurochem. 2010;112:389–396. doi: 10.1111/j.1471-4159.2009.06461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59•.Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. This comprehensive review discusses what is known about the normal function of TDP-43 and FUS/TLS and how mutations might cause neurodegeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weihl CC, Temiz P, Miller SE, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79:1186–1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olive M, Janue A, Moreno D, et al. TAR DNA-binding protein 43 accumulation in protein aggregate myopathies. J Neuropathol Exp Neurol. 2009;68:262–273. doi: 10.1097/NEN.0b013e3181996d8f. [DOI] [PubMed] [Google Scholar]
- 62••.Salajegheh M, Pinkus JL, Taylor JP, et al. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 2009;40:19–31. doi: 10.1002/mus.21386. This paper suggests that sarcoplasmic accumulations of TDP-43 are the most common abnormality seen in muscle fibers of patients with sBIM. These data suggest that TDP-43 aggregates are a sensitive and specific biomarker of this disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 65•.Ritson GP, Custer SK, Freibaum BD, et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010;30:7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010. This elegant genetic study implicates TDP-43 in VCP-mediated degeneration, suggesting that TDP-43 plays a role in the pathogenesis of familial IBM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66•.Caccamo A, Majumder S, Deng JJ, et al. Rapamycin rescues TDP-43 mis-localization and the associated low molecular mass neurofilament instability. J Biol Chem. 2009;284:27416–27424. doi: 10.1074/jbc.M109.031278. This study suggests that cytoplasmic TDP-43 is normally degraded via autophagy, and supports the hypothesis that accelerating autophagy may alleviate TDP-43-mediated toxicity. [DOI] [PMC free article] [PubMed] [Google Scholar]