

Abstract
Primary cilia are essential cellular organelles projecting from the cell surface to sense and transduce developmental signaling. They are tiny but have complicated structures containing microtubule (MT)-based internal structures (the axoneme) and mother centriole formed basal body. Intraflagellar transport (Ift) operated by Ift proteins and motors are indispensable for cilia formation and function. Mutations in Ift proteins or Ift motors cause various human diseases, some of which have severe bone defects. Over the last few decades, major advances have occurred in understanding the roles of these proteins and cilia in bone development and remodeling by examining cilia/Ift protein-related human diseases and establishing mouse transgenic models. In this review, we describe current advances in the understanding of the cilia/Ift structure and function. We further summarize cilia/Ift-related human diseases and current mouse models with an emphasis on bone-related phenotypes, cilia morphology, and signaling pathways.
Keywords: Ift proteins, Ift motors, Ciliopathies, Mouse models, bone, ENU, Gene trap, Knockout, Review
2. INTRODUCTION
Normal vertebrate skeletons, composed of cartilage and bone, are critical for locomotion, respiration, and protection of the vital organs (1). Both cartilage and bone are derived from embryonic mesenchyme (2, 3). Hyaline cartilage is the most widespread cartilage and prevails in the fetal skeleton, while bone is predominant at a later stage and persists postnatally throughout life (1). Most bones are formed by replacement of existing cartilage rudiments. This process is known as endochondral bone formation, which starts with embryonic mesenchymal cells condensing at the sites where the skeletal elements will form. The condensed mesenchymal cells result in the formation of the cartilaginous anlagen, which later is ossified to become bone by osteoblasts (4–6). In contrast, mesenchymal condensations that prefigure the clavicles, mandible, and certain bones of the skull, directly differentiate into osteoblasts. This process does not require a cartilaginous template and is called intramembranous ossification (1–3). Skeletal formation and homeostasis are strictly controlled by systemic and local regulators, such as Wnt, Hedgehog (Hh) (7), Notch (8), bone morphogenetic protein 2 (BMP2) (9), and thyroid hormones (10, 11). It has uncovered over 300 genetic diseases with disorders in the growth and development of the skeleton (12). Bone development abnormalities often lead to bony deformities that severely impact patients’ quality of life and general health and sometimes lead to premature death (1, 2, 13, 14). Among these genetic diseases, a certain groups are caused by disorders of the primary cilia (15).
Primary cilia are small microtubule (MT) based organs that are projected from the surface of nearly every mammalian cells and function as antennae. They sense a diverse range of extracellular signals, such as growth factors and hormones, through various surface receptors localized on the ciliary membrane, and transmit the signals back into the nucleus. Thus cilia are important for embryonic and postnatal development of many vital organs (16–18). Defects of cilia or cilia-related protein cause a wide variety of human disease symptoms, including polycystic kidneys, hydrocephalus, vision and hearing loss as well as severe bone abnormalities, suggesting that the role of cilia in multiple organ development is indispensable (18–25).
Intraflagellar transport (Ift), a bidirectional transport system run by Ift protein complexes and Ift motors, is found to be essential for building cilia and maintaining cilia function (26, 27). Ift protein complexes are formed with complex A and complex B containing about 20 Ift proteins (28). Mutations in Ift proteins (such as Ift80, Ift122, and Ift43) or Ift motors (such as DYNC2H1) usually lead to cilia defects and human diseases (29–31). Those groups of human diseases with primary cilia malfunction display disorders of multiple organs, including kidney, liver, brain, and limb (30) and are called ciliopathies (29, 32). A certain portion of ciliopathies, such as Jeune asphyxiating thoracic dystrophy (JATD) and short rib polydactyly syndromes (SRP), result in several bone defects, suggesting the important role of cilia in bone development (29, 33–35).
During the 1960s, scientists found cilia present in chondrocytes (36). Subsequent work has revealed that cilia are also present in osteoblasts, osteocytes, and mesenchymal stem cells (37–41). Bone is a sensitive and dynamic tissue that constantly remodels to meet the demands of its environment (42, 43). Cilia are considered to function as chemosensors and mechanosensors for bone formation and maintenance (43–45). Multiple bone developmental signaling pathways, such as Hh and Wnt signaling pathways, have been linked to primary cilia, Ift proteins and motors (46–49). Both Indian hedgehog (Ihh) and Sonic hedgehog (Shh) are required for skeletal development (50–53). Ihh is an important signaling molecule in endochondral ossification because Ihh null mice display shortened face and tail with extremely short limbs and narrow rib cages (51). Shh is required for the early stage of patterning of the limb (54, 55). Genetic screens for embryonic patterning mutations first uncovered the connection between mammalian Hh signaling and primary cilia (56). Since then, the Hh pathway has become the most intensively studied pathway among the cilia-related signaling pathways (see review (57) and (58)). It is now clearly established that the core components of Hh signaling pathway, including Hh receptor Patched 1 (Ptch1), Smoothened (Smo) protein, and Gli transcription factors are localized to cilia (57). Ptch1 is found at the base of primary cilia in the absence of Hh. Upon exposure to Hh, Ptch1 moves out of cilia (59, 60), and then Smo moves into the cilia and shifts the Gli process from a transcriptional repressor to transcriptional activator (61). Functional intact cilia are required for the Hh signaling transduction, and mutations in cilia/Ift proteins and motors disrupt Hh signaling pathway. However, mutations in different genes lead to different phenotypes, suggesting the distinct roles of individual Ift proteins and motors in Hh signaling or their involvement of other signaling pathways (57). Wnt pathway is another critical pathway for skeletal development and homeostasis (62, 63). However, the relationship between Wnt and primary cilia is controversial (58, 64). Wnt signaling cascades can be broadly divided into two general categories: 1) canonical Wnt/β-catenin pathway (stabilizing the transcription of co-activator β-catenin and therefore turning on gene expression) and 2) the non-canonical Wnt/planar cell polarity (PCP) pathway (65). Different mutations in cilia/Ift proteins cause different changes in Wnt signaling pathway (64, 66, 67). However, the role of cilia/Ift proteins and motors in Wnt signaling transduction remains largely unknown.
To study the fundamental biology of cilia/Ift proteins and motors, various animal models, including tetrahymena thermophila, zebrafish, and mouse, have been developed (68–70). Mouse is genetically and pathophysiologically similar to human and relatively easy to be genetically manipulated. Therefore, mouse has become the favorite and reliable mammalian model for studies of bone biology (71, 72). Within the last decade, significant progress in the understanding of the cilia/Ift proteins and motors in bone development and diseases has been made using various mouse genetic models and we summarize those advances here.
3. STRUCTURE OF IFT PROTEINS/MOTORS AND CILIA
The structure of the cilia is conserved throughout evolution. When cell is in G0 stage, primary cilium is generated from a “mother” centriole of the centrosome, which becomes the basal body in the cilia structure (73). Centriole is a cylinder comprising nine triplet MTs that provide the template for the formation of an MT-based internal structure (the axoneme) (74). Two of the most common axoneme architectures are “9 + 2” for motile cilia (9 peripheral MT doublets and 2 single central MTs) and “9 + 0” for primary cilia (only 9 peripheral MT doublets) (75). Since ribosome and membrane vesicles are absent from the cilia, ciliary proteins have to be synthesized in the cytoplasm and actively transported into the cilia along MTs (73). This highly conserved process is called Ift (76), which moves macromolecules from the base to the tip of a cilium (anterograde transport) and from the tip of the cilium back to the cell body (retrograde transport) (77). Effective Ift involves the Ift complexes and Ift motors. It is believed that the heterotrimeric kinesin-II motor protein drives the anterograde transport (78), whereas cytoplasmic dynein-2 moves the rafts back in the retrograde direction (79).
Ift complexes serve as adaptors to mediate the contacts between cargo proteins and motors (80). Ift was first reported in Chlamydomonas in 1993 by Kozminski et al (76, 81), and later Ift particles were identified in Chlamydomonas by biochemical purifications (82, 83). By biochemical purification, Cole et al first reported that Ift complex has two sub-complexes named complex A (Ift-A) and complex B (Ift-B) (82). Subsequently, four proteins in complex A (Ift144, Ift140, Ift139 and Ift122) and eleven proteins in complex B (Ift172, Ift88, Ift81, Ift80, Ift74, Ift72, Ift57/55, Ift52, Ift46, Ift27 and Ift20) have been identified (82). The proteins were named according to their size (in kD). Later, Ift43 and then Ift121 were identified in the complex A (82, 84, 85). Thus complex A is thought to be composed of six subunits including Ift144, Ift140, Ift139, Ift122, Ift43 and Ift121.
Ift-B complex is more complicated and contains 14 known proteins (Ift20, Ift22, Ift25, Ift27, Ift46, Ift52, Ift54, Ift57, Ift70, Ift74/Ift72, Ift80, Ift81, Ift88 and Ift172) (18, 82, 85, 86). Lucker et al used increased ionic strength to dissociate the Ift-B of Chlamydomonas reinhardtii and found that the biochemically intact core of Ift-B contains Ift88, Ift81, Ift74/72, Ift52, Ift46, and Ift27 (87). Ift72 and Ift74 are derived from the same gene and present in a 1:1 stoichiometry. So it is usually combined into a single term, Ift74/72 (87, 88). The other subunits including Ift172, Ift80, Ift57, and Ift20 are not required for the core and can be disassociated with the core by using a high salt concentration. Among those core proteins, Ift81 and Ift74/72 are believed to form a tetrameric complex to serve as a scaffold for the interaction with other subunits (87).
All of the Ift proteins are conserved in mammals. Ift proteins are typically found along the cilia and in the base of the cilia (89). Anterograde transport is regulated by Ift-B, while the retrograde transport is related to Ift-A (28, 90). Mutations in Ift-B usually block cilia assembly (91, 92), similar to kinesin-II mutation (93, 94), while defects in Ift-A protein(s) typically result in malformed cilia with accumulation and function defects of Ift proteins (95–97) that resemble the mutations in cytoplasmic dynein-2 (48) (Figure 1).
Figure 1.
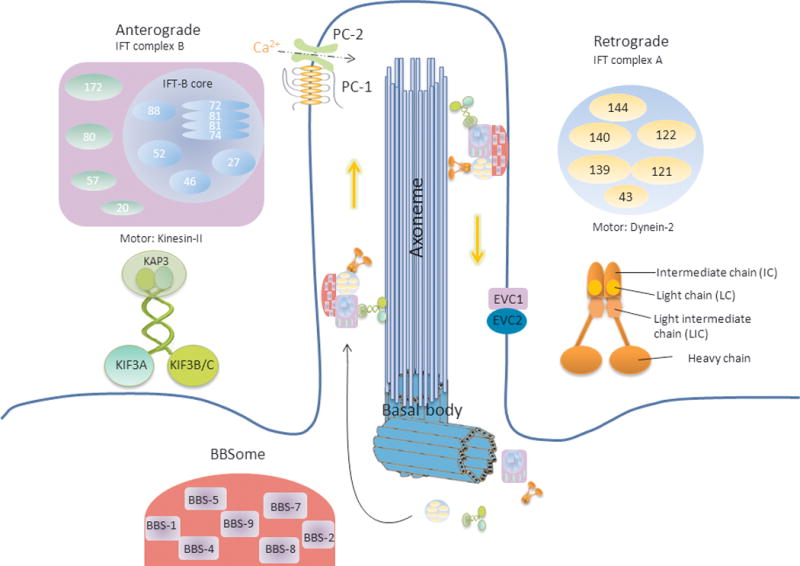
The primary cilium and ciliary proteins (adapted from (28, 87, 234, 235)). The structure of primary cilia is conserved throughout evolution with a basal body (“mother” centriole) and 9 peripheral MT doublets (the axoneme). The Ift-A contains 6 known proteins (Ift144, Ift140, Ift139, Ift122, Ift121 and Ift43) and in charge of retrograde transport (from ciliary tip back to the cell body) with cytoplasmic dynein-2 as motor. Ift-B contains 14 known proteins (Ift20, Ift22, Ift25, Ift27, Ift46, Ift52, Ift54, Ift57, Ift70, Ift74/Ift72, Ift80, Ift81, Ift88 and Ift172) and is involved in anterograde transport (from the base to ciliary tip) with heterotrimeric kinesin-II as motor. Ift81 and Ift74/72 form a tetrameric complex and interact with Ift88, Ift52, Ift46 and Ift27 formed the core of complex B. Besides Ift motors and Ift complexes, BBS proteins are also important for cilia formation and maintenance. Seven highly conserved BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8 and BBS9) formed BBSome. Evc and Evc-2 are located at the basal body and the base of the axoneme. Other proteins like OFD, NEK, Nephrocyustins and MKS proteins are found localized in the basal body. And mutation of these proteins caused diseases.
Besides Ift motors and Ift complexes, sixteen identified Bardet–Biedl syndrome proteins (BBS1-16) are also important for cilia formation and maintenance. Seven highly conserved BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8 and BBS9) and one newly defined interacting protein BBIP10 formed BBSome (98, 99). BBSome is indispensable for ciliary membrane biogenesis, microtubule stability, and acetylation (99). Other BBS proteins have a variety of functions including guanosine triphosphate hydrolases (GTPase) and ubiquitin ligase (See review (100)).
A large amount of cargo proteins have been found in the cilia or associated with cilia structures (75). Those proteins include the signaling components such as Gli and Smo in the Hh signaling pathway. Some of them are proteins located at a specific region of the cilium to regulate cilia formation or cilia-related signaling transduction such as Ellis-van Creveld syndrome protein homolog (Evc) and oral-facial-digital syndrome 1 (OFD1) (101, 102). Mutations of those proteins also cause ciliopathies.
4. CILIA/IFT PROTEIN AND MOTOR-RELATED HUMAN DISEASES
Cilia were first found in the kidneys and the thyroid gland one century ago (103). Subsequently, cilia have been found in many other organs and cell types (31). Further work revealed a dozen human genetic diseases caused by cilia-related gene mutation, which are commonly called as ciliopathies (31, 104). Those disorders includes BBS, OFD, Meckel-Gruber syndrome (MKS), Joubert syndrome (JBTS)/JBTS-related disorder (JSRD), JATD, Leber congenital amaurosis (LCA), Primary ciliary dyskinesia (PCD)/Kartagener syndrome (KS), Ellis-van Creveld syndrome (EVC), Nephronophthisis (NPHP), Senior-Loken syndrome (SLS), autosomal dominant and recessive polycystic kidney disease (ADPKD and ARPKD), Usher syndrome (US), and Alström syndrome (ALMS) (104, 105). Those ciliopathies share common clinical features and involve many major organs, including kidney, brain, limb, retina, liver, and bone (29, 32, 104, 105).
More than 30 different genes have been linked to the skeletal-related ciliopathies. These are dynein-2 motor (Dync2h1), Ift genes (Ift80, Ift122, Ift43, Ift140, WDR35, WDR19, and TTC21B) as well as the genes responsible for the basal body (NEK1, Evc, Evc2, BBS1-16, OFD1, MKS1, MKS3, TMEM216, CEP290, RPGRIP1L, CC2D2A, TCTN2, B9D1, B9D2, and NPHP3). Mutations of those genes result in common skeleton defects, including polydactyly, shortened bones, and craniofacial dysmorphology (Table 1) (29). Here, we only summarize the ciliopathies with bone phenotypes and their underlying genetic defects.
Table 1.
Currently known genes that are involved in skeletal ciliopathies
| Disease | Ift complex A | Ift complexes B | Ift Motors | others | References |
|---|---|---|---|---|---|
| JATD | TTC21B, WDR19, Ift140 | Ift80 | DYNC2H1 | (35, 108, 109, 111, 233) | |
| MSS | Ift140 | (111) | |||
| BBS | BBS1-BBS16 | (114) | |||
| EVC | Evc and Evc2 | (117–120) | |||
| WAD | Evc2 | (121) | |||
| OFD | OFD1 | (126) | |||
| SRP type II | DYNC2H1 | NEK1 | (128, 131) | ||
| SRP type III | Ift80 | DYNC2H1 | (35, 130) | ||
| SRP severe type | WDR35 | (34) | |||
| CED | Ift122, Ift43, WDR35, WDR19 | (109, 132, 135–137) | |||
| MKS | BBS2, BBS4, BBS6, MKS1/FLJ20345, MKS3/TMEM67, TMEM216, CEP290, RPGRIP1L, CC2D2A, TCTN2, B9D1, B9D2, and NPHP3 | (114) |
Jeune asphyxiating thoracic dystrophy (JATD), Intraflagellar transport (Ift), Mainzer-Saldino-syndrome (MSS), Bardet-Biedl syndrome (BBS), Ellis-van Creveld syndrome (EVC), WAD (Weyers acrofacial dysostosis), oralfacialdigital syndrome (OFD), short rib polydactyly syndromes (SRP), cranioectodermal dysplasia (CED), Meckel syndrome (MKS)
4.1. JATD and Mainzer-Saldino-Syndrome (MSS)
JATD, also known as asphyxiating thoracic dystrophy (ATD) was first reported in 1955 by Jeune et al. The patient with JATD had a narrow thoracic cavity and multiple cartilage anomalies, and died due to respiratory insufficiency during the early neonatal period (106). JATD is a rare autosomal recessive skeletal dysplasia with short limbs, narrow thorax, trident acetabular roof, and occasional polydactyly (107). Studies have suggested that mutations of Ift80, DYNC2H1, TTC21B (encoding Ift139) and WDR19 (encoding Ift144) are associated with JATD (33, 35, 40, 108–110). Respiratory failure is the usual cause of death in JATD in early infancy (110). MSS is related to JATD but has a milder rib phenotype and additional renal and eye defects (111).
Very recently, Ift140 mutation was identified in JATD and MSS (111, 112). It has been found that Ift140 patients display mild chest narrowing, end-stage renal failure, and ocular dysfunction (112). Ciliary abundance and localization of IFT-A proteins are changed in the fibroblasts from Ift40 mutant patients (111).
4.2. BBS
BS is a pleiotropic genetic disorder with the phenotypes of retinal dystrophy, obesity, post-axial polydactyly, renal dysfunction, learning difficulties and hypogonadism (113). Polydactyly, hepatic fibrosis, and kidney, genital and heart malformations can be observed at birth (114). Sixteen BBS gene (BBS1 to BBS16) mutations are account for 80% of BBS. The majority of mutations occur in BBS1 and BBS10 (114).
4.3. EVC and Weyers acrofacial dysostosis (WAD)
EVC was first reported by Richard Ellis and Simon van Creveld in 1940. Patients had retarded growth, narrow rib cage, supernumerary fingers and toes, orofacial abnormalities and heart abnormalities (115, 116). WAD was named after Helmut Weyers, who reported it in 1952. EVC and WAD are allelic disorders, but WAD has less severe symptoms and a different pattern of inheritance (117). They are both caused by the loss-of-function mutations of Evc or Evc2 genes (117–121). Valencia et al further domonstrated that Hh signal transduction is impaired when a mutant murine Evc2 Weyer variant is expressed in NIH 3T3 cells (121).
