

Abstract
Autism spectrum disorder (ASD) is a complex disorder that appears to be caused by interactions between genetic changes and environmental insults during early development. A wide range of factors have been linked to the onset of ASD, but recently both genetic associations and environmental factors point to a central role for immune- related genes and immune responses to environmental stimuli. Specifically, many of the proteins encoded by the major histocompatibility complex (MHC) play a vital role in the formation, refinement, maintenance, and plasticity of the brain. Manipulations of levels of MHC molecules have illustrated how disrupted MHC signaling can significantly alter brain connectivity and function. Thus, an emerging hypothesis in our field is that disruptions in MHC expression in the developing brain caused by mutations and/or immune dysregulation may contribute to the altered brain connectivity and function characteristic of ASD. This review provides an overview of the structure and function of the three classes of MHC molecules in the immune system, healthy brain, and their possible involvement in ASD.
Keywords: maternal infection, neuroimmunology, genetics, synapse formation, synaptic plasticity
Immune dysregulation during brain development can have dramatic effects on neuronal function (Kipnis et al., 2008; Boulanger, 2009; McAllister and van de Water, 2009) and there is increasing evidence that immune dysregulation is associated with neurodevelopmental disorders, including autism spectrum disorder (ASD)(Patterson, 2009; Brown and Patterson, 2011; Patterson, 2011). ASD affects 1 in 88 children (approximately one in 54 boys and one in 252 girls) in the United States (Wingate et al., 2012). Although the etiology of ASD remains for the most part unknown, it is clearly a complex disorder that appears to be caused by interactions between genetic changes and environmental insults during early development (Onore et al., 2011).
A wide range of factors have been linked to the onset of ASD, but recently both genetic associations and environmental factors point to a central role for immune-related genes and immune responses to environmental stimuli (Patterson, 2009; Garay and McAllister, 2010; Patterson, 2011). Patients with ASD and their relatives have an increased incidence of immune disorders (Warren et al., 1990; Singh et al., 1997; Goldsmith and Rogers, 2008; Enstrom et al., 2009; Onore et al., 2009). In addition, abnormalities in peripheral immune cells correlate with behavioral symptoms in ASD (Plioplys et al., 1994; Enstrom et al., 2009; Enstrom et al., 2010) and signs of neural inflammation are present in the brains of postmortem individuals with ASD (Vargas et al., 2005). Cytokine levels are altered in the blood, brain, and cerebrospinal fluid (CSF) (Vargas et al., 2005; Molloy et al., 2006; Bauer et al., 2007; Chez et al., 2007; Ashwood et al., 2008; Deverman and Patterson, 2009; Ashwood et al., 2010; Careaga et al., 2010; Ashwood et al., 2011) and CNS-reactive antibodies are also present in individuals who have ASD (Careaga et al., 2010; Onore et al., 2011). Finally, specific haplotypes of immune genes within the major histocompatibility complex (MHC) of human chromosome 6 – human leukocyte antigen (HLA) and complement – have been implicated in ASD (Careaga et al., 2010). Consistent with these findings, the expression of many immune-related genes in the brain is altered in ASD (Voineagu et al., 2011). Together, these data suggest that immune dysregulation resulting from either immunogenetic mutations or from environmental exposures may underlie the pathogenesis of ASD.
Since there have been several comprehensive reviews published recently that cover the role for cytokines in typical brain development and in disease, as well as maternal infection in neurodevelopmental disorders (Garay and McAllister, 2010; Patterson, 2011), we have chosen to focus this review specifically on describing the structure and function of genes within the MHC and their role in both typical brain development and in ASD.
MHC molecules: Expression, Structure, and Immune Function
The MHC is a highly polymorphic cluster of genes with some of the greatest allelic diversity in the genome. MHC genes are both polygenic (containing multiple genes) and polymorphic (containing multiple variants of each gene). This complexity is critical for ensuring sufficient diversity in MHC molecules to allow for peptide presentation from a wide range of microorganisms. The specific genes and variants that an individual expresses comprise her/his MHC haplotype (Neefjes et al., 2011). The MHC is found on chromosome 6 of humans, and on chromosome 17 and 20 of mice and rats, respectively. It is divided into two primary regions, MHC class I and II (MHCI and MHCII), which are integral to the adaptive and innate immune responses. A third region, MHC class III (MHCIII), encodes a diverse array of proteins, including members of the complement system and some cytokines. MHC molecules are subclassified as classical and nonclassical. Unlike classical MHC molecules, non-classical MHC molecules are not polymorphic and do not present peptides.
MHCI and II proteins share an important role in the immune response; their extracellular domains combine to create a cleft in which a single peptide fragment is displayed on the surface of the cell. The MHC/peptide complex is recognized by a T cell, which in turn regulates the activation state of immune cells. MHCI, but not MHCII, molecules also activate natural killer (NK) cells in the immune system. MHCI and II are distinguished by the source of peptides that they display, the T cell types that recognize the peptides, as well as their structure and molecular components (Neefjes et al., 2011).