4.4. OFD
OFD is an X-linked dominant inheritance disorders with 13 different described types (type I to type XIII) classified by their patterns of signs and symptoms (122, 123). The phenotype includes oral, facial, and digital anomalies (124, 125). OFD type I, which is also known as Papillon-Leage and Psaume syndrome, has the higheset incidence (122). Ferrante et al indentified the responsible gene OFD1 (formerly named Cxorf5/71–7a) for this syndrome in 2001 (126). OFD1 is found missense, nonsense, splice, and frameshift mutation in OFD1 patients (126). Further studies showed OFD1 is a centrosomal protein and localized in the basal body of primary cilia (102, 127). Whether OFD1 mutation is responsible for other types of OFD is not clear now.
4.5. SRP
SRP is a group of lethal skeletal dysplasia that are characterized by short limbs, narrow thorax, polydactyly, and multiple anomalies of major organs (128). Four lethal types have been described based on radiological findings, including Saldino-Noonan syndrome (type I), Majewski syndrome (type II), Verma-Naumoff syndrome (type III) and Beemer-Langer syndrome (Type IV) (34, 128). In addition, JATD and EVC are phenotypically related to SRP but are compatible with life. Recently, Ho et al suggested that JATD and SRP type III are variants of the same disorder and SRP type III has more severe phentype in bone and other organs (129). Mutation of Ift80 or DYNC2H1 causes those diseases (35, 70, 130). Most recently, mutation of either DYNC2H1 or Never-in-mitosis Kinase 1 (NEK1), which regulates cell cycle–associated ciliogenesis, was identified in SRP type II (128, 131). A severe form of SRP was reported with in-frame homozygous deletion of exon 5 in WDR35 (orthologous of Ift121 in mammals) (34).
4.6. Cranioectodermal dysplasia (CED, or sensenbrenner syndrome)
CED is a rare autosomal recessive skeletal dysplasia (132). The syndromes of CED patients are similar to JATD patients as they have short stature, limbs and ribs, narrow chest, brachydactyly, renal failure, and hepatic fibrosis (132). They also show dolichocephaly with frontal bossing and craniosynostosis (133, 134). CED is genetically heterogeneous and is caused by mutations in Ift122 (132), Ift43 (135), WDR35 (136, 137) or WDR19 (109).
4.7. Meckel syndrome (MKS, or Meckel–Gruber Syndrome, Gruber Syndrome, Dysencephalia Splanchnocystica)
MKS is a severe fetal developmental disorder characterized by occipital encephalocele, cystic kidney dysplasia, fibrotic changes of the liver and polydactyly (138). MKS is genetically heterogeneous and at least ten genes have been associated with MKS, including MKS1(Meckel syndrome, type 1)/BBS13 (encoding MKS1, localized at the ciliary body), MKS3/TMEM67 (Transmembrane protein 67, encoding MECKELIN, which localized to the cilium base), TMEM216 (encoding TMEM216, which localized to the base of the primary cilia), CEP290/BBS14 (encoding centrosomal protein CEP290/nephrocystin-6, which is involved in chromosome segregation and localized around the centrosome), RPGRIP1L (encoding RPGRIP1L, which colocalizes to the basal body and centrosomes), CC2D2A (encoding CC2D2A, which localizes to basal body and the transition zone), TCTN2 (encoding TCTN2, which localizes to transition zone), B9D1 (encoding B9D1, which localizes to the transition zone complex), B9D2 (encoding B9D2, which localizes to the transition zone complex), and NPHP3 (nephrocystin-3, encoding NPHP3, which localizes to the primary cilia) (139–144).
Karmous-Benailly discovered mutations in BBS2, BBS4, and BBS6 in MSK patients. They suggested that prenatal presentation of BBS may mimic MKS (114).
5. FUNCTION OF IFT PROTEINS OR MOTORS IN EXISTING MOUSE MODELS
To deeply understand the function of cilia-related proteins, a variety of mouse genetic models have been generated in which Ift proteins or motors are inactivated by using different genetic mutant approaches. To make the summary of Ift proteins and motors clearly understood, we first briefly introduce the most commonly used techniques to generate gene mutation models in mice as described in section 5.1., and then further summarize the published mouse models that have mutations in Ift proteins and motors. We focus on the bone-related phenotypes, ciliogenesis, and altered signaling pathways.
5.1. Methods to generate gene mutational mouse models
There are two main methods to generate gene mutational mouse models. One is directed and disease driven; and the other is non-directed, randomly generated mutation. The common directed techniques include most powerful method of transgenesis. The non-directed, random mutation-driven method is the application of radiation and chemicals such as N-ethyl-N-nitrosourea (ENU) to induce mutations (145).
Transgenesis can be accomplished by two methods. One is pronuclear injection of fertilized oocytes, which is predominantly used to overexpress endogenous or mutant proteins. The other approach is using homologous recombination to modify embryonic stem cells (ESCs). The modified ESCs are then injected into mouse blastocysts to create mutant mice (knockout mice) (146, 147). Knockout mouse models can be divided into two categories: conventional and conditional knockout. In the conventional knockout model, the target gene is permanent inactivation in all cells of the body. However, this usually causes prenatal death, which makes identification of phenotype difficult. The development of the Cre-LoxP recombination system for conditional gene knockout limits the deletion of a target gene to the desired tissue and has dramatically improved knockout mouse studies (148, 149). Osteoblast-, osteoclast-, chondrocyte-, and osteocyte-specific Cre expression has been created with various promoters to turn on the Cre expression at various differentiation stages and in different cell lineages (See reviews (150) and (72)). Although transgenic mouse models provide a powerful approach to study gene function, the Cre-LoxP recombination system is not perfect. The generation of these models is time and money consuming. Moreover, Cre expression leaking to other cell types and the toxicity of high Cre levels are still the issues that need to be resolved (148, 149, 151).
ENU-induced mutation is one of those approaches that allow genome-wide, high-throughput research (146). ENU is the most potent germ line mutation inducer in rodents (152). ENU treatment of male mice results in random point mutations in the main targets of ENU in the spermatogonial stem cells. By breeding these male mice with normal female mice, the generated F1 population carries unique random ENU-induced mutations. Both genotype-driven and phenotype-driven approaches can be used for screening (146). In addition, the point mutation introduced by ENU resembles the most common form of human genetic variation (146), which makes it a powerful tool to study the specific genes. Large ENU screens have already yielded several mutations in Ift-related genes showing skeletal abnormalities.
Gene trapping is another technique that randomly generates loss-of-function mutations, which takes the middle path between random (such as ENU induced) and molecularly defined mutations (such as knockout mutation) (153). Gene trapping is performed with gene-trap vectors, which usually contain a promoterless reporter and/or selectable marker gene and a downstream polyadenylation sequence. When an expressed gene is inserted into the gene-trap vector, the reporter/selectable marker gene is expressed with the upstream exons, but the gene is prematurely terminated due to the inserted polyadenylation sequence (153, 154). In this way, the gene inactivation and transcriptional activity report can be achieved at the same time. The libraries of ESC lines containing mutations in single genes have been created and about 70 percent of protein-coding genes in the mouse genome have been inactivated in this way (154).
5.2. Mouse models of Ift complex A proteins
Currently, there are six known Ift-A proteins, which are Ift139, Ift144, Ift122, WDR35, Ift143 and Ift140. The in vitro and in vivo studies of these proteins are described in the following sections (Table 2).
Table 2.
Current mouse models to study the function of Ift-A proteins in bone development
| Gene | Mouse models | Mutation | Cilia | Phenotype | Pathways | Ref |
|---|---|---|---|---|---|---|
| Ift139 | Ift139alien | ENU mutagenesis, complete loss-of-function | Short and bulb-like structure cilia with accumulation of complex B proteins in the cilia tip | Defects in limbs, eyes, skull and brain development. | Overactivated Shh pathway Ift139 is the negative regulator in the Shh pathway downstream of Smo and upstream of Gli2, no significant change in Wnt pathway. |
(95, 96) |
| Ift122 | Ift122- null | Exon 1–3 mutation of MED1, null mutation | Cilia loss in the homozygous and malformed cilia in heterozygous | Usually die between E10.5 to E13.5. Exencephaly, delay in limb development, defects of the ventral portion of the head, rostral neural tube, eye and branchial arches. | Impaired Shh due to reduced Gli2/Gli3 and Gli3 repressor functions. | (97) |
| Ift122sopb | ENU- mutagenesis, complete loss-of-function | Short and swollen cilia with complex B proteins accumulation | Die around E13.5. Neural defects, pre-axial polydactyly, enlarged bronchial arches, and ocular defects. |
Enhanced Shh with Gli2 and Gli3 accumulation in the cilia tips but absent of TULP3 in cilia. | (158) | |
| Ift144 | Ift144twt | ENU- mutagenesis, partial loss-of-function | Relatively normal cilia structure but reduced frequency | Die at E11.0. Polydactyly, small rib cages, cleft palate and severe craniofacial anomalies. |
Ift144twt MEFs respond weakly to stimulation with upstream Hh agonists, a general increase or ectopic activation of Hh in Ift144twt embryos, particularly in the facial prominences and limbs. | (160) |
| Ift144dmhd | ENU- mutagenesis complete loss-of-function | Short and had highly disrupted axoneme | Die after E13.0. Polydactyly and craniofacial abnormalities. Smaller forelimb paddle, with truncation in the PD and to a lesser extent the AP axes. |
Less activation of the Shh pathway in the caudal neural tube. | (160, 161) | |
| Ift144twtIft122sopb | Double mutation | Similar to Ift144dmhd | Similar to Ift144dmhd. | Less Shh pathway activity. | (161) | |
| Ift144twtDync2h1mmi | Double mutation | Reduced cilia number, cilia swollen in Dync2h1mmi | Partially rescued most ventral neural cell type, and the floor plate. | Partial rescue of Shh signaling. | (161) | |
| WDR35 (Ift121) | WDR35yet | ENU- mutagenesis, complete loss-of-function | Cilia loss with Ift88 accumulated in the basal body | Usually die at E12.5. Severe defects in cardiovascular, embryo turning, and limb outgrowth and patterning. Failure of the somite derivatives. |
Not reported | (34) |
| WDRtm2a | “targeted trap” null allele | Not reported | Phenocopies WDR35yet. | Not reported | (34) | |
| Ift140 | Ift140null | Germline deletion with C57Bl/6 Prm-Cre | Not reported | Die at mid-gestation. | Not reported | (162) |
| Ift140cauli | ENU- mutagenesis, “possibly damaging” mutation | Cilia morphology was severely disrupted with broader and bulbous appearance | Start to die at E13.5. Exencephaly, anophthalmia, severely disorganised ribs with extensive exostoses, vertebral and palatal defects, agenesis/hypoplasia of the craniofacial skeleton, and polydactyly of the hindlimbs. |
Ectopic Hh signaling activation and reduced Gli3 repressor production. | (163, 164) | |
| Ift140−/− | Germline deletion with CMV-Cre | Not reported | Similar to Ift140cauli. | Not reported | (164) |
5.2.1. Ift139
Ift139, also termed THM1 (Tetratricopeptide Repeat Containing Hedgehog Modulator 1), is a newly identified ciliary protein (96). The gene encoding this protein is named TTC21B (tetratricopeptide repeat domain 21B), or Thm 1(tetratricopeptide repeat-containing hedgehog modulator-1) (96). Ift139 contains 1,317 amino acids (about 150 kDa) and is predicted to contain 11 tetricopeptide repeat (TPR) domains (96). Davis et al reported that the TTC21B mutation in humans causes JATD and isolated NPHP. By performing in vitro and in vivo complementation assays, they further found there is a significant enrichment of pathogenic alleles in ciliapathy cases, suggesting that TTC21B mutation accounts for 5% of ciliopathies cases (108).
Ift139 is highly expressed in the maxillary prominence, branchialarches, limb buds, somites, and spinal cord at embryonic day 10.5. (E10.5). A null mutant of TTC21B/Ift139 named alien (aln) (Ift139alien) in A/J mice by ENU mutagenesis (95, 96) results in embryonic defects in limbs, eyes, skull, and brain development (95), which are correlated with its expression pattern. In vivo and in vitro studies showed that Ift139 accumulates in cilia and colocalizes with acetylated α-tubulin, which is similar to other Ift proteins such as Ift88 (96). Cilia in the Ift139alien limb cells display short and bulb-like structures in the distal tips. Interestingly, Ift88 largely accumulates at this bulb-like structure while undetectable at the proximal ends (96). However, Smo and full-length Gli proteins (key factors of Hh pathway) still can be transported into cilia. Those data suggest that mutation of Ift139 only impairs retrograde Ift, which leads the accumulation of complex B proteins in the cilia tip and forms the bulb-like structure (96).
Further studies show that Shh pathway was inappropriately activated in Ift139alien and that overall phenotype of Ift139alien was very similar to Rab23 (a negative modulator of Shh signaling) mutants (96). The phenotype of Smo null mice was partially rescued by Ift139 deletion. Additionally, Gli2 was overactive in Ift139 mutant, and the double mutant of Gli2 and Ift139 resembled the phenotype of Gli2 mutant. Those studies suggested that Ift139 is a negative regulator in Shh pathway downstream of Smo and upstream of Gli2 (96). Consistence to this study, Stottmann et al reported that Shh signaling was up-regulated in the rostral portion of the Ift139alien embryo, and the reduction of Shh ligand level (heterozygous for a null allele of Shh) partially rescued the phenotype (95). Besides, the levels of the β-catenin protein were not significantly altered in the mutant mice (95).
5.2.2. Ift122
Ift122 has a molecular weight of approximately 140 KD and contains seven WD40 domains at the N-terminus (132, 155). The missense mutation of Ift122 is found in CED patients, and the fibroblasts from CED patients had significantly reduced both the numbers of primary cilia in and their length (132). Currently, two Ift122 mutant mouse models have been generated (97, 156). One mouse model named Ift122-null was generated by deletion of exon 1–3 of DNA repair gene-mediator of RNA polymerase II transcription subunit 1 (Med1). Due to the genomic overlap of Ift122 and Med1, mutation in Ift122-null abrogates the expression of both Med1 and Ift122. (97, 157). Ift122-null embryos had defects as early as E9.5 and died from E10.5 to E13.5 due to heart defect and diffuse hemorrhagic lesions. Other phenotypes expressed by these mice included exencephaly, rostral neural tube defect, delay in limb development, defects in the ventral portion of the head with altered development of the eyes and branchial arches. Cilia loss was found in the homozygous mutant and malformed cilia were observed in heterozygous embryos, which associated with defects in left–right asymmetry. Additionally, mutation of Ift22 reduced Gli2/Gli3 and Gli3 repressor functions, suggesting that Shh signaling is impaired (97).
Another mouse model Ift122sopb was generated with ENU mutagenesis and named sister of open brain (sopb) due to the similar phenotype to Rab23/open brain mutants (158). Sopb is a null allele of Ift122 with a T-to-C transition mutation in the start codon of Ift122. Ift122sopb embryos died around E13.5, displayed neural defects, pre-axial polydactyly, enlarged bronchial arches, and ocular defects. Cilia frequency was normal in the neural tube of Ift122sopb mice but reduced in Ift122sopb MEFs. Cilia in Ift122sopb were shorter but swollen along their lengths and tips, which accumulated with complex B proteins as stained with Ift88 and Ift57. The complex A protein Ift140 was undetectable in the cilia of Ift122sopb cells, suggesting a role of Ift122 in facilitating complex A stability and retrograde Ift (158). Suppressor of Fused (SUFU) and Smo transportation to cilia were not affected while Gli2 and Gli3 accumulated in the cilia tips after disruption of Ift122. Ciliary localization of TULP3, a repressor of Shh pathways, was also blocked in Ift122sopb. These data suggested that Ift122 negatively regulates the Shh pathway downstream of Smo through differentially controlling the ciliary localization of the Hh pathway regulators (158). The different phenotypes observed in those two models may be caused by different mouse strain backgrounds (Ift122sopb of C3Heb/FeJ and Ift122-null of C57BL/6, respectively) and/or by the effect of MED1 deletion in Ift122-null mutant (158).
5.2.3. Ift144
Ift144, coded by the gene WDR19 (WD repeat domain 19), has WD40 regions and TPR protein–protein interaction domains (159). Ift144 localizes at the base and tip of the cilium in a punctate pattern along the axoneme in the mouse embryo fibroblasts (MEFs) (160). Mutation of Ift144 causes CED. The fibroblasts from the CED patients show undetectable Ift144 and the reduction in cilia abundance and length (109).
Currently, two ENU induced mouse mutant models of Ift144 (Ift144twt and Ift144dmhd) have been generated (160, 161). Mice with the Twinkle-toes mutation (Ift144twt), which introduces a leucine-to-proline substitution at amino acid position 750 (L750P, within the first TPR repeat), are partial loss-of-function mutations. Similar to the phenotype of CED patients, Ift144twt embryos die at E11.0 and display polydactyly, small rib cage, and severe craniofacial anomalies. The number of ciliated cells was reduced in Ift144twt mouse limbs and isolated MEFs, while the cilia structure appeared relatively normal. In addition, Ift144twt cells and tissues have enhanced ligand-independent Hh signaling, but reduced response to Hh ligands.
A more severe allele of the Ift144 mutation is known as diamondhead (Ift144dmhd), which represents a functional null mutation (161). These mutant embryos displayed different phenotypes when compared to Ift144twt mice. The cilia were shorter and showed highly disrupted axoneme in Ift144dmhd embryos. Unlike Ift144twt, Ift122sopb and Ift139alien, Ift144dmhd had less activity in Shh signaling, but greater numbers of cells with an intermediate level of Shh activity in the caudal neural tube (161). Ift144dmhd showed smaller forelimb paddle, with truncation in the proximal-distal (PD), and to a lesser extent, the anterior-posterior (AP) axes when compared to those Ift144twt mice revealed a dose-dependent effect of Ift144 on limb outgrowth (160).