MHCI molecules are found on all nucleated cells (Neefjes et al., 2011). They are trimeric proteins comprised of one transmembrane heavy chain (with three α1-3 domains), a β2-microglobulin (β2m) light chain, and a peptide bound within the groove formed by the α1 and α2 regions of the heavy chain (Figure 1). The majority of class I-bound peptides are generated in the cytosol and are subsequently translocated into the lumen of the endoplasmic reticulum (ER) through the ATP-dependent transporter associated with antigen processing 1/2 (TAP1/2). Assembled trimers are transported to the cell surface of all cells in the body; both β2m and a peptide are necessary for cell surface expression of the MHC class I heavy chain (Williams et al., 1989; Spiliotis et al., 2002). Peptides presented by MHCI heavy chain are derived from proteolysis of intracellular proteins and are monitored by cytotoxic CD8+ T lymphocytes (T-cells) (Machold and Ploegh, 1996). These T-cells identify the peptides as self and non-self; non-self peptides presented by MHCI are usually of viral origin. If identified as non-self, an immune response is initiated, resulting in eventual lysis of cells displaying foreign peptide (Neefjes et al., 2011).
Figure 1. Schematic representations of MHC Class I and II molecules.
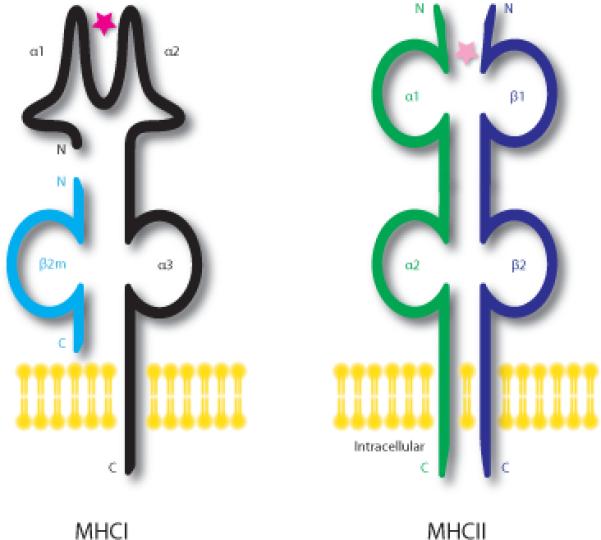
MHCI molecules are trimeric proteins comprised of one transmembrane heavy chain (α1-3 domains; black), a β2-microglobulin light chain (β2m; light blue), and a peptide (pink star) that is bound within the groove of α1 and α2 of the heavy chain. The MHCI peptide is generated in the cytosol. Trimers are assembled in the endoplasmic reticulum and all three components are required for transport to the cell surface of cells. MHCII molecules consist of two transmembrane chains an α (green) and a β (dark blue); each chain contains two domains (α1-α2, and β1-β2). The peptide is bound for display in the groove of the α1 and β1 domains and is derived from extracellular pathogens. MHC Class III genes encode such a diversity of proteins that examples are not shown in this figure.
MHCI molecules also bind to inhibitory receptors (IRs) on NK cells. The balance between activating and inhibitory receptor signaling on the NK-target cell interface determines the outcome of the interaction. Decreased MHCI on the target cell, caused by viral infection or tumorigenesis, favors formation of an activating immune synapse, resulting in apoptosis of the target cell. Conversely, normal levels of MHCI on the target cell engage NK-IRs and prevent the formation of the immune synapse required for NK activation and apoptosis of the target cell (Krzewski and Strominger, 2008; Long, 2008).
In humans, classical MHCI molecules are called human leukocyte antigens, HLA-A, -B, and –C (Figure 2). In rats, they include RT1-A and in mice, H2-B, -D, and –K (Figure 2). The nonclassical MHCI region in both rats and mice encodes numerous molecules, most of which are less polymorphic and many of which have not yet been characterized even in the immune system (Janeway et al., 2004). In humans, these are labeled HFE, HLAE, -F, -G, MICA, and MICB (Figure 2). In rats nonclassical MHC-I include RT1-CE and M, and in mice, H2-M, Q and –T (Figure 2). Finally, the gene encoding the invariant β2m light chain lies outside the MHC, on a separate chromosome (Neefjes et al., 2011).
Figure 2. Genomic maps of the human, rat, and mouse MHC.

This schematic identifies genes of MHC Class I classical (red) and non-classical (pink), MHC Class I pseudogenes (outlined in red), MHC Class II classical (dark blue) and non-classical (light blue), MHC Class III (black), and genes involved in processing and loading the MHC molecules. Classical MHC genes are typically polymorphic whereas the non-classical MHCI genes are not. These MHC genes are located on Chromosome 6, 20, and 17 in the human, rat, and mouse, respectively. The light chain of MHC Class I molecules, β2-microglobulin, is encoded on a separate chromosome (15 in humans, 3 in rats, and 2 in mice). Information for this figure was compiled from the UCSC Genome Browser on human Feb. 2009 (GRCh37/hg19), rat Mar. 2012 (RGSC 5.0/rn5), and mouse July 2007 (NCBI37/mm9) assemblies.
MHCII molecules are found in a more restricted subset of cells in the body than MHCI. They are expressed exclusively by antigen presenting cells such as macrophages, dendritic cells and B cells, as well as some endothelial cells and the epithelium of thymus (Janeway et al., 2004). MHCII molecules consist of two transmembrane chains (α and β); each chain contains two domains (α1-2 and β1-2) (Figure 1). The peptide is bound for display in the groove of the α1 and β1 domains and is derived from extracellular pathogens. Following endocytosis, these extracellular proteins are digested in lysosomes and then loaded onto MHCII molecules prior to trafficking to the plasma membrane. On the cell surface, MHCII presents exclusively to CD4+ helper T cells, which often either recruit phagocytes to increase local inflammation or initiate an antibody-based immune response through B cell activation (Janeway et al., 2004; Neefjes et al., 2011).
In humans, genes encoding MHCII include HLA-DP, -DQ, and –DR (Figure 2). Of special relevance to ASD, DR4 and DR13/14 haplotypes bear the DRβ1 gene that encodes the beta subunit of the HLA-DR which has been implicated in ASD. In rats, classical MHCII molecules include RT1-B and -D, and in mice, H2-A and –E (Figure 2). The non-classical MHCII molecules regulate transport and peptide loading onto classical MHCII molecules (Alfonso and Karlsson, 2000). In humans, nonclassical MHCII include HLA-DM and –DO (Figure 2). In rats, they include RT1-DM and DO, and in mice, H2-DM and -O (Alfonso and Karlsson, 2000) (Figure 2).
MHCIII molecules encode a large number of diverse proteins—some with important immune functions and others with no known immune function. Those with immune functions include complement proteins (C2 and C4)—components of the classical complement pathway—which opsonize or “tag” unwanted cells or debris for elimination (Song, 2006) (Figure 2). MHCIII also encodes the cytokines tumor necrosis factor (TNF) –α and –β (Figure 2). TNFα is a major pro-inflammatory cytokine that is responsible for early immunological responses (Garay and McAllister, 2010). Additionally, MHCIII encodes heat shock proteins, which mediate cellular responses to heat, stress and viral infection (Figure 2). Unlike MHCI and II, MHCIII molecules do not present peptides (Turturici et al., 2011). It is noteworthy that the HLA-DRβ1 gene of MHCII is located in close proximity to the C4B of MHCIII on human chromosome 6 (Warren et al., 1996) (Figure 2). Both regions (as discussed below) have been implicated in ASD.
MHC molecules in the healthy CNS
Although the brain and immune system are still considered to be mostly independent, there is increasing evidence for substantial neural-immune crosstalk starting in the fetal brain and continuing throughout life (McAllister and van de Water, 2009). Many proteins that were first described in the immune system have recently been discovered to have critical functions in the healthy CNS (Bhat and Steinman, 2009; Boulanger, 2009; Deverman and Patterson, 2009; McAllister and van de Water, 2009; Patterson, 2009; Garay and McAllister, 2010). Immune proteins such as antibodies, cytokines, and chemokines cross from the periphery into the CNS during early brain development, in the healthy aging brain, and during times of peripheral inflammation caused by disease (Bhat and Steinman, 2009). Moreover, “immune” proteins such as MHCI and II molecules, MHCI receptors, and components of the complement cascade (MHCIII) are expressed by cells of the healthy brain (Boulanger, 2009).
MHCI molecules were some of the first products of the MHC to be discovered as having essential roles in typical CNS development. The first evidence that MHCI might play a non-immune role in the brain came from the unexpected discovery that MHCI molecules are regulated by activity in the developing visual system during the period of activity-dependent remodeling of connections (Corriveau et al., 1998). Since this initial observation in 1998, several studies have revealed that MHCI mRNAs and protein are found throughout the developing and adult brain (Corriveau et al., 1998; Huh et al., 2000; McConnell et al., 2009; Needleman et al., 2010; Ribic et al., 2010; Glynn et al., 2011). Moreover, MHCI molecules are present at synapses on both the pre- and postsynaptic sides in the visual cortex from early development through adulthood (Needleman et al., 2010; Ribic et al., 2010).
MHCI molecules regulate many aspects of brain development, including the initial establishment of connectivity and activity-dependent refinement and plasticity (Corriveau et al., 1998; Shatz, 2009; Glynn et al., 2011). During the initial establishment of connections within the cerebral cortex in early postnatal mice, MHCI molecules negatively regulate synapse density (Glynn et al., 2011). Decreasing surface expression of MHCI using β2m knockout mice (β2m−/−) or by expressing an siRNA to β2m increases the density of both functional glutamatergic and GABAergic synapses in vitro and in vivo (Glynn et al., 2011), but has no effect on glutamatergic synapse density in hippocampal neurons (Goddard et al., 2007). Conversely, increasing surface expression of MHCI by overexpression of mouse MHCI H2-Kb in young cultured cortical neurons significantly decreases the density of functional glutamatergic synapses (Glynn et al., 2011). Interestingly, altering surface MHCI expression not only changes the frequency of miniature excitatory postsynaptic currents (mEPSCs), consistent with the effects on synapse density, but also alters mEPSC amplitude suggesting that MHCI negatively regulates synaptic strength through controlling AMPA receptor (AMPAR) trafficking at the synapse. Finally, changing MHCI surface expression also alters the balance of excitation to inhibition onto cortical neurons, which has serious implications for functional connectivity within the cerebral cortex (Glynn et al., 2011). Thus, MHCI molecules act to bidirectionally regulate the initial establishment of connections within the early postnatal cortex—a time thought to be critical for the developmental defects that cause ASD.