The cilia in double mutation of Ift144twt and Ift122sopb (Ift144twtIft122sopb) were shorter and more bulbous cilia than those in either single mutation and were similar to those in Ift144dmhd embryos (161). Also the phenotype of Ift144twtIft122sopb is close to that in Ift144dmhd, suggesting that both Ift144twtIft122sopb and Ift144dmhd result in a stronger disruption of the IFT-A complex. Double mutations of Ift144twt and Dync2h1mmi (Ift144twtDync2h1mmi) partially rescued the axoneme structure and reduced the level of cilia swollen in Dync2h1mmi (161). The authors concluded that Ift144 is also required for normal anterograde trafficking (161). Further studies on the Hh pathway showed that transportation of the Shh pathway proteins Gli2, SUFU, and Kif7 were normal in Ift144twt, Ift144twtIft122sopb and Ift144dmhd. But the membrane proteins Arl13b, adenylyl cyclase III (ACIII), and Smo failed to localize to primary cilia in the more complete Ift-A mutation (Ift144twtIft122sopb and Ift144dmhd). Reduced ACIII level in cilia led to the decrease in cAMP and protein kinase A (PKA) activity, which leads to the ligand independent activation of Shh. This can explain the up-regulated Shh observed in Ift144twt, but loss of Shh activity in Ift144dmhd with the null mutation of Ift144 due to severe disruptions of the cilia structure and membrane protein trafficking (161).
5.2.4. WDR35 (Ift121)
WDR35 (WR repeat domain 35), which is orthologous to Ift121 in mammals, has seven closely spaced WD40 repeats at the N-terminal and a TRP-like motif at the C-terminus (34). WD40 repeats are believed to have the functions of intracellular trafficking, cargo recognition, and binding. Mutation of this structure caused SRP with a ciliogenesis defect (34). Mutations in the C-terminal domains are more likely associated with CED and result in impairment, but not in complete inhibition of retrograde transport (34, 136).
Mill et al isolated a WDR35 null mutant mouse line yeti (WDR35yet) from a recessive ENU mutagenesis screen that was looking for genetic effects on embryogenesis (34). They found that a single G>A mutation in the splice acceptor site of exon 22 of WDR35 resulted in nonsense-mediated decay of mutant transcripts and complete loss of function (34). WDR35yet embryos died at E12.5 and exhibited severe defects in cardiovascular, embryo turning, and limb outgrowth and patterning. Additionally, WDR35yet displayed failure of the somite derivatives, including migration and differentiation of putative ribs (34).
WDR35 accumulates in the basal body and along the cilia axoneme. Both WDR35yet MEFs and fibroblasts from SRP patient (WDR35Δ5/Δ5) had cilia loss and less Ift88 accumulation around the basal body area (34). Interestingly, fibroblasts from CED patient (WDR35 mutation) displayed shorter cilia and accumulation of Ift88 and Ift57 in the distal ends of cilia (135). Those data suggest that WDR35 mutation in SRP may interfere with anterograde transportation or other vesicular trafficking, which leads to the cilia defect, while the WDR35 mutation in CED only affects retrograde transport and results in a typical Ift-A mutation phenotype like the Ift122 mutation (34). Mill et al also described WDRtm2a mice as an embryonic stem cell derived “targeted trap” null mutation (34). The loss of WDR35 by targeted mutation phenocopies the WDR35yet, but the details have not yet been revealed (34).
5.2.5. Ift43
Ift43 is the smallest member in Ift-A complex and it does not contain any identifiable domains (28). Ift43 was the third gene identified as mutation in patients of CED with a homozygote N-terminal deletion (135). A recent study suggested that there is the direct interaction between Ift43 and Ift121, and Ift121 is another gene responsible for CED (90). Fibroblasts from an Ift43 mutant CED patient had shorter cilia and an accumulation of complex B proteins in the distal ends of cilia indicated by Ift88 and Ift57 staining, which is similar to other Ift-A mutation (135). No Ift143 mutant mouse model has been published yet.
5.2.6. Ift140
The structure of Ift140 is similar to Ift144 (28). Mutation of Ift140 has been correlated with MSS and JATD (111). Fibroblasts from some MSS patients had fewer ciliated cells and a normal distribution of Ift140, but altered distributions of Ift88 and Ift46 (evenly distributed along the cilium instead of localizing in the base and tip), suggesting a defect in retrograde ciliary transport (111).
Jonassen et al generated Ift140flox/flox mice with two Loxp sites flanking exon 7 of Ift140 (162). Ift140null was generated by crossing these mice with C57Bl/6 Prm-Cre (Cre recombinase expressed in the germline). These mice died at mid-gestation, but the phenotype has not described yet (162). The function of Ift140 in kidney development has been studied by crossing Ift140flox/flox mice with HoxB7-Cre to delete Ift140 in the collecting ducts. Loss of Ift140 from the collecting ducts disrupted cilia assembly and caused renal cystic disease. Significant increases in tissue fibrosis and the expression of canonical Wnt pathway genes and Hh mediators was also found in highly cystic, but not precystic, kidneys in these mice, suggesting that abnormalities in these different pathways may influence cyst progression (162).
Another Ift140 mouse model was identified from a comprehensive phenotype-driven ENU screen and was named cauliflower (cauli) (163, 164). Cauli mutation is a recessive missense mutation with a T-to-A substitution in exon 19 at position 2564 (leading to an isoleucine to lysine amino acid change at position 855, p.I855K). Ift140cauli embryos died at E13.5 and exhibited exencephaly, anophthalmia, severely disorganized ribs with extensive exostoses, vertebral and palatal defects, agenesis/hypoplasia of the craniofacial skeleton, and polydactyly of the hindlimbs (163, 164). Cilia morphology in Ift140cauli limb buds was severely disrupted with a broader and bulbous appearance. Similar to other Ift-A mutants, ectopic Hh signaling activity has been observed in Ift140cauli embryos. The loss of Ift140 resulted in reduced Gli3 repressor production, and thus led to the polydactylous phenotype and rib exostoses (164). The null mutation of Ift140 (Ift140−/−) was also generated by Miller et al by crossing the Ift140flox/flox mice with CMV-Cre mice (Cre recombinase is expressed in all tissues including germ cells) (164). Ift140−/− embryos died at about E11.5 with the identical phenotype to that observed in Ift140cauli (164).
5.3. Mouse models of Ift complex B proteins
Ift complex B contains 14 known proteins named Ift20, Ift22, Ift25, Ift27, Ift46, Ift52, Ift54, Ift57, Ift70, Ift74/Ift72, Ift80, Ift81, Ift88, and Ift172. In vitro and in vivo studies examining these proteins are described in the following sections (Table 3).
Table 3.
Current mouse models to study the function of Ift-B proteins in bone development
| Gene | Mouse models | Mutation | Cilia | Phenotype | Pathways | Ref |
|---|---|---|---|---|---|---|
| Ift80 | Ift80gt/gt | Gene trap, hypomorph with low level wild-type transcript production | Normal cilia | Usually die perinatally. Severe growth retardation with short long bones, narrow ribcage and polydactyly |
Less Hh activity. | (70) |
| Ift88 | Ift88Tg737Rpw (ORPK) | Transgene insertion-induced hypomorphic allele | Stunted and malformed cilia | Survive to young adulthood. Scruffy fur, severe growth retardation, cystic renal phenotype, preaxial polydactyly, defects in skeletal patterning and growth with craniofacial abnormalities, cleft palate, unfused tibia and fibula, and significantly smaller growth plates. |
Defects in PDGFR-α/PDGF-A signaling. | (168–171) |
| Ift88Δ2–3βGal (Ift88tm1Rpw) | A Null mutation | Cilia loss | Arrested at mid-gestation with neural tube defects, enlargement of the pericardial sac and left-right asymmetry defects. | Reduced expression of Shh and Hnf3β in midline. No changes in Shh or its downstream genes in limb bud. |
(91, 170) | |
| Ift88fxo (Ift88hypo) | ENU- mutagenesis, null mutation | Lack nodal cilia | Dies at E12.5. Sharp angle of the mesencephalic flexure, abnormal brain morphology and preaxial polydactyly. |
Disrupted Hh signaling with loss of both Gli activator function and Gli repressor function in the limbs. | (56, 173) | |
| Ift80flox/flox; Prx1-Cre | Deletion in limb mesenchyme | Cilia loss | Severe polydactyly and defects in endochondral bone formation. | Aberrant Shh and Ihh signaling. | (174) | |
| Ift80flox/flox; Msx2-Cre | Deletion in limb ectoderm | Cilia loss | No overt effect on limb patterning. | Not reported. | (174) | |
| Ift80flox/flox; Col2α-Cre | Deletion in committed chondrocyte | Cilia loss | Postnatal dwarfism and premature loss of the growth plate with down-regulated proliferation and up-regulated hypertrophic differentiation of the chondrocytes. Thicker articular cartilage with increased cell density and OA symptoms. | Reduced Hh but elevated Wnt/β-catenin (reduced Sfrp5) signaling. Uprelated Hh signaling in articular cartilage with reduced Gli3 repressor/Gli3 activator. | (93) (176) (175) | |
| Ift172 | Ift172wim | ENU- mutagenesis, a null mutation | Lack nodal cilia | Arrested at E10.5–11.5. Open neural tube in the head that lacked the normal groove on the ventral midline. | Strong defect in Shh signaling. Normal Wnt signaling. |
(56) (178) |
| Ift172avc1 | ENU- mutagenesis, partial loss of function | Defects in cilia | Die at birth. Anomalies in vertebral, trancheoesophageal and limb, defects in cardiac, anal atresia, renal dysplasia and hydrocephalous. | Defects in Hh signaling with reduced Gli3 processing and Gli2 accumulation. | (92) | |
| Ift172Slb | Conventional knockout, a null mutation | Short and without visible microtubules | Die between E12.5 and 13.0. Severe craniofacial malformations, defects in brain patterning, failure to close the neural tube and exencephaly. | Shh and Gli1 decreased in the developing forebrain and midbrain. | (179) | |
| Ift172Δ/Δ | CMV-Cre–mediated germline deletion | Not reported | Embryonic lethality. Neural tube defects (Similar to Ift172wim and 172Slb) |
Not reported | (180) | |
| Ift172f/t; Prx1-Cre | One allele is germline deletion and another is only deleted in the developing limb bud mesenchyme | Not reported | Eight digits on each forelimb and a single extra digit on each hind limb. | Not reported | (180) | |
| Ift20 | Ift20Flox; Prm-Cre | Germline deletion | Not reported | Die before birth | Not reported | (183) |
| Ift25 | Ift25neo and Ift25null1 | Null mutation | Normal cilia | Die at birth. Omphaloceles, polydactyly with a preaxial digit duplication, micrognathia, cleft palate and malalignment of the sternal vertevbrae. | Ciliary accumulation of Shh components and attenuated Shh. | (185) |
| Ngd5 (Ift52) | Ift52hypo | Partial loss-of-function mutations | Not reported | Tight mesencephalic flexure, left-right and ventral midline defects, polydactyly and craniofacial defect. | Defects in Hh pathway in limb buds. | (173) |
| Traf3ip1/MIP-T3 (Ift54) | Traf3ip1GT | Gene Trap | Cilia disrupt | Die before E13.5. Neural developmental defects, cardiac edema and polydactyly. |
Defects in Hh pathway. Unchanged Wnt pathway. Elevated mTor pathway. |
(193) |
| Hippi (Ift57) | Hippi−/− | Conventional knockout | Defects in nodal cilia | Die predominantly prior to E10.5. Defects in left-right patterning and nervous system development. |
Down-regulated Shh pathway. | (196) |
5.3.1. Ift80
Ift80 is a 777-residue protein, which contains seven WD40 domains (165) and is the first Ift protein that was identified with mutation in human diseases -JATD and SRP type III, (130). Knockdown of ift80 in zebrafish resulted in cystic kidneys with down-regulated Ptch1, suggesting reduced Hh activity (165). Rix et al generated the mouse model named Ift80gt/gt using gene trap strategy (insert gene-trap cassette pGT0lxr in the intronic region downstream of exon 9 in Ift80) (70). The homozygotes Ift80gt/gt are hypomorphs with the low level of wild-type transcript production, but most died perinatally. Two percent of homozygotes survived to postnatal stages and had severe growth retardation with short long bones, a narrow rib cage and polydactyly. MEFs from Ift80gt/gt E14.5 embryos had normal cilia but reduced Hh activity in response to smoothened agonist purmorphamine. These data suggested that a low level of Ift80 expression is enough for ciliogenesis, but not sufficient for Hh signaling transduction (70).
The results from our laboratory showed that Ift80 was highly expressed in mouse long bone and skull, as well as during osteoblast and chondrocyte differentiation (166, 167). We used RNA interference (RNAi) to silence Ift80 in the murine mesenchymal progenitor cell line C3H10T1/2 and bone marrow derived stromal cells (BMSCs) and found that loss of Ift80 led to shorten or loss of cilia and significantly blocked osteogenesis. Gli2 expression was significantly decreased in Ift80-silenced cells, suggesting the impaired Hh pathway activity (167). Silence of Ift80 in BMSCs also impaired the chondrogenic differentiation by down-regulating Hh signaling, whereas up-regulating Wnt signaling (166). Those data suggested the important role of Ift80 in skeletal development. Currently, we have generated Ift80flox/flox conditional knockout mouse alleles, and are identifying the role and mechanism of Ift80 in different bone cells by using specific bone cell Cre transgenic lines to delete Ift80 in various bone cells.
5.3.2. Ift88
Ift88 (Tg737 or Polaris), a TPR-containing protein, was first identified in ARPKD (168). The Ift88Tg737Rpw (also known as ORPK, FVB/N strain) mouse model was generated 20 years ago as a model for human recessive polycystic kidney disease (168). Ift88Tg737Rpw has a transgene insertion-induced hypomorphic allele of Ift88 (continued expression of alternatively spliced transcripts and presence of low levels Ift88), which allows homozygous mutant mice to survive into young adulthood. Ift88Tg737Rpw mice have scruffy fur, severe growth retardation, cystic renal phenotype, and preaxial polydactyly in all limbs (168, 169). Ift88Tg737Rpw mice also have defects in skeletal patterning and growth with craniofacial abnormalities, cleft palate, unfused tibia and fibula (170). The cilia are not completely abolished in Ift88Tg737Rpw but are stunted and malformed (169). Fibroblasts isolated from Ift88Tg737Rpw mice do not respond to platelet-derived growth factor A (PDGF-A) and fail to conduct chemotaxis toward PDGF-A (169). The growth plates of Ift88Tg737Rpw mice are significantly smaller in length and width, suggesting the role of Ift88/cilia in chondrocyte differentiation and hypertrophy (171). Chondrocytes from Ift88Tg737Rpw mice have a defect in the compression-induced Ca2+ signaling but not ATP release, suggesting that cilia are involved in ATP reception instead of initiation of mechanotransduction (172).
To further study the function of Ift88, Murcia et al (91) generated a null allele (Tg737Δ2–3βGal or Ift88tm1Rpw, FVB/N strain) by homologous recombination in ESCs. The initial coding exons (exon 2 and part of exon 3) were replaced with the β-galactosidase (β-Gal) reporter gene. Tg737Δ2–3βGal embryos that are arrested in development at mid-gestation exhibit neural tube defects, enlargement of the pericardial sac, and significant left-right asymmetry defects. Cilia were absent in Tg737Δ2–3βGal ventral node cells. Expression of both Shh and Hnf3β is reduced in the midline in E8.0 Ift88Δ2–3βGal embryos, contributing to the alterations in midline development (91). However, Zhang et al found that no changes in Shh or its downstream genes occurred in the limb buds of Tg737Δ2–3βGal embryos (170).
Huangfu et al identified Ift88fxo (Flexo, also called Ift88hypo) from the screen of ENU-induced embryonic abnormalities (56). Ift88fxo is a null allele of Ift88 with deletion of exon 16 (including part of one of the ten predicted TRP motif). Ift88fxo embryos lack nodal cilia and display an unusually sharp angle of the mesencephalic flexure at E9.5.–10.5. Those embryos are arrested at E12.5–13.5 with abnormal brain morphology and preaxial polydactyly (56, 173). Liu et al studied the Hh signaling in the limbs of Ift88fxo embryos and proposed that both Gli activator function (down-regulation of Ptch1 and Gli1 expression) and Gli repressor function (Hand2 misexpression and polydactyly) are compromised in the limbs of Ift88fxo (173).
Haycraft et al generated Ift88flox/flox conditional allele with LoxP sites flanking exons 4–6 (174). The deletion of these exons with Cre-line is predicted to result in a translational frame shift and loss of all Ift88 function (a null mutation). Prx1-Cre (Ift88flox/flox; Prx1-Cre, limb mesenchyme with both chondrocytes and perichondrium) and Msx2-Cre (Ift88flox/flox; Msx2-Cre, limb ectoderm) were used to delete Ift88 on different cell populations within the developing limb (174). Disrupted Ift88 using both Cre lines caused cilia loss (93, 174). Ift88flox/flox; Msx2-Cre mice did not exhibit an overt effect on limb patterning. However, Ift88flox/flox; Prx1-Cre displayed severe polydactyly and defects in endochondral bone formation with aberrant Shh and Ihh signaling (174). Song et al crossed Ift88flox/flox mice with Col2α1-Cre to delete Ift88 in committed chondrocyte lineage (Ift88flox/flox; Col2α1-Cre) (93). The phenotype of Ift88flox/flox; Col2α1-Cre is similar to Kif3aflox/flox; Col2α-Cre with postnatal dwarfism and complete loss of the growth plate, resulting from down-regulated the proliferation and up-regulated hypertrophic differentiation of the chondrocytes. They further demonstrated that Ift88 regulated chondrocyte rotation to maintain the columnar organization of the growth plate (93). Chang et al studied the signaling pathways involved in the growth plate abnormality by comparing gene expression profiles in normal and Ift88flox/flox; Col2α-Cre growth plates (175). They found that the expression of Hh and secreted frizzled-related sequence protein 5 (Sfrp5) (an extracellular antagonist of Wnt signaling pathways) was significantly lower in Ift88flox/flox; Col2α-Cre. These data support the increase of Wnt/β-catenin signaling in flat columnar cells of the growth plate in Ift88flox/flox; Col2α-Cre as measured by expression of Axin2 and Lef1, as well as nuclear localization of β-catenin (175). In addition, Ift88flox/flox; Col2α-Cre mice showed thicker articular cartilage with increased cell density, which probably results from the reduced apoptosis rate in cartilage remodeling (176). The mechanical properties of cartilage from Ift88flox/flox; Col2α-Cre mice significantly decreased, particularly in the deeper zones (172). Up-regulation of Hh signaling as measured by expression of Ptch1 and Gli1 was observed in Ift88flox/flox; Col2α-Cre articular cartilage, which contributes to the osteoarthritis (OA) symptoms. A reduction in the Gli3 repressor to activator ratio also occurred, which is an explanation for the elevated Hh signaling (176).
5.3.3. Ift172
Ift172 is the largest Ift protein. It is predicted to have N-terminal WD-40 repeats and C-terminal α-helical TPRs (28).