In addition to their involvement in the development of cortical connections, MHCI molecules are also required for synaptic function and plasticity in the more mature CNS (Fourgeaud and Boulanger, 2010). Although synapse density is not altered in the hippocampus of mice deficient in surface MHCI (β2m/TAP1−/− mice), mEPSC frequency is increased in β2m/TAP1−/− hippocampal cultures (Goddard et al., 2007). A recent report in marmoset monkeys indicates that acute application of antibodies that bind to MHCI molecules alter synaptic transmission in hippocampal neurons—decreasing mEPSC frequency and increasing mEPSC amplitude (Ribic et al., 2010). Mice deficient in surface MHCI (β2m/TAP −/− mice) or a subunit of the T-cell receptor that binds MHCI in immune cells, CD3ζ, also exhibit altered hippocampal synaptic plasticity. In both knockout mice, long-term depression (LTD) is absent and long-term potentiation (LTP) can be induced at lower frequencies than in wild-type mice (Huh et al., 2000). Knockout mice deficient for two MHCI molecules, H2-Kb and H2-Db, also have a lowered threshold for the induction of LTD in the parallel fibers of the cerebellum, leading to enhanced motor learning (McConnell et al., 2009).
MHCI may modulate synaptic plasticity through altering NMDA receptor (NMDAR) function and AMPAR trafficking in the hippocampus during plasticity (Fourgeaud et al., 2010). The AMPAR/NMDAR ratio is decreased in hippocampal neurons from mice deficient in surface MHCI (β2m/TAP −/− mice). This decreased ratio reflects an increase in NMDAR-mediated current, which then alters AMPAR trafficking and thereby mediates the strength of glutamatergic synapses (Fourgeaud et al., 2010). Thus, MHCI molecules negatively regulate NMDAR function and subsequently NMDAR-mediated AMPAR trafficking to control synaptic strength and plasticity (Fourgeaud and Boulanger, 2010).
MHCI molecules also alter activity-dependent plasticity of the developing visual system (Corriveau et al., 1998; Huh et al., 2000; Boulanger and Shatz, 2004; Datwani et al., 2009). MHCI mRNA and protein levels are regulated by neuronal activity (Corriveau et al., 1998; Santos et al., 2004; Goddard et al., 2007; Glynn et al., 2011), and the surface expression of MHCI molecules in the plasma membrane is also dynamically regulated by activity (Glynn et al., 2011). In young cultured cortical neurons, activity blockade using tetrodotoxin (TTX) decreases surface MHCI, while increasing total MHCI protein levels (Glynn et al., 2011). Similarly, in adolescent animals, total MHCI mRNA levels decrease following activity blockade, and increase after kainic-acid induced seizures (Corriveau et al., 1998). At this adolescent age, MHCI regulates refinement of visual connections from the retina to the lateral geniculate nucleus (LGN) of the thalamus (Corriveau et al., 1998). Retinal axons fail to segregate into eye-specific layers in the LGN in transgenic mice lacking surface MHCI (β2m/TAP1−/−) (Huh et al., 2000). Retinogeniculate refinement and retinal connectivity are also impaired in mice lacking CD3ζ−/− (Huh et al., 2000; Xu et al., 2010) and in mice lacking two MHCI genes, H2-Kb and H2-Db (Datwani et al., 2009). MHCI molecules also control ocular dominance (OD) plasticity in the developing visual cortex. OD plasticity is enhanced in both β2m/TAP1−/− and H2-KbDb −/− mice (Datwani et al., 2009) as well as in mice lacking one of the many MHCI receptors, paired-immunoglobulin-like receptor B (PirB) (Syken et al., 2006). Together, these studies clearly demonstrate that MHCI molecules are required for refinement of retinal connections within the thalamus and to limit the extent and timing of OD plasticity in the visual cortex.
MHCII molecules may also play a role in development of the healthy brain, but much less is known about their function. MHCII proteins are expressed primarily by microglial cells, astrocytes, and perivascular monocytes in the healthy developing and adult brain (Peudenier et al., 1991; Perlmutter et al., 1992; Wierzba-Bobrowicz et al., 2001). HLADR, -DP, and –DQ are also expressed in the white matter of neurologically normal and diseased patients (Luber-Narod and Rogers, 1988; Rezaie and Male, 1999). The most common immunoreactive cells are microglia, which express HLA-DR at increased levels during typical development. Expression of MHCII molecules is dramatically altered in disease, such as Alzheimer's disease, which is characterized by greater microglial expression of MHCII and changes in the morphology of MHCII-expressing cells (Perlmutter et al., 1992). Further research must be conducted to determine if there is a non-immune role for MHCII molecules in the healthy brain. Several MHCII-deficient transgenic mouse lines have been created so the tools are available to address this question.
MHCIII molecules clearly play important roles in brain development and function. Complement components C2 and C4 are found throughout the brain on astrocytes, microglia, and neurons from development into adulthood (Haga et al., 1996; Davies et al., 1998; Walker et al., 1998; Thomas et al., 2000; Zhou et al., 2008). Complement C2 and C4 are members of the classical complement pathway, which has been implicated in mediating synapse elimination in the healthy developing CNS (Schafer and Stevens, 2010). This classical pathway is initiated by C1q, which is upregulated on neurons in response to signals from glia. C1q and C3 are found at developing synapses where they are thought to tag those synapses for elimination. Consistent with this hypothesis, mice deficient in C1q or C3 have pronounced defects in synapse elimination (Stevens et al., 2007).
Also encoded by MHCIII, TNFα plays a critical role in neural development, synaptic transmission, and homeostatic plasticity (Stellwagen, 2011). TNF-α is found on glial cells of the healthy CNS and its expression induces MHCI transcription in neurons (Neumann et al., 1997; Bauer et al., 2007). TNF-α regulates the surface expression of AMPARs, increases the growth of dendrites, and enhances synaptic connectivity (Beattie et al., 2002; Stellwagen et al., 2005; Lee et al., 2010). TNF-α also mediates the balance between excitation and inhibition in neurons, may alter long-term synaptic plasticity, and clearly alters homeostatic plasticity (Albensi and Mattson, 2000; Tancredi et al., 2000; Stellwagen et al., 2005; Stellwagen and Malenka, 2006). Finally, the heat shock protein 70 (HSP70) family, also encoded by the MHCIII region, is expressed by neurons, glia, and endothelial cells in the healthy brain (Foster and Brown, 1996; Yenari, 2002). HSPs are molecular chaperones that assist in the refolding of misfolded proteins and are implicated in a wide range of functions including synaptic transmission, autophagy, ER stress response, and protein kinase and cell death signaling (Stetler et al., 2010). HSP70 is thought to be particularly important for preventing the accumulation of protein aggregates in neurodegenerative disease (Turturici et al., 2011).
Clearly, the many proteins encoded by the MHC play a vital role in the formation, refinement, maintenance, and plasticity of the brain. Manipulations of levels of MHC molecules have illustrated how disrupted MHC signaling can significantly alter brain connectivity and function. Thus, an emerging hypothesis is that disruptions in MHC expression in the developing brain caused by mutations and/or immune dysregulation contribute to altered brain connectivity characteristic of ASD.
Genetic mutations in MHC genes and ASD
Since autism was initially identified and described in 1943 (Kanner, 1943), scientists and clinicians have struggled to identify its cause. It is clear that no single genetic locus is the sole cause of this disorder. In the past few years, a surprisingly small number of genes conferring ASD risk have been identified through genome-wide association studies, and a rapidly growing number of rare mutations have been identified as causal for ASD (Geschwind, 2011). While many of these genes encode proteins integral to formation, function, and/or plasticity of synapses in the CNS, others encode proteins traditionally thought to play roles exclusively in the immune system, including MHC genes.
MHCI genes
Reports of associations between MHC genes and ASD are controversial due primarily to small sample sizes and differing results between different research groups. Several reports have shown a significant association between HLA haplotypes and ASD. 75% of children with ASD were reported to share at least one HLA antigen with their parents, while the control group shared only 22% (Stubbs, 1981; Stubbs et al., 1985). Specifically, mothers and children with ASD more often had similar HLA types than did typically developing mothers and their children (Stubbs et al., 1985). In addition, another study showed an increase of certain HLA-A2 (but not HLA-B) allele haplotypes in subjects with autism (Torres et al., 2002; Torres et al., 2006). Several reports have not reproduced these effects, but the sample sizes in these reports were too low to be conclusive (Stubbs and Magenis, 1980). Overall, these results suggest a potential role for specific types of MHCI in increasing risk for ASD.
MHCII genes
The MHCII region has also been implicated in ASD. A strong association of HLA-DR4 with ASD has been replicated in several studies (Warren et al., 1992; Daniels et al., 1995; Torres et al., 2002; Johnson et al., 2009). Specifically, the HLADRβ1 alleles are linked to ASD (Warren et al., 1996); the DRβ1 gene is encoded by the DR4 and DR13/14 haplotypes. Interestingly, some MHCII genes might also be protective, as suggested by the observation that HLA-DR13/14 is found with decreased frequency in children with ASD compared to non-ASD controls (Daniels et al., 1995; Warren et al., 1996; Torres et al., 2002). Finally, autistic children with large decreases in CD4+ naïve T cells and increased CD4+ memory T cells are more likely to express a higher frequency of both the HLA-A2 and DR11 alleles (Ferrante et al., 2003). However, contrary to previous reports, these researchers did not find an association of HLA-DR4 with ASD (Ferrante et al., 2003). These findings suggest that some of the genes within the MHCII region might be pathogenic factors in ASD.
To address the possibility that the disparities in results described above could result from sampling genetically and geographically mixed populations, family members and autistic children from the genetically-isolated Italian island Sardinia were genotyped for HLA alleles. In 50% of the families, ASD was linked to HLA but no single particular allelic polymorphism was identified (Guerini et al., 2006). The same researchers subsequently found a positive association with ASD for an allele in the HLA-A region and a negative association in the HLA-B region (Guerini et al., 2009). The same HLA loci were linked to ASD both in Sardinian and in peninsular Italian individuals with ASD, suggesting a genetic link that is not entirely geographically or genetically-isolated. (Guerini et al., 2011). Alternatively, another investigation focused on families within Tennessee and also across the USA. The HLA-DR4 frequencies of mothers and their sons were significantly higher than typically developing subjects in Tennessee, but not across the USA. This result is consistent with the hypothesis that specific MHCII genes may alter fetal brain development within a population residing in a geographically-defined region and may contribute to the inconsistencies from study to study where location was not considered (Lee et al., 2006).