Huangfu et al identified a wimple (wim) mutation of Ift172, a null mutation, in a screen for recessive ENU-induced mutations (56). The T to C mutation in the wim allele causes a leucine to proline substitution (Leu1564Pro) near the C-terminus. Ift172wim embryos have morphological abnormalities at E9.5 and arrest at E10.5–11.5 due to strong defect in Shh signaling. Ift172wim mice lack nodal cilia and have an open neural tube in the head, which does not have the normal groove on the ventral midline (56). Ptch1 mutant mice (177) (partial exon 1, and exon 2 of ptch1 were replaced with lacZ and a neomycin (neo) resistance gene through homologous recombination in ESCs) were crossed with Ift172wim to generate double mutants, which showed the similar phenotype as Ift172wim. Loss of Ptch1 caused the activation of Hh pathway, but Ift172 mutation blocked this activation, suggesting Ift172 acts downstream of Ptch1 in Shh signaling pathway (56). The loss of cilia did not affect the response of the Ift172wim mid-gestation embryos or MEFs to Wnt ligands (178).
Friedland-Little et al identified Ift172avc1 from a screen of ENU-induced mutations (92). This mutation is an A-to-G transition in the splice donor site downstream of exon 24 of Ift172 to alter Ift172 splicing. Ift172avc1 allele maintains some wild-type transcript, which is a partial loss of function mutation and dies at birth. Ift172avc1 mice show anomalies in vertebral, trancheoesophageal and limb, and defects in cardiac, anal atresia, renal dysplasia and hydrocephalous. But left-right axis formation is normal in Ift172avc1 (92). Ift172avc1 cells have truncated cilia (including disrupted IFT88 localization) both in vivo and in vitro. Ift172avc1 embryos display a global reduction in the expression of Ptch1 and Gli1. A 70.7% reduction of Gli3 processing (from full-length to transcriptional repressor) in Ift172avc1 embryos and disrupted Hh-dependent Gli2 localization in Ift172avc1 MEFs has been observed, which contributes to the Hh signaling defect in Ift172avc1. Quantitative analysis wild-type, Ift172avc1/avc1 and Ift172wim/avc1 revealed the dose-dependent requirement for Ift172 in ciliogenesis and Hh signaling (92).
Another mouse model named Slb mutant (Ift172Slb) was generated by Gorivodsky et al (179). A neo cassette was inserted to replace the first three exons of Ift172 and cause the loss of function mutation. Ift172Slb mice die between E12.5 and E13.0 with severe craniofacial malformations, defects in brain patterning, failure to close neural tube and exencephaly. Cilia are initiated but shorter and without visible microtubules. Expression of Shh and Gli1 is lower in the developing forebrain and midbrain of Ift172Slb. In addition, Ift172 is required for early regulation of Fgf8 at the midbrain-hindbrain boundary (179).
To further study the role of Ift172 in the later stage of development, Howard et al generated mice with a conditional knockout of Ift172 (Ift172f/f) using LoxP sites flanking exons 2–3 (180). Deletion with the Cre line can remove about a third of a WD40 repeat domain and cause a frame shift, resulting in a stop codon. Similar to Ift172wim and 172Slb, germline deletion with CMV-Cre (Ift172Δ/Δ) leads to embryonic lethality due to neural tube defects. To examine tissue-specific function of Ift172, Prx1-Cre was used to delete Ift172 in the developing limb. Ift172f/t; Prx1-Cre mice (one allele is a germ line deletion and another is only deleted in the developing limb bud mesenchyme) have eight digits on each forelimb and a single extra digit on each hind limb (180).
5.3.4. Ift20
Ift20 is the smallest Ift protein in the Ift family and has several unique features. It is the only Ift protein that localizes to the Golgi complex in mammalian cells, suggesting its role in the ciliary protein sorting (165, 166). Ift20 is anchored to the Golgi complex by the Golgi protein-Golgi Microtubule Associated Protein 210 (GMAP210)/thyroid hormone receptor interacting protein 11 (TRIP11) (166). Without GMAP210, Ift20 is no longer localized to the Golgi structure and cilia are shorter with reduced ciliary localization of membrane protein polycystin-2. GMAP210 mutant mice die at birth and display growth restriction, ventricular septal defects of the heart, omphalocele, and lung hypoplasia (166). Strong knockdown of Ift20 in mammalian cells blocks cilia formation while moderate knockdown does not block cilia assembly, but reduces the amount of polycystin-2 to localize to the cilia (165). Those data demonstrated GMAP210 and Ift20 work together at Golgi in sorting or transporting proteins like polycystin-2. Besides GMAP210/ TRIP11, Ift20 also exhibits a strong interaction with Ift57/Hippi and kinesin II subunit, Kif3b (181). Ift20 is tethered by Ift57 to the Ift particle, physically interacts with Kif3b and mediates the ATP-dependent dissociation of kinesin. Loss of Ift57 leads to the disassociation of Ift20 from Ift particles but does not affect the association of kinesin to the Ift particle (182).
In addition, Ift20 is expressed in lymphoid and myeloid cells, which do not have primary cilia. Ift20 takes part in the polarized recycling of the TCR/CD3 complex, which is the first connection of Ift proteins and membrane trafficking in cells without cilia (167). But whether Ift20 functions in membrane trafficking in the ciliated cells remains unknown.
The mouse floxed allele of Ift20 (Ift20Flox), which has LoxP sites flanking exons 2 and 3 of Ift20, has been established by Jonassen et al (183). Germline deletion of Ift20 using Prm-Cre led to embryonic lethality (183). So far, HoxB7-Cre and human red/green pigment gene promoter (HRGP)-Cre have been used to study the function of Ift20 in kidney and photoreceptor cells (183, 184). But the function of Ift20 has yet to be reported in bone, and our lab is currently trying to understand the function and mechanism of Ift20 in bone development and remodeling.
5.3.5. Ift25
Ift25 is one of the two Ift proteins (Ift25 and Ift27) that are absent from C. elegans and Drosophila, suggesting a role of Ift25 beyond ciliogenesis (185). Keady et al generated the Ift25neo mice by using homologous recombination to modify ESCs containing a splice acceptor, a promoterless β-galactosidase-neo resistance gene fusion and LoxP sites flanked exon 3 (185). By crossing these mice with the germ line Cre Prm-Cre mice, they generated Ift25null1, which lacks exon 3 of Ift25. Both Ift25neo and Ift25null1 are null mutations without Ift25 expression. These mice die at birth with severe heart defects but have normal cilia. These are different from other Ift-B null mutants that usually fail to form cilia and die around mid-gestation. In addition, mutation of Ift25 leads to omphaloceles, polydactyly with a preaxial digit duplication, micrognathia, cleft palate, and malalignment of the sternal vertebrae. Ift25 is needed for the exit of Smo and Ptch1 from cilia (not import) and Gli2 accumulation at the ciliary tip in response to activation of the Shh pathway. Loss of Ift25 leads to a failure to activate the Hh pathway upon stimulation (185).
5.3.6. Ift27
Ift27 belongs to the small G protein family (close to Rab subfamily) with Rab-like small GTPase activity (186, 187). It binds weakly with GDP and GTP, and displays very low, but significant intrinsic GTPase activity (186). In addition, it forms a sub-complex with Ift25 in green algae, Chlamydomonas reinhardtii, mice and humans (186, 188). Ift25 and Ift27 seem to be preassembled before entering the cilia and associating with other Ift-B members, suggesting the role of Ift25/27 in Ift initiation (186, 188). However, the roles of the Ift27 in cilia formation and bone development are not yet known and no Ift27 mutant mouse model has been published yet.
5.3.7. RabL5 (Ift22)
Ift22, another Ift protein with significant sequence similarity to Rab-like GTPases, is known as Rab-like 5 (RabL5) in mammals (28). Mutation of the Ift22 ortholog in C.elegans (Rab-like 5) did not alter the cilia (189). But knock-down of Ift22 by RNAi in the trypanosome flagellum surprisingly resulted in short flagella filled with a high amount of IFT proteins, which is a typical retrograde Ift defect phenotype (190). No mouse model or related human diseases have been reported yet.
5.3.8. Ift46
Even though Ift46 does not contain any recognizable domains or motifs, it directly interacts with Ift52, Ift70 and Ift88 in the core of Ift-B (28). Study showed that Ift46 is necessary for Ift-B stability (191). Recently, Ift46 has been identified as a BMP-2 sensitive protein in mouse chondrocytes (192). Ift46 is located in the mouse primary cilium of chondrocytes and is preferentially expressed in early hypertrophic chondrocytes in the growth plate. Knockdown of Ift46 in cultured chondrocytes by small interfering RNA (siRNA) oligonucleotides does not affect the expression of the Col2α1, Col10α1, BMP receptors, and Smads. Instead, it up-regulates Msx1, fibroblast growth factor receptor-1 (Fgfr1), type XII collagen (Col12α1), biglycan (Bgn), matrix metalloproteinase-10 (Mmp10), and heat shock protein 47 (Hsp47) expression (192). Antisense morpholino oligonucleotides were injected into Danio rerio embryos to deplete zebrafish Ift46, which led to severe defects in embryonic development. These data highlight the role of Ift46 in chondrocyte function and vertebrates development (192). However, whether Ift46 is involved in cilia formation in chondrocytes and how it affects bone development and remodeling have not been evaluated yet.
5.3.9. Ngd5 (Ift52)
Ift52 is predicted to have an N-terminal ‘GIft’-domain (GldG/Ift) and proline-rich stretch (28). Ift52 can bind Ift70 and Ift88 directly through the proline-rich stretch (28). Mouse Ngd5 is the mammalian ortholog of Ift52 (181). Liu et al generated Ift52hypo from two mutant ESC lines in which the Ngd5 gene was disrupted by replacing it with the lacZ gene (173). The C-terminal 99 amino acids of the Ngd5 protein were replaced with lacZ in the first line (XL826). In the second allele (RRJ295), the last 47 amino acids of the Ngd5 protein were replaced with lacZ. Mice derived from both lines are termed as Ift52hypo. They are partial loss-of-function mutations and have similar phenotypes, including tight mesencephalic flexure, left-right and ventral midline defects, polydactyly and craniofacial defects at embryonic stage (173). In addition, ptch1 is not detected in the limb buds of Ift52hypo, suggesting the disruption of Hh signaling (173).
5.3.10. Traf3ip1 (Ift54)
The homology of Ift54 in mammalian systems is tumor necrosis factor alpha receptor 3 interacting protein 1 (Traf3ip1)/microtubule interacting protein that associates with Traf3 (MIP-T3, hereafter called Traf3ip1), which was initially characterized through its interactions with tubulin, actin, TNFR-associated factor-3 (Traf3), interleukin-13 receptor α1 (IL-13R1), and Disrupted-In-Schizophrenia 1 (DISC1). Traf3ip1 inhibits IL-13–mediated phosphorylation of signal transducer and activator of transcription-6 (STAT6). Traf3ip1 also regulates Traf3 and DISC1 by sequestration of these factors to the cytoskeleton in mammals (193, 194).
Berbari et al generated the Traf3ip1GT mouse model with embryonic stem cell line harboring a gene trap insertion in intron 11 of Traf3ip1 (193). MEFs from Traf3ip1GT had nearly 90% less transcript levels than the level in wild-type MEFs. Traf3ip1GT mouse is lethal at the embryonic stage before E13.5, having neural developmental defects, cardiac edema, and polydactyly. Hh signaling was dramatically reduced in both the neural and endoderm tubes in E10.5 Traf3ip1GT embryos as determined by crossing with Ptch1-lacZ Hh reporter mice. By crossing those mice with BAT-Gal Wnt reporter mouse line, the author found that there is no overt alternation in canonical Wnt signaling in the Traf3ip1GT mutant embryos. MEFs from Traf3ip1GT embryos have defective cilia formation, up-regulated acetylated microtubules, and increased cell size. Interestingly, deletion of Traf3ip 1 increases the activity of the mammalian target of rapamycin (mTor) pathway, which likely causes the change of cell size in Traf3ip1 mutant cells (193).
5.3.11. Hippi (Ift57)
Ift57 has a coiled-coil domain at the C-terminus and interacts with Ift20 in a yeast two-hybrid assay (181). The ortholog of Ift57 in mouse is Hippi (huntingtin interacting protein-1 protein interactor), which is involved in neuronal apoptotic cell death in Huntington disease (195). Houde et al generated Hippi-null mice (Hippi−/−, C57BL/6J-129S1/SvImJ genetic background) by replacing the first 95 codons of the Hippi open reading frame with the neo gene or neo plus β-Gal (196). Hippi−/− embryos predominantly die prior to E10.5 and have defects in nodal cilia, left–right patterning, and nervous system development. In addition, the expression levels of Shh and ptch1 in the neural tube are down-regulated, suggesting that deletion of Hippi likely disrupts Hh signaling pathway (196).
5.3.12. Ift70
Ift70 is predicted to contain a number of TPRs (28). Mutations of the Ift70 orthologs dyf-1 in C. elegans or fleer in zebrafish led to defective cilia formation (197–199). The roles of Ift70 in mouse and human disease have not been reported yet.
5.3.13. Ift74/72 and Ift81
Ift74 and Ift72 are derived from the same gene and often named as Ift74/72 (88). Both Ift74/72 and Ift81 are core proteins in complex B. Ift74/Ift72 interact with the Ift81. They all have coiled-coil regions in their structure (28). Very recently, Bhogaraju et al demonstrated that Ift74 and Ift81 form a tubulin-binding module, which is required for ciliogenesis in human RPE-1 cells (retinal pigment epithelium cells), suggesting Ift74/Ift81 play a role in the tubulin transportation within cilia (200). Further studies are needed to determine whether these Ift proteins are involved in bone development.
5.4. Mouse models of Ift motors
Anterograde and retrograde Ift are operated by two types of MT-based motors, kinesin-2 (plus end-directed MT motor) and dynein-2 (minus end-directed MT motor). Although significant efforts have been contributed to elucidate the function of Ift motors (See review (201)), their roles in ciliogenesis and signaling transduction are still poorly characterized (Tables 4=5).
Table 4.
Current mouse models to study the function of Ift retrograde motors in bone development
| Gene | Mouse models | Mutation | Cilia | Phenotype | Pathways | Ref |
|---|---|---|---|---|---|---|
| Dync2h1 | Dnchc2lln | ENU- mutagenesis, missense mutation | Abnormal cilia with reduced length and bulge along their length | Die at approximately E12.5, abnormal brain morphology, randomization of heart-looping polarity and polydactylous. | Defects in Hh pathways with high level of Smo, Gli2 and Ptch1 accumulation along the cilia with or without Shh stimulation. | (48, 204) |
| Dnchc2GT | Gene trap insertion, null allele | Not reported | Similar to Dnchc2lln. | Not reported | (48) | |
| Dync2h1ttn | ENU- mutagenesis, deletion in AAA domain 4 | Abnormal cilia with reduced length and bulges along the axoneme | Not reported | Transportation of activated Gli2 out of cilia is blocked. Wnt signaling is normal in midgestation embryo or embryo-derived fibroblasts. |
(67, 205) | |
| Dnchc2Q397Stop | ENU- mutagenesis, null mutation | Shorter and wider cilia | Die at E12.0 or earlier with pericardial edema and heart failure. Severe defects in dorsoventral patterning of the forebrain and patterning of the spinal cord. |
Both Gli activator and Gli repressor activity were significantly reduced. Disruption in Gli3 proteolytic processing and Smo localization to cilia. |
(207) | |
| Dnchc2W2502R | ENU- mutagenesis, null mutation | Shorter and wider cilia | Similar to Dnchc2Q397Stop. | Similar to Dnchc2Q397Stop. | (207) | |
| Dync2h1mmi | ENU- mutagenesis, missense mutation | Swollen and filled with electron-dense particles and defects in axonemal | Defects in neural tube and motor neurons. | Not reported | (161, 206) | |
| Dync2h1lln Ift172wim | Double mutation | Absent of cilia | Similar to Ift172wim. | Not reported | (204) | |
| Dync2h1lln Ift172avc1 and Dync2h1lln/llnIft172avc1/+ | Double mutation | Normal morphology in Dync2h1lln/llnIft172avc1/+ | Dync2h1lln phenotype can be rescued by reducing Ift172 expression, more dramatic rescue of the Dync2h1 phenotype in Dync2h1lln/llnIft172avc1/+ (survived to at least E16.5 with nearly normal neural patterning in the caudal neural tube). | No Ift or Hh pathway proteins accumulate in cilia, and Hh target gene expression appears to be normal. | (204) | |
| Ift122sopbDync2h1lln | Double mutation | Similar to Ift122sopb | Similar to Ift122sopb. | Ectopic activity of the Shh pathway in the neural tube. Gli2 limited to the tips of cilia and Smo was not in cilia without Shh stimulation. | (204) | |
| Dync2h1lln/llnIft122sopb/+ | Double mutation | Partially rescued the cilia morphology | Partially rescued the phenotype of Dync2h1lln/lln including ventral neural development. | Partially rescued Shh-dependent protein trafficking in cilia. | (204) | |
| mD2LIC | mD2LIC−/− | Gene targeting, null mutation | Cilia fail to form in the node | mD2LIC−/− died before E11.5 with defects in notochord and floorplate formation and a reduction in definitive endoderm. | Reduction in Hh activity. | (210) |
Table 5.