More recently, a significant increased frequency of HLA-DR4 was found in mothers of individuals with ASD as compared to mothers of typically developing children (Johnson et al., 2009). This led to the idea that enrichment in HLA-DR4 of the mothers may be responsible for ASD in the child and is consistent with the hypothesis that prenatal maternal-fetal immune interaction can alter fetal brain development (as described below; (Johnson et al., 2009). Finally, subjects with ASD exhibit enhanced astroglial and microglial activation; increased HLA-DR expression by activated microglia is observed most prominently in the cerebellum, but also in cortical regions and white matter of postmortem autistic patients (Vargas et al., 2005). The increased expression of HLA-DR during development and disease is considered to be a marker of astroglial and microglial activation and their capacity to present antigen to T-helper cells.
MHCIII genes
Only a few genes encoded within the MHCIII region have been analyzed for association with ASD. Interestingly, the genes encoding complement gene C4 have been linked to ASD (Warren et al., 1991; Warren et al., 1994; Odell et al., 2005; Corbett et al., 2007; Mostafa and Shehab, 2010). Although there are no reports to date linking ASD and MHCIII genes encoding TNFα or β, there are several reports of increased TNFα protein levels in the CSF, blood, and post-mortem brain tissue from individuals with ASD (Vargas et al., 2005; Chez et al., 2007). To date, there is no reported association of HSP70 with ASD.
Taken together, these genetic studies have identified a potential link between the MHC region of the genome and ASD. Despite inconsistencies and small sample sizes, the HLA-A and HLA-B regions of MHC-I, the HLA-DR region of MHC-II, and the complement C4 gene, are the genes within the MHC most associated with ASD. Importantly, findings may vary depending on the subjects’ geographic location, parents’ genetic background and age, birth rank, disorder phenotype, and many other variables (Lee et al., 2006; Johnson et al., 2009; Shelton et al., 2010; Nordahl et al., 2011; Turner et al., 2011). Nevertheless, because so many studies suggest an immunological role for the onset of ASD, it will be important to continue focusing on the MHC region of the genome and to design studies that unequivocally identify the contribution of genes within the MHC to ASD. Interestingly, there is also a reproducibly strong link between genes within the MHC and schizophrenia (Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009; Bergen et al., 2012; Hamshere et al., 2012; Jia et al., 2012), consistent with the increasingly large number of genetic and environmental associations shared specifically by these two disorders (Patterson, 2009; Gejman et al., 2011; Meyer et al., 2011; Brown, 2012; Reichelt et al., 2012).
Viral infection and immune activation in ASD
In addition to rare genetic mutations in MHC genes, proteins encoded by the MHC could contribute to the etiology of ASD by mediating systemic immune dysregulation during early brain development in gestation or in early postnatal life. Increasing evidence suggests that environmental contributions to ASD and viral infection during gestation has, until recently, been one of the most controversial but persistent factors associated with this disorder (Patterson, 2011). Recently, compelling evidence indicates that maternal viral infection is one of the few documented risk factors for ASD (Atladottir et al., 2010).
Many of the environmental insults linked to ASD involve the maternal-fetal environment. ASD twin studies highlight the fact that concordance for dizygotic twins is much greater than that for siblings, implying a significant role for the fetal environment (Rosenberg et al., 2009; Hallmayer et al., 2011; Szatmari, 2011). Maternal exposure to valproic acid or thalidomide greatly increases incidence of ASD (Kinney et al., 2008). The risk for ASD is also higher in offspring conceived in the winter months, consistent with a role for respiratory infections (Zerbo et al., 2011). In addition to other kinds of infection (Patterson, 2009; Patterson, 2011), maternal viral infection during the first trimester, in particular, is associated with increased risk of ASD (Atladottir et al., 2010; Patterson, 2011). Rubella infection has been repeatedly associated with increased incidence of ASD features (Chess, 1977; Deykin and MacMahon, 1979; Libbey et al., 2005) and autistic children also have an increased incidence of congenital cytomegalovirus infections (Stubbs, 1978; Deykin and MacMahon, 1979; Yamashita et al., 2003; Sweeten et al., 2004; Libbey et al., 2005). Despite these correlations, most of these studies relied on semi-quantitative or subjective reports of infections and so, were considered controversial. However, a recent report of a large-scale study from a well-documented population of patients in Denmark clearly indicates an association between increased incidence of maternal viral infection in the first trimester and maternal bacterial infection in the second trimester requiring hospitalization and diagnosis of ASDs in offspring (Stubbs and Magenis, 1980; Atladottir et al., 2010). Together, these reports indicate that maternal infection is indeed a risk factor for ASD (Patterson, 2011; Hagberg et al., 2012).
How maternal immune activation (MIA) increases the incidence of ASD in offspring remains unknown. One hypothesis is that the high levels of cytokines in the maternal bloodstream induced by infection enables some of them to cross the placenta and enter the brain of the fetus and alter brain development. Consistent with this hypothesis, a recent case report found that elevated maternal serum concentrations of IFN-γ, and two interleukins (ILs), IL-4 and IL-5, are associated with a 50% increased risk of ASD, while a distinct cytokine profile of increased IL-2, IL-4 and IL-6 is associated with an increased risk of developmental delay without ASD (Goines et al., 2011). The amniotic fluid of mothers of children later diagnosed with ASD also shows increased levels of cytokines, including IL-4, TNFα and β (Abdallah et al., 2011; Abdallah et al., 2012). Moreover, there are many reports of altered cytokines in the blood, brain, and CSF of individuals with ASD of various ages (Vargas et al., 2005; Molloy et al., 2006; Chez et al., 2007; Ashwood et al., 2008; Ashwood et al., 2010; Ashwood et al., 2011; Manzardo et al., 2011).
The development of mouse models of the maternal infection risk factor for ASD (Patterson, 2009; Meyer and Feldon, 2010; Patterson, 2011) has strengthened the link between MIA and much of the neuropathology and abnormal behaviors characteristic of ASD. Adult offspring of pregnant mice given intra-nasal influenza virus exhibit behavioral abnormalities and changes in gene expression, neuroanatomy, and neurochemistry consistent with those found in ASD (Fatemi et al., 1998; Fatemi et al., 2002; Shi et al., 2003). Because these symptoms can also be elicited in the absence of infection by maternal injection of synthetic dsRNA (polyI:C), which mimics the acute phase response to viral infection (Traynor et al., 2004), it is likely that the maternal inflammatory response drives the changes in fetal brain development (Shi et al., 2003). Importantly, the polyI:C MIA model is one of the few mouse autism models that exhibits all three core symptoms of ASD: stereotyped and repetitive behaviors, deficits in social interaction, and deficits in communication (Smith et al., 2007; Malkova, 2012). Adult MIA offspring also exhibit abnormalities in gene expression, neurochemistry, and neuropathology similar to those found in ASD (Shi et al., 2003; Meyer et al., 2008; Patterson, 2009; Shi et al., 2009; Meyer and Feldon, 2010)(see also Michel et al., this issue).
Despite recent progress in developing and characterizing mouse models of MIA, little is known about how MIA alters fetal brain development. Current evidence indicates that maternal cytokines secreted immediately after poly(I:C) injection cross the placenta and enter the fetus (Smith et al., 2007; Patterson, 2009; Hsiao and Patterson, 2011). IL -6 is necessary and sufficient to mediate these effects since the effects of MIA on neuropathology and behavior are prevented by co-injection of an anti-IL-6 antibody and are mimicked by injection of pregnant mice with IL-6 (Smith et al., 2007; Patterson, 2009). These maternal cytokines induce endocrine changes in the placenta (Hsiao and Patterson, 2011) and change expression of other cytokines in the fetal brain, including IL-1β and IL-6, hours after maternal poly(I:C) injection (Meyer et al., 2006). However, it remains unknown if MIA causes chronic changes in brain cytokines in offspring during multiple stages of postnatal development and whether chronically altered cytokines regulate connectivity in the developing brain.
The cytokine hypothesis of MIA and ASD may also involve the MHC. Since cytokines control expression of MHC proteins in the immune system (Meraz et al., 1996; Schindler et al., 2007; Li, 2008), the elevated fetal brain cytokines following MIA may well alter the expression profiles of other immune molecules, including MHCI molecules on neurons and glial cells, MHCII molecules on microglia, and MHCIII molecules on neurons and microglia. Because all of these MHC molecules are present in the developing brain, where they regulate connectivity and plasticity, any alteration in their expression could contribute to the aberrant behaviors characteristic of ASD. Future work testing this hypothesis will be critical for determining if MHC molecules mediate the effects of immune dysregulation on brain development, thereby contributing to neurodevelopmental disorders, including ASD.
Conclusions
Given the dogma of CNS immune-privilege that prevailed just over a decade ago, it is remarkable how far the field of neuroimmunology has come in such a short time. We now know that many of the diverse families of molecules encoded within the MHC are expressed in the healthy CNS, where they play critical roles in brain development, function, and plasticity. Moreover, there is growing evidence for both MHC mutations and immune dysregulation in brain development, which may indirectly alter CNS MHC molecule expression, and contribute to ASD. However, there is not yet a clear role for MHC proteins in ASD. Future work in clarifying changes in MHC genes and in other brain immune-related molecules in individuals with ASD, as well as clarifying affected molecular pathways in mouse models, is essential to test the hypothesis that alterations in expression of MHC molecules in the developing brain contribute to the pathogenesis of ASD. If true, then MHC molecules represent a family of exciting and untapped, potential targets for therapeutic intervention for ASD.
Footnotes
Conflict of Interest:
The authors have no conflict of interest to declare.