Current mouse models to study the function of Ift anterograde motors in bone development
| Gene | Mouse models | Mutation | Cilia | Phenotype | Pathways | Ref |
|---|---|---|---|---|---|---|
| Kif3a | Kif3a-Null | Removal exon 2 in ESCs | Cilia loss | Structural abnormalities, defects in left and right asymmetry and cilia loss in embryonic node. | Not reported | (94) |
| Kif3aflox/flox; Wnt1-Cre | Deletion in neural crest lineage cells | Not reported | Die shortly after birth due to respiratory failure, severe craniofacial defects with clefting and widened frontonasal prominence. | Enhanced Hh signaling in facial mesenchyme. Impaired Hh signaling in Kif3a-deficient neural crest-derived mesenchyme. |
(218, 219) | |
| Kif3aflox/flox; Dermo1-Cre | Deletion in trunk and limb skeletal progenitor cells | Not reported | Die at birth, split sternum, polydactyly and defects in ribs, knee joints and limbs. | Not reported | (218) | |
| Kif3aflox/flox; Prx1-Cre | Deletion in the lateral plate mesoderm | Cilia loss | Forelimb polydactyly and profound shortening of limb bones. | Disrupted Hh signaling in long bones. | (174, 218) | |
| Kif3aflox/flox; Col2α1-Cre | Deletion in cartilage | Cilia disrupted (93, 220) | Postnatal dwarfism. Abnormal cranial base development and altered synchondrosis growth plate organization and function at postnatal stages. |
Hh signaling significantly reduced within growth plates but enhanced and widespread all along perichondrial tissues. Chondrocytes proliferation reduced but the hypertrophic differentiation is unregulated in growth plate, Hh signaling is unchanged. |
(93, 220) | |
| Kif3aflox/flox; Oc-Cre | Deletion in mature osteoblast | Reduced cilia number and length | Osteopenia by 6 weeks of age and partially recovered at 16 weeks. | Impaired osteoblastic differentiation, impaired intracellular calcium response to fluid flow shear stress, and reduced Hh and Wnt response. | (221) | |
| Kif3a; Col3a; I 2.3-Cre | Deletion in osteoblast progenitor | Not reported | No obvious skeletal defects. Reduced bone formation in response to mechanical ulnar loading. | Not reported | (222) | |
| Kif3b | Kif3b-Null | Null mutation with first exon replaced by neo cassette | Cilia loss | Die at mid-gestation and exhibited randomized left–right asymmetry, growth retardation, pericardial sac ballooning and neural tube disorganization. | Not reported | (225) |
| Kif17 | Kif17−/−(231) | Frame shift with function loss | Not reported | No gross defects | Not reported | (231) |
| Kif7 | Kif7flox/flox; NLS-Cre | Ubiquitous deletion with NLS-Cre | Not reported | Die at birth with severe malformations, including exencephaly, polydactyly and sternal defects. | Up-regulated full-length Gli2 while down-regulated truncated 83-kD repressor Gli3 in total embryo lysates. | (215) |
| Kif7maki | ENU- mutagenesis, Loss of function | Normal cilia | Die at the end of gestation with abnormal motor neurons and preaxial polydactyly. | Reduced processed Gli3 proteins and the ratio of processed Gli3/full-length Gli3. | (206) | |
| Ift172wimKif7maki | Double mutants (206) | Disrupt cilia | Similar to Ift172wim. | Not reported | (206) | |
| Dync2h1mmiKif7maki | Double mutants (206) | Not reported | Similar to Kif7maki with greater loss of ventral neural cell types. | A stronger loss of Shh signaling than Dync2h1mmi. | (206) | |
| Kif7-KO | Knockout allele | Normal cilia | Spinal cord defects, preaxial polydactyly, exencephaly, and microphthalmia, which mimic the Gli3 mutants phenotypes. | Reduced proteolytic processing of Gli3. Reduce Gli2 and Gli3 accumulation at the tip of cilia in response to Hh. |
(216) |
5.4.1. Dynein-2
The dynein motor protein family contains axonemal dynein, cytoplasmic dynein-1, and cytoplasmic dynein-2, which are involved in a wide range of functions in eukaryotic cells (See review (202)). Dynein-2 only exists in the organisms that have cilia or eukaryotic flagella and is believed to function primarily as the retrograde motor for Ift (202). Dynein-2 contains a heavy chain (DYNC2H1), an intermediated chain (D2IC), a light intermediate chain (D2LIC, or LIC3), and a light chain (LC) (201, 203). The mouse homology of LC and D2IC has not been reported, and how the Dynein-2 motor interacts with Ift protein complexes is largely unknown.
5.4.1.1. DYNC2H1
Dync2h1 encodes the heavy chain of the cytoplasmic dynein-2 motor (DYNC2H1, or DHC1b and DHC2) (35). The Dync2h1 mutation has been linked to the JATD and SRP (33, 35, 131). Chondrocytes from patients with Dync2h1 mutation had short cilia with bulbous distal ends (33).
Huangfu et al generated two mouse lines with Dync2h1 mutation named Dnchc2lln and Dnchc2GT (48). Dnchc2lln is ENU induced mutation with a phenylalanine-to-serine (F3890S) mutation in a conserved residue of the Dnchc2 motor domain. Dnchc2GT is generated by using the ESC gene trap insertion approach, which produces a truncated Dync2h1 (lacking the entire dynein motor domain) fused to β-geo (48). Homozygous Dnchc2lln, Dnchc2GT, and transheterozygous Dnchc2lln/Dnchc2GT mice all die at approximately E12.5 and have similar phenotypes, including abnormal brain morphology, randomization of heart-looping polarity and polydactylous. Cilia were present on the Dnchc2lln embryos with altered morphology. These cilia are similar to those seen in the Ift-A defect embryos, with reduced length and a bulge along their length (48, 204). Among Dync2h1lln, Dync2h1ttn and Dync2h1Gt, cilia in Dync2h1lln are less affected (204). Hh signaling is disrupted, and the Ptch1 expression is reduced in Dnchc2lln neural tube. Besides, Hh pathway is fully activated in Ptch1 mutants. Dnchc2lln Ptch1 double mutant mice has the similar phenotypes as Dnchc2lln (48). Smo, Gli2, and Ptch1 accumulate along the Dnchc2lln cilia with or without Shh stimulation (204). These results suggest that Dync2h1 acts downstream of Ptch1.
Dync2h1ttn was generated by Ocbina et al also using ENU mutation method (205). Dync2h1ttn carries an A-to-C substitution in the splice donor site upstream of exon 51, which deletes 38 amino acids from the ATPase-activator (AAA) domain 4 (205). Cilia morphology is altered on the Dync2h1ttn MEFs with reduced length and bulges along the axoneme (67). Smo accumulates in the primary cilia of Dync2h1ttn MEFs even in the absence of Shh. But the presence of Smo in cilia is not sufficient to activate the Gli-luciferase reporter, suggesting that retrograde Ift blocks activated Gli2 to move out of the cilia (205). Wnt signaling is normal in mid-gestation Dync2h1ttn embryo or embryo-derived fibroblasts (67).
Besides the above mentioned models, Liem et al generated another ENU-induced Dync2h1 mutation mouse line Dync2h1mmi (206). Dync2h1mmi has a missense mutation (I1789N) in the motor domain. Dync2h1mmi has defects in neural tube and motor neurons similar to other Dync2h1 mutants (206). Transmission electron microscopy (TEM) analysis revealed that cilia in Dync2h1mmi are swollen and filled with electron-dense particles (161). Axonemal microtubules in Dync2h1mmi cilia are defective having a reduced number of cilia and singlets instead of doublets (161).
May et al identified two independent ENU-induced null mutations, Dnchc2Q397Stop and Dnchc2W2502R (207). Dnchc2Q397Stop carries an early nonsense mutation at amino acid position 397 (an early stop codon). Dnchc2W2502R carries a missense mutation in a conserved AAA domain (position 2502) that is predicted to be required for motor activity. Both Dnchc2Q397Stop and Dnchc2W2502R show similar developmental defects with severe defects in dorsoventral patterning of the forebrain and patterning of the spinal cord (207). Most of the mutant embryos die at E12.0 or earlier with pericardial edema and heart failure. Cilia in mutant embryos are shorter but wider than the normal cilia. Both Gli activator (Gli1 expression) and Gli repressor activity (Hoxd13 and gremlin expression) are significantly lower. In addition, mutant embryos have a disruption of the Gli3 proteolytic processing and Smo localized to the cilia (207).
Ocbina et al studied the relationships between Dync2h1 and other genes required for ciliary trafficking. To investigate the relationship between anterograde ciliary trafficking and Dync2h1, they generated double mutant allele of Ift172, an Ift-B gene and Dync2h1. The phenotype of Dync2h1llnIft172wim double mutant is similar to that of Ift172wim (a null mutation allele), showing an absent of cilia and defects in all ventral neural cell types (204). Double mutation of Dync2h1 and Ift172 using Ift172avc1, a partial loss-of-function allele of Ift172, can partially rescue the Dync2h1lln phenotype. Interestingly, a more dramatic rescue in Shh signaling and cilia morphology is found in the Dync2h1llnIft172avc1/+ double mutants. Dync2h1llnIft172avc1/+ embryos survive to at least E16.5 without having polydactyly. Thus, these results suggest that a modest reduction of Ift-B expression and anterograde trafficking can rescue ciliogenesis and Shh signaling transduction in the absence of Dync2h1 (204). To further elucidate the relationship between retrograde motors and Ift-A proteins, Ift122 from Ift-A was used to generate Ift122 and Dync2h1 double mutant mice. Interestingly, mutants that are homozygous for both Dync2h1lln and Ift122sopb, resemble the Ift122sopb phenotype with similar cilia morphology and ectopic activity of the Shh pathway in the neural tube (204). Ift88 is enriched at the cilia, and Gli2 is limited to the tips of cilia in Ift122sopbDync2h1lln. However, Smo is only in cilia with Shh stimulation in Ift122sopbDync2h1lln. Deletion of Ift122 relieves the accumulation of proteins within the cilia caused by loss of Dync2h1, suggesting Ift122 or Ift-A complex plays a role in anterograde as well as retrograde trafficking (204). Ift122sopb leads to the lack of Dync2h1 along the cilia, which may explain why the phenotypes in Ift122sopbDync2h1lln double mutants are similar to that in the Ift122sopb single mutants. Dync2h1llnIft122sopb/+, which has lowered expression of Ift122, can partially rescue the phenotype of Dync2h1lln, including the cilia morphology, ventral neural development and Shh-dependent protein trafficking in cilia (204), indicating that normal Shh-dependent protein trafficking in cilia can take place in the absence of Dync2h1 if the Ift122 level is reduced.
5.4.1.2. mD2LIC
Mouse dynein-2 light intermediate chain (mD2LIC) was recently discovered. Both reciprocal immunoprecipitations and sedimentation assays showed that mD2LIC interacts specifically with DYNC2H1, suggesting mD2LIC is also involved in retrograde transport (208, 209).
Rana et al generated a null mD2LIC mutant mice (mD2LIC−/−) by disrupting both the transcriptional and translational start sites in mD2LIC (210). mD2LIC−/− mice die before E11.5 with defects in notochord and floorplate formation, a reduction in definitive endoderm and loss of cilia in the node of mD2LIC−/− embryos. mD2LIC−/− embryos displayed reduction in Hh signaling activity, which is similar to the phenotype observed in Smo−/− and Shh−/−Ihh−/− double mutants.
5.4.2. Kinesin-2
Kinesins are ATP-dependent microtubule-based molecular motors that carry cargo along microtubules in an anterograde direction (211, 212). There are more than 40 members in the kinesin superfamily that can be divided into 14 distinct families (kinesin-1 to -14) (213). Accumulating evidence shows heterotrimeric kinesin-2 motor is the “core” of IFT motor while other proteins including kinesin-2, -3 and -4 function as “accessory” motors for the transport of specific cargo dependent on cell types (211). In vertebrates, two distinct heterotrimeric kinesin-2 complexes, named kinesin-2 family protein A (Kif3a)/Kif3b/ kinesin-associated protein (KAP3) and Kif3A/Kif3C/ KAP3, which may play different roles in the ciliogenesis and function. In addition, heterotrimeric kinesin-2 and homodimeric Kif17 in the Kinesin-2 family also transport exogenously expressed cyclic nucleotide-gated channels into the cilium (214). Kif7 in the kinesin-4 family is localized to cilia base and the tip in mouse embryos and plays important roles in the ciliary trafficking and the response of Hh signaling (206, 215, 216).
5.4.2.1. Kif3a
Marszalek et al generated the Kif3a-Null mouse by removing exon 2 in ESCs, which completely disrupted Kift3a protein expression (94). The Kif3a-Null mouse usually dies at E10 with structural abnormalities, defects in left and right asymmetry, and cilia loss in embryonic node, suggesting that a kinesin-based transport is required for cilia formation (94).
Kif3aflox/flox was generated by inserting two LoxP sites, which flanks exon 2 of Kif3a in the intronic DNA sequences (94, 217). By crossing these mice with Wnt1-Cre (Kif3aflox/flox; Wnt1-Cre or Kif3a CKO), Kif3a is inactive in neural crest lineage cells. Kif3aflox/flox ; Wnt1-Cre mice die shortly after birth due to respiratory failure and display severe craniofacial defects, including clefting of the secondary palate and widened frontonasal prominence (218, 219). Kolpakova-Hart et al reported the Kif3aflox/flox; Wnt1-Cre mice have the similar phenotype as Smoflox/flox; Wnt1-Cre mice (Shh signaling is disrupted in neural crest cells due to deletion of Smo). In addition, Gli1 is not detectable in the mesenchymal cells. Those data demonstrate that Hh signaling is impaired in Kif3a-deficient neural crest–derived mesenchyme (218). In contrast, Brugmann et al found that Kif3a-deficient neural crest–derived facial mesenchyme lacks cilia and has enhanced Hh signaling, including enhanced Gli1, Ptch1 and Shh, but reduced Gli3, which accounts for the excessive proliferation in the facial prominences (219). The opposite Hh signaling response after deletion of Kif3a may result from inconsistent expression of Gli proteins in different tissues.
To study the role of Kif3a in axial and appendicular skeleton development, Dermo1-Cre (mesoderm-specific) was used to inactivate Kif3a in the trunk and limb skeletal progenitor cells (Kif3aflox/flox; Dermo1-Cre) (218). Kif3aflox/flox; Dermo1-Cre mice die immediately after birth, which likely result from the defects in ribcage. Besides, Kif3aflox/flox; Dermo1-Cre mice have a split sternum, polydactyly and defects in knee joints and limbs (218).
Restricted deletion of Kif3a in the lateral plate mesoderm with Prx1-Cre (Kif3aflox/flox; Prx1-Cre), which is limb/sternum-specific and does not affect the somitic mesoderm skeletal derivatives, such as ribs and vertebrae, leads to abnormal rib pairing, forelimb polydactyly and profound shortening of limb bones (174, 218). Ihh signaling is inactive (reduced Gli1 expression and normal Gli3 expression) in the long bone in Kif3aflox/flox; Prx1-Cre, which contributes to defects in chondrocyte differentiation and the loss of the bone collar adjacent to the metaphysis (174, 218).
Specific deletion of Kif3a in cartilage using Col2α1-Cre (Kif3aflox/flox; Col2α1-Cre) resulted in abnormal cranial base development and altered synchondrosis growth plate organization and function at the postnatal stage. Kif3a deficient synchondroses display a delay in chondrocyte hypertrophy and enhanced intramembranous ossification. Hh signaling is significantly reduced within mutant growth plates but enhanced and widespread in all of the perichondrial tissues in Kif3aflox/flox; Col2α1-Cre mice as detected the expression of Ptch1, Gli1, and syndecan-3 (hedgehog-binding cell-surface component) (220). Song et al reported that Kif3aflox/flox; Col2α1-Cre mice have postnatal dwarfism. Chondrocytes proliferation is reduced, but the hypertrophic differentiation is up-regulated, which leads to premature loss of the growth plate in Kif3aflox/flox; Col2α1-Cre mice (93). Moreover, cilia, chondrocyte rotation, actin cytoskeleton and localization of activated FAK are all disrupted, suggesting that an important role of Kif3a or cilia in maintaining the columnar organization of the growth plate and chondrocyte differentiation. In addition, Ptch1 expression of the Hh signaling pathway is not changed (93).
To analyze the function of Kif3a in osteoblast formation and function, Qiu et al used Oc-Cre (osteocalcin promoter, limited in mature osteoblast) to specifically delete Kif3a in the osteoblast linage (Kif3aflox/flox; Oc-Cre). Kif3aflox/flox; Oc-Cre mice exhibit osteopenia at six weeks of age, which is partially recovered at 16 weeks. Kif3a deficiency leads to a reduction in the number of cilia and their length. Deletion of Kif3a increases cell proliferation but impairs osteoblastic differentiation. Qiu et al also found that deletion of Kif3a impairs intracellular calcium response to fluid flow shear stress and reduces Hh and Wnt responses, which may account for the defect of osteoblast-mediated bone formation in Kif3aflox/flox; Oc-Cre mice (221). Temiyasathit et al generated another osteoblast linage mutant mouse line (Kif3aflox/flox; Kif3a; Colα1(I)) using the Col1α 2.3-Cre (2.3kb fragment of the α1(I)-collagen promoter, deletion in osteoblast progenitor cells) mice (222). Kif3a; Colα1(I)-Cre mice have no obvious skeletal defects. However, Kif3a; Colα1(I)-Cre mice exhibited reduced bone formation in response to mechanical ulnar loading, which linked the Kif3a/cilia to mechanosensor function in the bone (222).
Kinumatsu et al studied the role of Kif3a/cilia in temporoman-dibular joint (TMJ) development (223). Three-month-old Kif3a; Col2α1-cre mice had narrow and flat condyles with an irregular bony surface that was often fused with the articular disc. Alteration of Ihh signaling topography was associated with ectopic bone formation in condyles, implicating Kif3a/cilia are required for the coordination of Hh in chondrogenic condylar growth (223).
5.4.2.2. Kif3b
Kif3b is the other motor subunit in kinesin-2. A recent study suggested the Kif3b serves as a link between polycystin-2 (PC2, encoded by PKD2) and fibrocystin/polyductin (FPC, encoded by PKHD1, which is responsible for ARPKD), and mediates the regulation of PC2 channel function by FPC (224).
Nonaka et al generated the Kif3b-Null mutant mouse model by replacing most of the first exon (encoding the entire kinesin motor domain) with neo cassette (225). Kif3b-Null embryos die at mid-gestation and exhibit randomized left–right asymmetry, growth retardation, pericardial sac ballooning, and neural tube disorganization. Cilia is completely absent in the nodes of Kif3b-Null embryos.
5.4.2.3. Kif7
Kif7 is the mammalian homologue of Drosophila Costal2 (Cos2), which plays a central role in the Hh pathway in Drosophila (216, 226). Kif7 belongs to the kinesin-4 family and has an N-terminal motor domain (227). Knockdown of Kif7 in zebrafish resulted in ectopic Hh signaling, suggesting that Kif7 also acts in Hh signal in vertebrates (228).
Cheung et al generated the conditional allele of Kif7 by flanking exons 2–4 with LoxP sites (Kif7flox/flox) (215). By crossing Kif7flox/flox with NLS-Cre transgenic mice (ubiquitous Cre expression), they created the null mutation of Kif7 (Kif7flox/flox; NLS-Cre), and found that Kif7flox/flox; NLS-Cre mice die at birth with severe malformations, including exencephaly, polydactyly, and sternal defects, a phenotype similar to the phenotype of Gli3 mutant mice. They further found that Kif7 physically interacts with Gli transcription factors and controls their proteolysis and stability (215). The deletion of Kif7 up-regulates full-length Gli2 while it down-regulates truncated 83-kD repressor Gli3 in total embryo lysates. To fully understand the role of Kif7 in Hh signaling pathway, they further generated double mutants by using Smo mutant, Gli2 mutant, or Gli3 mutant mice to cross with Kif7flox/flox; NLS-Cre, and compared these mutants with each single mutant. Their results suggested that Kif7 plays a critical role in the balance of Gli activators and repressors downstream of Smo (215).
Liem et al identified an ENU-induced matariki (maki) mutation of Kif7 (Kif7maki) (206). Kif7maki is a missense mutation (L130P) in a conserved region of the motor domain of Kif7, which led to the loss of Kif7 function. Kif7maki mice die at the end of gestation with abnormal motor neurons, preaxial polydactyly and elevated Shh signaling. The results from Smo and Kif7 double mutation result in Shh-dependent ventral neural cell types at E10.5, suggesting that Kif7 functions downstream of Smo and that loss of Kif7 causes ligand-independent activation of Shh pathway. Loss of Kif7 leads to a reduction in processed Gli3 proteins and in the ratio of processed Gli3/full-length Gli3, confirming the role of Kif7 in Gli3 proteolysis or stability. Kif7maki mice have normal cilia. To study the relationship between Kif7 and cilia, Ift172wimKif7maki double mutants were generated by Liem et al (206). They found that Ift172wimKif7maki is similar to Ift172wim mutants, suggesting that the activity of Kif7 depends on the presence of cilia. To analyze the role of Kif7 in ciliary trafficking (206), Liem et al also generated Dync2h1mmiKif7maki double mutants and found that Dync2h1mmiKif7maki phenocopies Kif7maki mutants with greater loss of ventral neural cell types, suggesting that Kif7 functions before retrograde Ift, and that DYNC2H1 has additive and positive roles in Shh signaling (206).
Endoh-Yamagami et al generated Kif7 knockout mice (Kif7-KO) by replacing the initiation codon and following N-terminus motor domain with a neo cassette (216). Kif7-KO embryos display spinal cord defects, preaxial polydactyly, exencephaly, and microphthalmia, which mimic the phenotypes of Gli3 mutants. The Gli3 repression targets Gremlin and HoxD13 are elevated in Kif7-KO. Meanwhile, Gli1 and Ptch1 expression are normal in both Kif7-KO embryos and MEFs. Those data suggest that Kif7 regulates Hh signaling mainly through controlling the formation or function of the Gli3 repressor. Cilia morphology is normal in MEFs from Kif7-KO embryos. Kif7 accumulates at the tip of the cilia in an Hh-dependent manner. Kif7 physically interacts with Gli3 and is required for efficient Gli3 processing to its repressor form, indicating that Kif7 functions principally as a negative regulator of the Hh pathway in mice.
5.4.2.4. Kif17
Kif17 belongs to the kinesin-2 family, and a pair of Kif17 forms a homodimer with their head domain (229). Kif17 has diverse functions in different species and different kind of cells. Dysfunctions of Kif17 are linked to a number of pathologies (See review (229)). Kif17 or its homolog mediates Ift cargos to the ciliary tip, therefore, it plays a role in ciliogenesis and function (229). Recently, a new peptide named TTC308 has been identified in Ift complex B in mouse pituitary cells that interacted with mouse Kif17 in co-immunoprecipitation studies (230). This finding confirmed the possible functional interaction between Kif17 and cargo transport. Whether Kif7 takes part in the ciliogenesis in mice is currently unknown.
Yin et al generated Kif7 mutant mice (Kif17−/−) by replacing exon 4 of Kif17 with a poly (A)-less PGK-neo cassette (231) resulting in a frame shift, and disruption of Kif17 expression. Kif17−/− mice grow normally without any gross defects. They found that Kif7 is important for maintenance of N-methyl-D-aspartate (NMDA) receptor subunit 2B (NR2B) and NR2A level in hippocampal neurons, which is involved in memory and the learning process. However, whether Kif17−/− mice have the bone related phenotype has not been evaluated yet (231).
6. SUMMARY AND PERSPECTIVES
The development of transgenic and mutagenesis technology provides powerful tools to study the function of cilia, Ift proteins, and motors. Some of the Ift proteins and motors already have the mouse models established. A few of them have more than one model with different degrees of loss of function, allowing the study of dosage dependent effects of these genes. The loss of Ift complex A protein usually leads to the “swollen” cilia, which are filled with complex B proteins, suggesting that the complex B, which is in charge of the anterograde transport, can function without the complex A. Mutations of most Ift complex B proteins lead to cilia disruption due to the totally blocked anterograde transport. This raises the problems to distinguish the function of individual Ift proteins and to identify whether the phenotype results from a deficient Ift protein alone or is secondary to cilia loss. It is also possible that the severe consequence of loss of cilia masks the other function of Ift proteins independent of cilia.
Over the past few years, much attention has been paid to elucidating the gene functions and mimicking the human diseases using mouse models. However, the treatment of Ift- and cilia-related diseases in humans is still at the initial developmental stages. For the patients with the Ift protein or Ift motor mutation, exogenous introduction of the Ift or Ift-related proteins or gene to treat these diseases are worth trying. However, before doing so, it is necessary to figure out which genes are defective in each individual patient and how these genes interact with other genes in the cells. Currently, several candidate modulators for ciliogenesis and cilium length have been found from one high-throughput functional screening (232). Further studies should elucidate whether these modulators can rescue ciliogenesis and cilia function in Ift proteins/motors defective or mutant cells. Proper dosage and delivery methods could be tested with the described mouse models. We anticipate that those small molecules will provide us novel strategies to treat ciliopathies. More work is needed to achieve the comprehensive understanding of the downstream pathways of cilia. Much attention has been paid to the Hh signaling pathways, but the interactions of Hh signaling and cilia/Ift, and the relationship of cilia/Ift with Wnt, Notch, PDGF and many other pathways are still ambiguous or largely unknown. With better knowledge of these pathways and how they interact with cilia/Ift, proper agonists or antagonists can be selected or developed to regulate the downstream signaling. These mouse models provide the needed tools to test the target candidates for the treatment of the ciliopathies patients.
Acknowledgments
We thank Dr. Kristina Wasson-Blader for editing our manuscript. We apologize to the many researchers whose work could not be cited due to space limitations. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and National Institute of Dental and Craniofacial Research under Award Numbers AR061052, AR066101 and DE023105 to S.Y. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- MTs
microtubules
- Ift
intraflagellar transport
- JATD
Jeune asphyxiating thoracic dystrophy
- SRP
short rib polydactyly syndromes
- Hh
hedgehog
- Ihh
Indian hedgehog
- Shh
Sonic hedgehog
- Ptch1
Patched 1
- Smo
Smoothened
- PCP
planar cell polarity
- BBS
Bardet–Biedl syndrome
- Evc
Ellis-van Creveld syndrome protein homolog
- OFD1
oral-facial-digital syndrome 1
- MKS
Meckel-Gruber syndrome
- JBTS
Joubert syndrome
- JSRD
JBTS-related disorder
- LCA
Leber congenital amaurosis
- PCD
primary ciliary dyskinesia
- KS
Kartagener syndrome
- NPHP
nephronophthisis
- SLS
Senior-Loken syndrome
- ADPKD
Autosomal dominant polycystic kidney disease
- ARPKD
autosomal recessive polycystic kidney disease
- US
Usher syndrome
- ALMS
Alström syndrome
- MSS
Mainzer-Saldino-Syndrome
- ATD
asphyxiating thoracic dystrophy
- WAD
Weyers acrofacial dysostosi
- NEK1
never-in-mitosis Kinase 1
- CED
Cranioectodermal dysplasia
- MKS
Meckel syndrome
- ENU
N-ethyl-N-nitrosourea
- PDGF-A
platelet-derived growth factor A
- ESCs
embryonic stem cells
- THM1
Tetratricopeptide Repeat Containing Hedgehog Modulator 1
- MEFs
mouse embryo fibroblasts
- PD
proximal-distal
- AP
anterior-posterior
- ACIII
adenylyl cyclase III
- SUFU
Suppressor of Fused
- BMSCs
bone marrow derived stromal cells
- Sfrp5
secreted frizzled-related sequence protein 5
- OA
osteoarthritis
- neo
neomycin
- GMAP210
Golgi Microtubule Associated Protein 210
- TRIP11
thyroid hormone receptor interacting protein 11
- HRGP
human red/green pigment gene promoter
- GTPase
guanosine triphosphate hydrolases
- Fgfr1
fibroblast growth factor receptor-1
- Col12α1
type XII collagen
- Bgn
biglycan
- Mmp10
matrix metalloproteinase-10
- Hsp47
heat shock protein 47 kDa
- Traf3
TNFR-associated factor-3
- IL-13R1
interleukin-13 receptor α1
- DISC1
Disrupted-In-Schizophrenia 1
- STAT6
signal transducer and activator of transcription-6
- AAA
ATPase-activator
- PC2
polycystin-2
- TMJ
temporoman-dibular joint
- FPC
fibrocystin/polyductin
- OSM-3
osmotic avoidance abnormal protein 3
- NMDA
N-methyl-D-aspartate
- NR2B
NMDA receptor subunit 2B
References
- 1.Teti A. Bone development: overview of bone cells and signaling. Curr Osteoporos Rep. 2011;9(4):264–73. doi: 10.1007/s11914-011-0078-8. [DOI] [PubMed] [Google Scholar]
- 2.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2(4):389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 3.Cooper C, Harvey N, Javaid K, Hanson M, Dennison E. Growth and bone development. Nestle Nutr Workshop Ser Pediatr Program. 2008;61:53–68. doi: 10.1159/000113170. [DOI] [PubMed] [Google Scholar]
- 4.Cancedda R, Descalzi Cancedda F, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265–358. doi: 10.1016/s0074-7696(08)62109-9. [DOI] [PubMed] [Google Scholar]
- 5.Colnot C. Cellular and molecular interactions regulating skeletogenesis. J Cell Biochem. 2005;95(4):688–97. doi: 10.1002/jcb.20449. [DOI] [PubMed] [Google Scholar]
- 6.van der Eerden BC, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24(6):782–801. doi: 10.1210/er.2002-0033. [DOI] [PubMed] [Google Scholar]
- 7.Day TF, Yang Y. Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am. 2008;90(Suppl 1):19–24. doi: 10.2106/JBJS.G.01174. [DOI] [PubMed] [Google Scholar]
- 8.Zanotti S, Canalis E. Notch regulation of bone development and remodeling and related skeletal disorders. Calcif Tissue Int. 2012;90(2):69–75. doi: 10.1007/s00223-011-9541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen V. BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev. 2009;20(5–6):475–80. doi: 10.1016/j.cytogfr.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Combs CE, Nicholls JJ, Duncan Bassett JH, Williams GR. Thyroid hormones and bone development. Minerva Endocrinol. 2011;36(1):71–85. [PubMed] [Google Scholar]
- 11.Smirnov SV, Vasil’eva AB. The role of thyroid hormones in skull bone development in the ribbed newt Pleurodeles waltl (Urodela: Salamandridae) Dokl Biol Sci. 2001;379:396–8. doi: 10.1023/a:1011681003652. [DOI] [PubMed] [Google Scholar]
- 12.Barlow-Stewart K. The Australasian genetics resource book : a source of current and accurate information for health professionals, individuals and families, teachers and students. Centre for Genetics Education; 2007. [Google Scholar]
- 13.Zelzer E, Olsen BR. The genetic basis for skeletal diseases. Nature. 2003;423(6937):343–8. doi: 10.1038/nature01659. [DOI] [PubMed] [Google Scholar]
- 14.Makitie O. Molecular defects causing skeletal dysplasias. Endocr Dev. 2011;21:78–84. doi: 10.1159/000328131. [DOI] [PubMed] [Google Scholar]
- 15.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–48. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 16.Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–73. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michaud EJ, Yoder BK. The primary cilium in cell signaling and cancer. Cancer Res. 2006;66(13):6463–7. doi: 10.1158/0008-5472.CAN-06-0462. [DOI] [PubMed] [Google Scholar]
- 18.Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol. 2011;12(4):222–34. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, Blanco MJ, Wilson A, Liu YN, Miles C, Peters H, Goodship JA. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development. 2007;134(16):2903–12. doi: 10.1242/dev.007542. [DOI] [PubMed] [Google Scholar]
- 20.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A. 2003;100(9):5286–91. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134(2):307–16. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 22.Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R, Pacifici M. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 2007;134(11):2159–69. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pazour GJ, Rosenbaum JL. Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol. 2002;12(12):551–5. doi: 10.1016/s0962-8924(02)02410-8. [DOI] [PubMed] [Google Scholar]
- 24.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18(5):1381–8. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 25.Pan J, Wang Q, Snell WJ. Cilium-generated signaling and cilia-related disorders. Lab Invest. 2005;85(4):452–63. doi: 10.1038/labinvest.3700253. [DOI] [PubMed] [Google Scholar]
- 26.KozMINSKI KG, Johnson KA, Forscher P, Rosenbaum JL. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proceedings of the National Academy of Sciences. 1993;90(12):5519–5523. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbaum JL, Witman GB. Intraflagellar transport. Nature Reviews Molecular Cell Biology. 2002;3(11):813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 28.Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012;83(2):S12–S22. doi: 10.1016/j.diff.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber C, Cormier-Daire V. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Wiley Online Library; 2012. Ciliary disorder of the skeleton. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz RS, Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. New England Journal of Medicine. 2011;364(16):1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 32.Baker K, Beales PL. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Wiley Online Library; 2009. Making sense of cilia in disease: the human ciliopathies. [DOI] [PubMed] [Google Scholar]
- 33.Merrill AE, Merriman B, Farrington-Rock C, Camacho N, Sebald ET, Funari VA, Schibler MJ, Firestein MH, Cohn ZA, Priore MA. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. The American Journal of Human Genetics. 2009;84(4):542–549. doi: 10.1016/j.ajhg.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MA, Keighren M, Bahlo M, Bromhead CJ, Budd P. Human and Mouse Mutations in WDR35 Cause Short-Rib Polydactyly Syndromes Due to Abnormal Ciliogenesis. The American Journal of Human Genetics. 2011;88(4):508–515. doi: 10.1016/j.ajhg.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dagoneau N, Goulet M, Geneviève D, Sznajer Y, Martinovic J, Smithson S, Huber C, Baujat G, Flori E, Tecco L. DYNC2H1 Mutations Cause Asphyxiating Thoracic Dystrophy and Short Rib-Polydactyly Syndrome, Type III. The American Journal of Human Genetics. 2009;84(5):706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scherft JP, Daems WT. Single cilia in chondrocytes. J Ultrastruct Res. 1967;19(5):546–55. doi: 10.1016/s0022-5320(67)80080-7. [DOI] [PubMed] [Google Scholar]
- 37.Tummala P, Arnsdorf EJ, Jacobs CR. The role of primary cilia in mesenchymal stem cell differentiation: a pivotal switch in guiding lineage commitment. Cell Mol Bioeng. 2010;3(3):207–212. doi: 10.1007/s12195-010-0127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malone AM, Anderson CT, Tummala P, Kwon RY, Johnston TR, Stearns T, Jacobs CR. Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc Natl Acad Sci U S A. 2007;104(33):13325–30. doi: 10.1073/pnas.0700636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao Z, Zhang S, Mahlios J, Zhou G, Magenheimer BS, Guo D, Dallas SL, Maser R, Calvet JP, Bonewald L. Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. Journal of Biological Chemistry. 2006;281(41):30884–30895. doi: 10.1074/jbc.M604772200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uzbekov R, Maurel D, Aveline P, Pallu S, Benhamou C, Rochefort G. Centrosome Fine Ultrastructure of the Osteocyte Mechanosensitive Primary Cilium. Microscopy and Microanalysis. 2012;18(6):1430. doi: 10.1017/S1431927612013281. [DOI] [PubMed] [Google Scholar]
- 41.Federman M, Nichols G., Jr Bone cell cilia: vestigial or functional organelles? Calcif Tissue Res. 1974;17(1):81–5. doi: 10.1007/BF02547216. [DOI] [PubMed] [Google Scholar]
- 42.Temiyasathit S, Jacobs CR. Osteocyte primary cilium and its role in bone mechanotransduction. Ann N Y Acad Sci. 2010;1192(1):422–428. doi: 10.1111/j.1749-6632.2009.05243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson CT, Castillo AB, Brugmann SA, Helms JA, Jacobs CR, Stearns T. Primary cilia: cellular sensors for the skeleton. Anat Rec (Hoboken) 2008;291(9):1074–8. doi: 10.1002/ar.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitfield J. The solitary (primary) cilium–a mechanosensory toggle switch in bone and cartilage cells. Cell Signal. 2008;20(6):1019–1024. doi: 10.1016/j.cellsig.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 45.Hoey DA, Tormey S, Ramcharan S, O’Brien FJ, Jacobs CR. Primary Cilia-Mediated Mechanotransduction in Human Mesenchymal Stem Cells. Stem Cells. 2012;30(11):2561–2570. doi: 10.1002/stem.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christensen ST, Pedersen LB, Schneider L, Satir P. Sensory cilia and integration of signal transduction in human health and disease. Traffic. 2007;8(2):97–109. doi: 10.1111/j.1600-0854.2006.00516.x. [DOI] [PubMed] [Google Scholar]
- 47.Wong SY, Reiter JF. The primary cilium: at the crossroads of mammalian hedgehog signaling. Curr Top Dev Biol. 2008;85:225–260. doi: 10.1016/S0070-2153(08)00809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102(32):11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lancaster MA, Gleeson JG. The primary cilium as a cellular signaling center: lessons from disease. Curr Opin Genet Dev. 2009;19(3):220–229. doi: 10.1016/j.gde.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karp SJ, Schipani E, St-Jacques B, Hunzelman J, Kronenberg H, McMahon AP. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and-independent pathways. Development. 2000;127(3):543–548. doi: 10.1242/dev.127.3.543. [DOI] [PubMed] [Google Scholar]
- 51.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan A, Chang L, Nguyen A, James AW. A review of hedgehog signaling in cranial bone development. Frontiers in physiology. 2013;4 doi: 10.3389/fphys.2013.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spinella-Jaegle S, Rawadi G, Kawai S, Gallea S, Faucheu C, Mollat P, Courtois B, Bergaud B, Ramez V, Blanchet AM. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J Cell Sci. 2001;114(11):2085–2094. doi: 10.1242/jcs.114.11.2085. [DOI] [PubMed] [Google Scholar]
- 54.Drossopoulou G, Lewis K, Sanz-Ezquerro J, Nikbakht N, McMahon A, Hofmann C, Tickle C. A model for anteroposterior patterning of the vertebrate limb based on sequential long-and short-range Shh signalling and Bmp signalling. Development. 2000;127(7):1337–1348. doi: 10.1242/dev.127.7.1337. [DOI] [PubMed] [Google Scholar]
- 55.Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418(6901):979–983. doi: 10.1038/nature01033. [DOI] [PubMed] [Google Scholar]
- 56.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426(6962):83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 57.Goetz SC, Ocbina PJ, Anderson KV. The primary cilium as a Hedgehog signal transduction machine. Methods in cell biology. 2009;94:199–222. doi: 10.1016/S0091-679X(08)94010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tasouri E, Tucker KL. Primary cilia and organogenesis: is Hedgehog the only sculptor? Cell and tissue research. 2011;345(1):21–40. doi: 10.1007/s00441-011-1192-8. [DOI] [PubMed] [Google Scholar]
- 59.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 60.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 61.Robbins DJ, Fei DL, Riobo NA. The hedgehog signal transduction network. Sci Signal. 2012;5(246):re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 63.Regard JB, Zhong Z, Williams BO, Yang Y. Wnt signaling in bone development and disease: making stronger bone with Wnts. Cold Spring Harb Perspect Biol. 2012;4(12) doi: 10.1101/cshperspect.a007997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.May-Simera HL, Kelley MW. Cilia, Wnt signaling, and the cytoskeleton. Signal transduction. 2012;11:19. [Google Scholar]
- 65.Sugimura R, Li L. Noncanonical Wnt signaling in vertebrate development, stem cells, and diseases. Birth Defects Research Part C: Embryo Today: Reviews. 2010;90(4):243–256. doi: 10.1002/bdrc.20195. [DOI] [PubMed] [Google Scholar]
- 66.Wang C, Yuan X, Yang S. IFT80 is essential for chondrocyte differentiation by regulating Hedgehog and Wnt signaling pathways. Exp Cell Res. 2013;319(5):623–632. doi: 10.1016/j.yexcr.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ocbina PJR, Tuson M, Anderson KV. Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS One. 2009;4(8):e6839. doi: 10.1371/journal.pone.0006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Awan A, Bell AJ, Satir P. Kin5 knockdown in Tetrahymena thermophila using RNAi blocks cargo transport of Gef1. PloS one. 2009;4(3):e4873. doi: 10.1371/journal.pone.0004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lunt SC, Haynes T, Perkins BD. Zebrafish ift57, ift88, and ift172 intraflagellar transport mutants disrupt cilia but do not affect hedgehog signaling. Developmental Dynamics. 2009;238(7):1744–1759. doi: 10.1002/dvdy.21999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rix S, Calmont A, Scambler PJ, Beales PL. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum Mol Genet. 2011;20(7):1306–1314. doi: 10.1093/hmg/ddr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vanhooren V, Libert C. The mouse as a model organism in aging research: Usefulness, pitfalls and possibilities. Ageing research reviews. 2013;12(1):8–21. doi: 10.1016/j.arr.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 72.Elefteriou F, Yang X. Genetic mouse models for bone studies—Strengths and limitations. Bone. 2011;49(6):1242–1254. doi: 10.1016/j.bone.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marshall WF. Basal bodies platforms for building cilia. Curr Top Dev Biol. 2008;85:1–22. doi: 10.1016/S0070-2153(08)00801-6. [DOI] [PubMed] [Google Scholar]
- 74.Ringo DL. Flagellar motion and fine structure of the flagellar apparatus in Chlamydomonas. The Journal of cell biology. 1967;33(3):543–571. doi: 10.1083/jcb.33.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 76.Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A. 1993;90(12):5519–23. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Orozco JT, Wedaman KP, Signor D, Brown H, Rose L, Scholey JM. Movement of motor and cargo along cilia. Nature. 1999;398(6729):674–674. doi: 10.1038/19448. [DOI] [PubMed] [Google Scholar]
- 78.Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 1998;141(4):993–1008. doi: 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pazour GJ, Wilkerson CG, Witman GB. A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT) J Cell Biol. 1998;141(4):979–92. doi: 10.1083/jcb.141.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mizuno N, Taschner M, Engel BD, Lorentzen E. Structural studies of ciliary components. J Mol Biol. 2012;422(2):163–80. doi: 10.1016/j.jmb.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kozminski KG. Intraflagellar transport—the “new motility” 20 years later. Mol Biol Cell. 2012;23(5):751–753. doi: 10.1091/mbc.E11-11-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II–dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 1998;141(4):993–1008. doi: 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Piperno G, Mead K. Transport of a novel complex in the cytoplasmic matrix of Chlamydomonas flagella. Proceedings of the National Academy of Sciences. 1997;94(9):4457–4462. doi: 10.1073/pnas.94.9.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cole DG. The Chlamydomonas Sourcebook. Ed G. B. Witman; 2009. Intraflagellar transport. [Google Scholar]
- 85.Piperno G, Siuda E, Henderson S, Segil M, Vaananen H, Sassaroli M. Distinct mutants of retrograde intraflagellar transport (IFT) share similar morphological and molecular defects. J Cell Biol. 1998;143(6):1591–1601. doi: 10.1083/jcb.143.6.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 87.Lucker BF, Behal RH, Qin H, Siron LC, Taggart WD, Rosenbaum JL, Cole DG. Characterization of the intraflagellar transport complex B core: direct interaction of the IFT81 and IFT74/72 subunits. J Biol Chem. 2005;280(30):27688–96. doi: 10.1074/jbc.M505062200. [DOI] [PubMed] [Google Scholar]
- 88.Qin H, Diener DR, Geimer S, Cole DG, Rosenbaum JL. Intraflagellar transport (IFT) cargo IFT transports flagellar precursors to the tip and turnover products to the cell body. J Cell Biol. 2004;164(2):255–266. doi: 10.1083/jcb.200308132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Follit JA, Xu F, Keady BT, Pazour GJ. Characterization of mouse IFT complex B. Cell Motil Cytoskeleton. 2009;66(8):457–468. doi: 10.1002/cm.20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cole DG, Snell WJ. SnapShot: Intraflagellar transport. Cell. 2009;137(4):784. doi: 10.1016/j.cell.2009.04.053. [DOI] [PubMed] [Google Scholar]
- 91.Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127(11):2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- 92.Friedland-Little JM, Hoffmann AD, Ocbina PJ, Peterson MA, Bosman JD, Chen Y, Cheng SY, Anderson KV, Moskowitz IP. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum Mol Genet. 2011;20(19):3725–37. doi: 10.1093/hmg/ddr241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song B, Haycraft CJ, Seo H-s, Yoder BK, Serra R. Development of the post-natal growth plate requires intraflagellar transport proteins. Developmental biology. 2007;305(1):202–216. doi: 10.1016/j.ydbio.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proceedings of the National Academy of Sciences. 1999;96(9):5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stottmann RW, Tran PV, Turbe-Doan A, Beier DR. Ttc21b is required to restrict sonic hedgehog activity in the developing mouse forebrain. Dev Biol. 2009;335(1):166–78. doi: 10.1016/j.ydbio.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, Yoder BK, Beier DR. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet. 2008;40(4):403–10. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cortellino S, Wang C, Wang B, Bassi MR, Caretti E, Champeval D, Calmont A, Jarnik M, Burch J, Zaret KS, Larue L, Bellacosa A. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev Biol. 2009;325(1):225–37. doi: 10.1016/j.ydbio.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 99.Loktev AV, Zhang Q, Beck JS, Searby CC, Scheetz TE, Bazan JF, Slusarski DC, Sheffield VC, Jackson PK, Nachury MV. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell. 2008;15(6):854–65. doi: 10.1016/j.devcel.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 100.Sung CH, Leroux MR. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nature cell biology. 2013;15(12):1387–1397. doi: 10.1038/ncb2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, Blanco MJ, Wilson A, Liu YN, Miles C, Peters H, Goodship JA. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development. 2007;134(16):2903–2912. doi: 10.1242/dev.007542. [DOI] [PubMed] [Google Scholar]
- 102.Romio L, Wright V, Price K, Winyard PJ, Donnai D, Porteous ME, Franco B, Giorgio G, Malcolm S, Woolf AS, Feather SA. OFD1, the gene mutated in oral-facial-digital syndrome type 1, is expressed in the metanephros and in human embryonic renal mesenchymal cells. J Am Soc Nephrol. 2003;14(3):680–9. doi: 10.1097/01.asn.0000054497.48394.d2. [DOI] [PubMed] [Google Scholar]
- 103.Zimmermann KW. Beiträge zur Kenntniss einiger Drüsen und Epithelien. Archiv für mikroskopische Anatomie. 1898;52(3):552–706. [Google Scholar]
- 104.Novarino G, Akizu N, Gleeson JG. Modeling human disease in humans: the ciliopathies. Cell. 2011;147(1):70–79. doi: 10.1016/j.cell.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee JE, Gleeson JG. A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 2011;3:59. doi: 10.1186/gm275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jeune M, Beraud C, Carron R. Asphyxiating thoracic dystrophy with familial characteristics. Arch Fr Pediatr. 1955;12(8):886–91. [PubMed] [Google Scholar]
- 107.De Vries J, Yntema J, Van Die C, Crama N, Cornelissen E, Hamel B. Jeune syndrome: description of 13 cases and a proposal for follow-up protocol. European journal of pediatrics. 2010;169(1):77–88. doi: 10.1007/s00431-009-0991-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, Muzny DM, Young AC, Wheeler DA, Cruz P, Morgan M, Lewis LR, Cherukuri P, Maskeri B, Hansen NF, Mullikin JC, Blakesley RW, Bouffard GG, Program NCS, Gyapay G, Rieger S, Tonshoff B, Kern I, Soliman NA, Neuhaus TJ, Swoboda KJ, Kayserili H, Gallagher TE, Lewis RA, Bergmann C, Otto EA, Saunier S, Scambler PJ, Beales PL, Gleeson JG, Maher ER, Attie-Bitach T, Dollfus H, Johnson CA, Green ED, Gibbs RA, Hildebrandt F, Pierce EA, Katsanis N. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43(3):189–96. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbo M, Filhol E, Bole-Feysot C, Nitschke P, Gilissen C, Haugen OH, Sanders JS, Stolte-Dijkstra I, Mans DA, Steenbergen EJ, Hamel BC, Matignon M, Pfundt R, Jeanpierre C, Boman H, Rodahl E, Veltman JA, Knappskog PM, Knoers NV, Roepman R, Arts HH. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;89(5):634–43. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baujat G, Huber C, El Hokayem J, Caumes R, Do Ngoc Thanh C, David A, Delezoide AL, Dieux-Coeslier A, Estournet B, Francannet C, Kayirangwa H, Lacaille F, Le Bourgeois M, Martinovic J, Salomon R, Sigaudy S, Malan V, Munnich A, Le Merrer M, Le Quan Sang KH, Cormier-Daire V. Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. J Med Genet. 2013;50(2):91–8. doi: 10.1136/jmedgenet-2012-101282. [DOI] [PubMed] [Google Scholar]
- 111.Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, Salih MA, Gerber S, Delphin N, Bigot K, Orssaud C, Silva E, Baudouin V, Oud MM, Shannon N, Le Merrer M, Roche O, Pietrement C, Goumid J, Baumann C, Bole-Feysot C, Nitschke P, Zahrate M, Beales P, Arts HH, Munnich A, Kaplan J, Antignac C, Cormier-Daire V, Rozet JM. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet. 2012;90(5):864–70. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schmidts M, Frank V, Eisenberger T, al Turki S, Bizet AA, Antony D, Rix S, Decker C, Bachmann N, Bald M. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Human mutation. 2013;34(5):714–724. doi: 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Forsythe E, Beales PL. Bardet–Biedl syndrome. European Journal of Human Genetics. 2012;21(1):8–13. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Karmous-Benailly H, Martinovic J, Gubler MC, Sirot Y, Clech L, Ozilou C, Augé J, Brahimi N, Etchevers H, Detrait E. Antenatal presentation of Bardet-Biedl syndrome may mimic Meckel syndrome. Am J Hum Genet. 2005;76(3):493. doi: 10.1086/428679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ellis RW, Van Creveld S. A Syndrome Characterized by Ectodermal Dysplasia, Polydactyly, Chondro-Dysplasia and Congenital Morbus Cordis Report of Three Cases. Arch Dis Child. 1940;15(82):65–84. doi: 10.1136/adc.15.82.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ruiz-Perez VL, Goodship JA. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Wiley Online Library; 2009. Ellis–van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to hedgehog ligands. [DOI] [PubMed] [Google Scholar]
- 117.D’Asdia MC, Torrente I, Consoli F, Ferese R, Magliozzi M, Bernardini L, Guida V, Digilio MC, Marino B, Dallapiccola B, De Luca A. Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. European journal of medical genetics. 2013;56(2):80–7. doi: 10.1016/j.ejmg.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 118.Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, Marino B, Dallapiccola B, Wright M, Meitinger T, Polymeropoulos MH, Goodship J. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 2000;24(3):283–6. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- 119.Ruiz-Perez VL, Tompson W Stuart, Blair J Helen, Espinoza-Valdez C, Lapunzina P, Silva EO, Hamel B, Gibbs JL, Young ID, Wright MJ. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. The American Journal of Human Genetics. 2003;72(3):728–732. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tompson SW, Ruiz-Perez VL, Blair HJ, Barton S, Navarro V, Robson JL, Wright MJ, Goodship JA. Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis–van Creveld syndrome patients. Hum Genet. 2007;120(5):663–670. doi: 10.1007/s00439-006-0237-7. [DOI] [PubMed] [Google Scholar]
- 121.Valencia M, Lapunzina P, Lim D, Zannolli R, Bartholdi D, Wollnik B, Al-Ajlouni O, Eid SS, Cox H, Buoni S. Widening the mutation spectrum of EVC and EVC2: ectopic expression of Weyer variants in NIH 3T3 fibroblasts disrupts Hedgehog signaling. Hum Mutat. 2009;30(12):1667–1675. doi: 10.1002/humu.21117. [DOI] [PubMed] [Google Scholar]
- 122.Naiboglu B, Oysu C, Gokceer T. Orofaciodigital syndrome. Ear, nose, & throat journal. 2012;91(1):E8. doi: 10.1177/014556131209100115. [DOI] [PubMed] [Google Scholar]
- 123.Gurrieri F, Franco B, Toriello H, Neri G. Oral–facial–digital syndromes: review and diagnostic guidelines. American Journal of Medical Genetics Part A. 2007;143(24):3314–3323. doi: 10.1002/ajmg.a.32032. [DOI] [PubMed] [Google Scholar]
- 124.Baraitser M. The orofaciodigital (OFD) syndromes. Journal of medical genetics. 1986;23(2):116. doi: 10.1136/jmg.23.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Burn J, Dezateux C, Hall C, Baraitser M. Orofaciodigital syndrome with mesomelic limb shortening. Journal of medical genetics. 1984;21(3):189–192. doi: 10.1136/jmg.21.3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ferrante MI, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, Sylvie O. Identification of the gene for oral-facial-digital type I syndrome. The American Journal of Human Genetics. 2001;68(3):569–576. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. Journal of the American Society of Nephrology. 2004;15(10):2556–2568. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- 128.Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, Zenker M, Zahnleiter D, Stöss H, Beinder E. NEK1 Mutations Cause Short-Rib Polydactyly Syndrome Type Majewski. The American Journal of Human Genetics. 2011;88(1):106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ho NC, Francomano CA, van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am J Med Genet. 2000;90(4):310–314. doi: 10.1002/(sici)1096-8628(20000214)90:4<310::aid-ajmg9>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 130.Cavalcanti DP, Huber C, Sang KHLQ, Baujat G, Collins F, Delezoide AL, Dagoneau N, Le Merrer M, Martinovic J, Mello MFS. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J Med Genet. 2011;48(2):88–92. doi: 10.1136/jmg.2009.069468. [DOI] [PubMed] [Google Scholar]
- 131.El Hokayem J, Huber C, Couvé A, Aziza J, Baujat G, Bouvier R, Cavalcanti DP, Collins FA, Cordier M-P, Delezoide A-L. NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in Beemer Langer cases. J Med Genet. 2012;49(4):227–233. doi: 10.1136/jmedgenet-2011-100717. [DOI] [PubMed] [Google Scholar]
- 132.Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A. Cranioectodermal Dysplasia, Sensenbrenner Syndrome, Is a Ciliopathy Caused by Mutations in the IFT122 Gene. The American Journal of Human Genetics. 2010;86(6):949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Konstantinidou AE, Fryssira H, Sifakis S, Karadimas C, Kaminopetros P, Agrogiannis G, Velonis S, Nikkels PG, Patsouris E. Cranioectodermal dysplasia: a probable ciliopathy. American Journal of Medical Genetics Part A. 2009;149(10):2206–2211. doi: 10.1002/ajmg.a.33013. [DOI] [PubMed] [Google Scholar]
- 134.Levin LS, Perrin J, Ose L, Dorst JP, Miller JD, McKusick VA. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: cranioectodermal dysplasia. J Pediatr. 1977;90(1):55–61. doi: 10.1016/s0022-3476(77)80764-6. [DOI] [PubMed] [Google Scholar]
- 135.Arts HH, Bongers EM, Mans DA, van Beersum SE, Oud MM, Bolat E, Spruijt L, Cornelissen EA, Schuurs-Hoeijmakers JH, de Leeuw N. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011;48(6):390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 136.Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant SG, Roepman R, Knoers NV, Veltman JA, Brunner HG. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87(3):418–23. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hoffer JL, Fryssira H, Konstantinidou AE, Ropers HH, Tzschach A. Novel WDR35 mutations in patients with cranioectodermal dysplasia (Sensenbrenner syndrome) Clin Genet. 2013;83(1):92–5. doi: 10.1111/j.1399-0004.2012.01880.x. [DOI] [PubMed] [Google Scholar]
- 138.Chen CP. Meckel syndrome: genetics, perinatal findings, and differential diagnosis. Taiwanese Journal of Obstetrics and Gynecology. 2007;46(1):9–14. doi: 10.1016/S1028-4559(08)60100-X. [DOI] [PubMed] [Google Scholar]
- 139.Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39(7):875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 140.Roume J, Genin E, Cormier-Daire V, Ma H, Mehaye B, Attie T, Razavi-Encha F, Fallet-Bianco C, Buenerd A, Clerget-Darpoux F. A gene for Meckel syndrome maps to chromosome 11q13. The American Journal of Human Genetics. 1998;63(4):1095–1101. doi: 10.1086/302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Leightner AC, Hommerding CJ, Peng Y, Salisbury JL, Gainullin VG, Czarnecki PG, Sussman CR, Harris PC. The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kyttälä M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestilä M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38(2):155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- 143.Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Sharif S Malik, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, Algazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH, 2nd, Harris PC, Johnson CA. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38(2):191–6. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 144.Szymanska K, Berry I, Logan CV, Cousins SR, Lindsay H, Jafri H, Raashid Y, Malik-Sharif S, Castle B, Ahmed M. Founder mutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia. 2012;1(1):18. doi: 10.1186/2046-2530-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hardouin SN, Nagy A. Mouse models for human disease. Clinical genetics. 2000;57(4):237–244. doi: 10.1034/j.1399-0004.2000.570401.x. [DOI] [PubMed] [Google Scholar]
- 146.Hofker MH, van Deursen JM. Transgenic mouse: methods and protocols. Springer; 2002. [Google Scholar]
- 147.Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51(3):503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 148.Kühn R, Torres RM. Transgenesis Techniques. Springer; 2002. Cre/loxP recombination system and gene targeting. [DOI] [PubMed] [Google Scholar]
- 149.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26(2):99–109. [PubMed] [Google Scholar]
- 150.VanKoevering KK, Williams BO. Transgenic mouse strains for conditional gene deletion during skeletal development. IBMS BoneKEy. 2008;5(5):151–170. [Google Scholar]
- 151.Loonstra A, Vooijs M, Beverloo HB, Al Allak B, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proceedings of the National Academy of Sciences. 2001;98(16):9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Russell W, Kelly E, Hunsicker P, Bangham J, Maddux S, Phipps E. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proceedings of the National Academy of Sciences. 1979;76(11):5818–5819. doi: 10.1073/pnas.76.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stanford WL, Cohn JB, Cordes SP. Gene-trap mutagenesis: past, present and beyond. Nature reviews genetics. 2001;2(10):756–768. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- 154.De-Zolt S, Altschmied J, Ruiz P, von Melchner H, Schnütgen F. Gene Knockout Protocols. Springer; 2009. Gene-trap vectors and mutagenesis. [DOI] [PubMed] [Google Scholar]
- 155.Gross C, De Baere E, Lo A, Chang W, Messiaen L. Cloning and characterization of human WDR10, a novel gene located at 3q21 encoding a WD-repeat protein that is highly expressed in pituitary and testis. DNA and cell biology. 2001;20(1):41–52. doi: 10.1089/10445490150504684. [DOI] [PubMed] [Google Scholar]
- 156.Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci U S A. 2011;108(4):1456–61. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proceedings of the National Academy of Sciences. 2003;100(25):15071–15076. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proceedings of the National Academy of Sciences. 2011;108(4):1456–1461. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Efimenko E, Blacque OE, Ou G, Haycraft CJ, Yoder BK, Scholey JM, Leroux MR, Swoboda P. Caenorhabditis elegans DYF-2, an orthologue of human WDR19, is a component of the intraflagellar transport machinery in sensory cilia. Mol Biol Cell. 2006;17(11):4801–11. doi: 10.1091/mbc.E06-04-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ashe A, Butterfield NC, Town L, Courtney AD, Cooper AN, Ferguson C, Barry R, Olsson F, Liem KF, Jr, Parton RG, Wainwright BJ, Anderson KV, Whitelaw E, Wicking C. Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum Mol Genet. 2012;21(8):1808–23. doi: 10.1093/hmg/ddr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liem KF, Ashe A, He M, Satir P, Moran J, Beier D, Wicking C, Anderson KV. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. The Journal of cell biology. 2012;197(6):789–800. doi: 10.1083/jcb.201110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jonassen JA, SanAgustin J, Baker SP, Pazour GJ. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. Journal of the American Society of Nephrology. 2012;23(4):641–651. doi: 10.1681/ASN.2011080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Caruana G, Farlie PG, Hart AH, Bagheri-Fam S, Wallace MJ, Dobbie MS, Gordon CT, Miller KA, Whittle B, Abud HE, Arkell RM, Cole TJ, Harley VR, Smyth IM, Bertram JF. Genome-wide ENU mutagenesis in combination with high density SNP analysis and exome sequencing provides rapid identification of novel mouse models of developmental disease. PLoS One. 2013;8(3):e55429. doi: 10.1371/journal.pone.0055429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Miller KA, Ah-Cann CJ, Welfare MF, Tan TY, Pope K, Caruana G, Freckmann ML, Savarirayan R, Bertram JF, Dobbie MS. Cauli: A Mouse Strain with an Ift140 Mutation That Results in a Skeletal Ciliopathy Modelling Jeune Syndrome. PLoS Genet. 2013;9(8):e1003746. doi: 10.1371/journal.pgen.1003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Follit JA, Tuft RA, Fogarty KE, Pazour GJ. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Molecular biology of the cell. 2006;17(9):3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Follit JA, San Agustin JT, Xu F, Jonassen JA, Samtani R, Lo CW, Pazour GJ. The Golgin GMAP210/TRIP11 anchors IFT20 to the Golgi complex. PLoS genetics. 2008;4(12):e1000315. doi: 10.1371/journal.pgen.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nature cell biology. 2009;11(11):1332–1339. doi: 10.1038/ncb1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Moyer JH, Lee-Tischler MJ, Kwon HY, Schrick JJ, Avner ED, Sweeney WE, Godfrey VL, Cacheiro N, Wilkinson J, Woychik RP. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science. 1994;264(5163):1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- 169.Lehman JM, Michaud EJ, Schoeb TR, Aydin-Son Y, Miller M, Yoder BK. The Oak Ridge Polycystic Kidney mouse: modeling ciliopathies of mice and men. Developmental Dynamics. 2008;237(8):1960–1971. doi: 10.1002/dvdy.21515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang Q, Murcia NS, Chittenden LR, Richards WG, Michaud EJ, Woychik RP, Yoder BK. Loss of the Tg737 protein results in skeletal patterning defects. Developmental Dynamics. 2003;227(1):78–90. doi: 10.1002/dvdy.10289. [DOI] [PubMed] [Google Scholar]
- 171.McGlashan S, Haycraft C, Jensen C, Yoder B, Poole C. Articular cartilage and growth plate defects are associated with chondrocyte cytoskeletal abnormalities in Tg737orpk mice lacking the primary cilia protein polaris. Matrix biology: journal of the International Society for Matrix Biology. 2007;26(4):234. doi: 10.1016/j.matbio.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 172.Irianto J, Ramaswamy G, Serra R, Knight MM. Depletion of primary cilia reduces the compressive modulus of articular cartilage. J Biomech. 2013 doi: 10.1016/j.jbiomech.2013.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132(13):3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 174.Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134(2):307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 175.Chang CF, Serra R. Ift88 regulates Hedgehog signaling, Sfrp5 expression, and beta-catenin activity in post-natal growth plate. J Orthop Res. 2013;31(3):350–6. doi: 10.1002/jor.22237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chang CF, Ramaswamy G, Serra R. Depletion of primary cilia in articular chondrocytes results in reduced Gli3 repressor to activator ratio, increased Hedgehog signaling, and symptoms of early osteoarthritis. Osteoarthritis and Cartilage. 2012;20(2):152–161. doi: 10.1016/j.joca.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277(5329):1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 178.Ocbina PJ, Tuson M, Anderson KV. Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS One. 2009;4(8):e6839. doi: 10.1371/journal.pone.0006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gorivodsky M, Mukhopadhyay M, Wilsch-Braeuninger M, Phillips M, Teufel A, Kim C, Malik N, Huttner W, Westphal H. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev Biol. 2009;325(1):24–32. doi: 10.1016/j.ydbio.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Howard PW, Howard TL, Maurer RA. Generation of mice with a conditional allele for Ift172. Transgenic research. 2010;19(1):121–126. doi: 10.1007/s11248-009-9292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Baker SA, Freeman K, Luby-Phelps K, Pazour GJ, Besharse JC. IFT20 links kinesin II with a mammalian intraflagellar transport complex that is conserved in motile flagella and sensory cilia. Journal of Biological Chemistry. 2003;278(36):34211–34218. doi: 10.1074/jbc.M300156200. [DOI] [PubMed] [Google Scholar]
- 182.Krock BL, Perkins BD. The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle–kinesin-II dissociation in vertebrate photoreceptors. J Cell Sci. 2008;121(11):1907–1915. doi: 10.1242/jcs.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jonassen JA, San Agustin J, Follit JA, Pazour GJ. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. The Journal of cell biology. 2008;183(3):377–384. doi: 10.1083/jcb.200808137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Keady BT, Le YZ, Pazour GJ. IFT20 is required for opsin trafficking and photoreceptor outer segment development. Molecular biology of the cell. 2011;22(7):921–930. doi: 10.1091/mbc.E10-09-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ. IFT25 links the signal-dependent movement of hedgehog components to intraflagellar transport. Developmental cell. 2012;22(5):940–951. doi: 10.1016/j.devcel.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bhogaraju S, Taschner M, Morawetz M, Basquin C, Lorentzen E. Crystal structure of the intraflagellar transport complex 25/27. EMBO J. 2011;30(10):1907–1918. doi: 10.1038/emboj.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Qin H, Wang Z, Diener D, Rosenbaum J. Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr Biol. 2007;17(3):193–202. doi: 10.1016/j.cub.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wang Z, Fan ZC, Williamson SM, Qin H. Intraflagellar transport (IFT) protein IFT25 is a phosphoprotein component of IFT complex B and physically interacts with IFT27 in Chlamydomonas. PLoS One. 2009;4(5):e5384. doi: 10.1371/journal.pone.0005384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Schafer JC, Winkelbauer ME, Williams CL, Haycraft CJ, Desmond RA, Yoder BK. IFTA-2 is a conserved cilia protein involved in pathways regulating longevity and dauer formation in Caenorhabditis elegans. J Cell Sci. 2006;119(Pt 19):4088–100. doi: 10.1242/jcs.03187. [DOI] [PubMed] [Google Scholar]
- 190.Adhiambo C, Blisnick T, Toutirais G, Delannoy E, Bastin P. A novel function for the atypical small G protein Rab-like 5 in the assembly of the trypanosome flagellum. J Cell Sci. 2009;122(6):834–841. doi: 10.1242/jcs.040444. [DOI] [PubMed] [Google Scholar]
- 191.Hou Y, Qin H, Follit JA, Pazour GJ, Rosenbaum JL, Witman GB. Functional analysis of an individual IFT protein: IFT46 is required for transport of outer dynein arms into flagella. J Cell Biol. 2007;176(5):653–665. doi: 10.1083/jcb.200608041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gouttenoire J, Valcourt U, Bougault C, Aubert-Foucher E, Arnaud E, Giraud L, Mallein-Gerin F. Knockdown of the intraflagellar transport protein IFT46 stimulates selective gene expression in mouse chondrocytes and affects early development in zebrafish. Journal of Biological Chemistry. 2007;282(42):30960–30973. doi: 10.1074/jbc.M705730200. [DOI] [PubMed] [Google Scholar]
- 193.Berbari NF, Kin NW, Sharma N, Michaud EJ, Kesterson RA, Yoder BK. Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev Biol. 2011;360(1):66–76. doi: 10.1016/j.ydbio.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Niu Y, Murata T, Watanabe K, Kawakami K, Yoshimura A, Inoue J, Puri RK, Kobayashi N. MIP-T3 associates with IL-13Ralpha1 and suppresses STAT6 activation in response to IL-13 stimulation. FEBS Lett. 2003;550(1–3):139–43. doi: 10.1016/s0014-5793(03)00860-3. [DOI] [PubMed] [Google Scholar]
- 195.Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nature cell biology. 2002;4(2):95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- 196.Houde C, Dickinson RJ, Houtzager VM, Cullum R, Montpetit R, Metzler M, Simpson EM, Roy S, Hayden MR, Hoodless PA. Hippi is essential for node cilia assembly and Sonic hedgehog signaling. Developmental biology. 2006;300(2):523–533. doi: 10.1016/j.ydbio.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pathak N, Austin CA, Drummond IA. Tubulin tyrosine ligase-like genes ttll3 and ttll6 maintain zebrafish cilia structure and motility. Journal of Biological Chemistry. 2011;286(13):11685–11695. doi: 10.1074/jbc.M110.209817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pathak N, Obara T, Mangos S, Liu Y, Drummond IA. The zebrafish fleer gene encodes an essential regulator of cilia tubulin polyglutamylation. Molecular biology of the cell. 2007;18(11):4353–4364. doi: 10.1091/mbc.E07-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ou G, Blacque OE, Snow JJ, Leroux MR, Scholey JM. Functional coordination of intraflagellar transport motors. Nature. 2005;436(7050):583–587. doi: 10.1038/nature03818. [DOI] [PubMed] [Google Scholar]
- 200.Bhogaraju S, Cajanek L, Fort C, Blisnick T, Weber K, Taschner M, Mizuno N, Lamla S, Bastin P, Nigg EA. Molecular Basis of Tubulin Transport Within the Cilium by IFT74 and IFT81. Science. 2013;341(6149):1009–1012. doi: 10.1126/science.1240985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Scholey JM. Intraflagellar transport motors in cilia: moving along the cell’s antenna. J Cell Biol. 2008;180(1):23–29. doi: 10.1083/jcb.200709133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rajagopalan V, D’Amico JP, Wilkes DE. Cytoplasmic dynein-2: from molecules to human diseases. Frontiers in Biology. 2013;8(1):119–126. [Google Scholar]
- 203.Pfister KK, Fisher EM, Gibbons IR, Hays TS, Holzbaur EL, McIntosh JR, Porter ME, Schroer TA, Vaughan KT, Witman GB. Cytoplasmic dynein nomenclature. J Cell Biol. 2005;171(3):411–413. doi: 10.1083/jcb.200508078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ocbina PJR, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43(6):547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ocbina PJR, Anderson KV. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Developmental Dynamics. 2008;237(8):2030–2038. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Liem KF, He M, Ocbina PJR, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proceedings of the National Academy of Sciences. 2009;106(32):13377–13382. doi: 10.1073/pnas.0906944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287(2):378. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 208.Grissom PM, Vaisberg EA, McIntosh JR. Identification of a novel light intermediate chain (D2LIC) for mammalian cytoplasmic dynein 2. Mol Biol Cell. 2002;13(3):817–829. doi: 10.1091/mbc.01-08-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Perrone CA, Tritschler D, Taulman P, Bower R, Yoder BK, Porter ME. A novel dynein light intermediate chain colocalizes with the retrograde motor for intraflagellar transport at sites of axoneme assembly in Chlamydomonas and mammalian cells. Mol Biol Cell. 2003;14(5):2041–2056. doi: 10.1091/mbc.E02-10-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rana AA, Barbera JPM, Rodriguez TA, Lynch D, Hirst E, Smith JC, Beddington RS. Targeted deletion of the novel cytoplasmic dynein mD2LIC disrupts the embryonic organiser, formation of the body axes and specification of ventral cell fates. Development. 2004;131(20):4999–5007. doi: 10.1242/dev.01389. [DOI] [PubMed] [Google Scholar]
- 211.Verhey KJ, Dishinger J, Kee HL. Kinesin motors and primary cilia. Biochem Soc Trans. 2011;39(5):1120. doi: 10.1042/BST0391120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Scholey JM. Kinesin-2: A Family of Heterotrimeric and Homodimeric Motors with Diverse Intracellular Transport Functions. Annu Rev Cell Dev Biol. 2013;29(1) doi: 10.1146/annurev-cellbio-101512-122335. [DOI] [PubMed] [Google Scholar]
- 213.Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15(9):467–76. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 214.Jenkins PM, Hurd TW, Zhang L, McEwen DP, Brown RL, Margolis B, Verhey KJ, Martens JR. Ciliary targeting of olfactory CNG channels requires the CNGB1b subunit and the kinesin-2 motor protein, KIF17. Current biology. 2006;16(12):1211–1216. doi: 10.1016/j.cub.2006.04.034. [DOI] [PubMed] [Google Scholar]
- 215.Cheung HO-L, Zhang X, Ribeiro A, Mo R, Makino S, Puviindran V, Law KK, Briscoe J, Hui C-c. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci Signal. 2009;2(76):ra29. doi: 10.1126/scisignal.2000405. [DOI] [PubMed] [Google Scholar]
- 216.Endoh-Yamagami S, Evangelista M, Wilson D, Wen X, Theunissen JW, Phamluong K, Davis M, Scales SJ, Solloway MJ, de Sauvage FJ. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Current biology. 2009;19(15):1320–1326. doi: 10.1016/j.cub.2009.06.046. [DOI] [PubMed] [Google Scholar]
- 217.Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102(2):175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 218.Kolpakova-Hart E, Jinnin M, Hou B, Fukai N, Olsen BR. Kinesin-2 controls development and patterning of the vertebrate skeleton by Hedgehog-and Gli3-dependent mechanisms. Dev Biol. 2007;309(2):273–284. doi: 10.1016/j.ydbio.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Brugmann SA, Allen NC, James AW, Mekonnen Z, Madan E, Helms JA. A primary cilia-dependent etiology for midline facial disorders. Hum Mol Genet. 2010;19(8):1577–1592. doi: 10.1093/hmg/ddq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 2007;134(11):2159–2169. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Qiu N, Xiao Z, Cao L, Buechel MM, David V, Roan E, Quarles LD. Disruption of Kif3a in osteoblasts results in defective bone formation and osteopenia. J Cell Sci. 2012;125(8):1945–1957. doi: 10.1242/jcs.095893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Temiyasathit S, Jacobs CR. Osteocyte primary cilium and its role in bone mechanotransduction. Annals of the New York Academy of Sciences. 2010;1192(1):422–428. doi: 10.1111/j.1749-6632.2009.05243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kinumatsu T, Shibukawa Y, Yasuda T, Nagayama M, Yamada S, Serra R, Pacifici M, Koyama E. TMJ development and growth require primary cilia function. Journal of dental research. 2011;90(8):988–994. doi: 10.1177/0022034511409407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wu Y, Dai XQ, Li Q, Chen CX, Mai W, Hussain Z, Long W, Montalbetti N, Li G, Glynne R. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum Mol Genet. 2006;15(22):3280–3292. doi: 10.1093/hmg/ddl404. [DOI] [PubMed] [Google Scholar]
- 225.Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95(6):829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 226.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304(5678):1755–1759. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 227.Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15(9):467–476. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 228.Tay SY, Ingham PW, Roy S. A homologue of the Drosophila kinesin-like protein Costal2 regulates Hedgehog signal transduction in the vertebrate embryo. Development. 2005;132(4):625–634. doi: 10.1242/dev.01606. [DOI] [PubMed] [Google Scholar]
- 229.Wong-Riley MT, Besharse JC. The kinesin superfamily protein KIF17: one protein with many functions ( 2012 doi: 10.1515/bmc-2011-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Howard PW, Jue SF, Maurer RA. Interaction of mouse TTC30/DYF-1 with multiple intraflagellar transport complex B proteins and KIF17. Exp Cell Res. 2013 doi: 10.1016/j.yexcr.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yin X, Takei Y, Kido MA, Hirokawa N. Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron. 2011;70(2):310–325. doi: 10.1016/j.neuron.2011.02.049. [DOI] [PubMed] [Google Scholar]
- 232.Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, Ideker T, Aza-Blanc P, Gleeson JG. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464(7291):1048–1051. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet. 2007;39(6):727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 234.Hao L, Scholey JM. Intraflagellar transport at a glance. Journal of cell science. 2009;122(7):889–892. doi: 10.1242/jcs.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Arts HH, Knoers NV. Current insights into renal ciliopathies: what can genetics teach us? Pediatric Nephrology. 2013;28(6):863–874. doi: 10.1007/s00467-012-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]