References
- Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen EL, Hougaard DM. Amniotic fluid inflammatory cytokines: Potential markers of immunologic dysfunction in autism spectrum disorders. World J Biol Psychiatry. 2011 doi: 10.3109/15622975.2011.639803. [DOI] [PubMed] [Google Scholar]
- Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen EL, Hougaard DM. Amniotic fluid chemokines and autism spectrum disorders: an exploratory study utilizing a Danish Historic Birth Cohort. Brain Behav Immun. 2012;26:170–176. doi: 10.1016/j.bbi.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Alfonso C, Karlsson L. Nonclassical MHC class II molecules. Annual review of immunology. 2000;18:113–142. doi: 10.1146/annurev.immunol.18.1.113. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Enstrom A, Krakowiak P, Hertz-Picciotto I, Hansen RL, Croen LA, Ozonoff S, Pessah IN, Van de Water J. Decreased transforming growth factor beta1 in autism: a potential link between immune dysregulation and impairment in clinical behavioral outcomes. J Neuroimmunol. 2008;204:149–153. doi: 10.1016/j.jneuroim.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011;25:40–45. doi: 10.1016/j.bbi.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J Neuroimmunol. 2010 doi: 10.1016/j.jneuroim.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Kwong C, Hansen R, Hertz-Picciotto I, Croen L, Krakowiak P, Walker W, Pessah IN, Van de Water J. Brief report: plasma leptin levels are elevated in autism: association with early onset phenotype? J Autism Dev Disord. 2008;38:169–175. doi: 10.1007/s10803-006-0353-1. [DOI] [PubMed] [Google Scholar]
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40:1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bergen SE, O'Dushlaine CT, Ripke S, Lee PH, Ruderfer DM, Akterin S, Moran JL, Chambert KD, Handsaker RE, Backlund L, Osby U, McCarroll S, Landen M, Scolnick EM, Magnusson PK, Lichtenstein P, Hultman CM, Purcell SM, Sklar P, Sullivan PF. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R, Steinman L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron. 2009;64:123–132. doi: 10.1016/j.neuron.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64:93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Shatz CJ. Immune signalling in neural development, synaptic plasticity and disease. Nature reviews. Neuroscience. 2004;5:521–531. doi: 10.1038/nrn1428. [DOI] [PubMed] [Google Scholar]
- Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012 doi: 10.1002/dneu.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Patterson PH. Maternal infection and schizophrenia: implications for prevention. Schizophr Bull. 2011;37:284–290. doi: 10.1093/schbul/sbq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga M, Van de Water J, Ashwood P. Immune dysfunction in autism: a pathway to treatment. Neurotherapeutics. 2010;7:283–292. doi: 10.1016/j.nurt.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess S. Follow-up report on autism in congenital rubella. Journal of autism and childhood schizophrenia. 1977;7:69–81. doi: 10.1007/BF01531116. [DOI] [PubMed] [Google Scholar]
- Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. 2007;36:361–365. doi: 10.1016/j.pediatrneurol.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Corbett BA, Kantor AB, Schulman H, Walker WL, Lit L, Ashwood P, Rocke DM, Sharp FR. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol Psychiatry. 2007;12:292–306. doi: 10.1038/sj.mp.4001943. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- Daniels WW, Warren RP, Odell JD, Maciulis A, Burger RA, Warren WL, Torres AR. Increased frequency of the extended or ancestral haplotype B44-SC30-DR4 in autism. Neuropsychobiology. 1995;32:120–123. doi: 10.1159/000119223. [DOI] [PubMed] [Google Scholar]
- Datwani A, McConnell MJ, Kanold PO, Micheva KD, Busse B, Shamloo M, Smith SJ, Shatz CJ. Classical MHCI molecules regulate retinogeniculate refinement and limit ocular dominance plasticity. Neuron. 2009;64:463–470. doi: 10.1016/j.neuron.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BJ, Pickard BS, Steel M, Morris RG, Lathe R. Serine proteases in rodent hippocampus. J Biol Chem. 1998;273:23004–23011. doi: 10.1074/jbc.273.36.23004. [DOI] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Deykin EY, MacMahon B. Viral exposure and autism. Am J Epidemiol. 1979;109:628–638. doi: 10.1093/oxfordjournals.aje.a112726. [DOI] [PubMed] [Google Scholar]
- Enstrom AM, Onore CE, Van de Water JA, Ashwood P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav Immun. 2010;24:64–71. doi: 10.1016/j.bbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enstrom AM, Van de Water JA, Ashwood P. Autoimmunity in autism. Curr Opin Investig Drugs. 2009;10:463–473. [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Sidwell RW, Kist DA, Stary JM, Earle JA, Thuras P. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol Psychiatry. 2002;7:633–640. doi: 10.1038/sj.mp.4001046. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Sidwell R, Kist D, Akhter P, Meltzer HY, Bailey K, Thuras P, Sedgwick J. Differential expression of synaptosome-associated protein 25 kDa [SNAP-25] in hippocampi of neonatal mice following exposure to human influenza virus in utero. Brain Res. 1998;800:1–9. doi: 10.1016/s0006-8993(98)00450-8. [DOI] [PubMed] [Google Scholar]
- Ferrante P, Saresella M, Guerini FR, Marzorati M, Musetti MC, Cazzullo AG. Significant association of HLA A2-DR11 with CD4 naive decrease in autistic children. Biomed Pharmacother. 2003;57:372–374. doi: 10.1016/s0753-3322(03)00099-4. [DOI] [PubMed] [Google Scholar]
- Foster J, Brown I. Basal expression of stress-inducible hsp70 mRNA detected in hippocampal and cortical neurons of normal rabbit brain. Brain Res. 1996;724:73–83. doi: 10.1016/0006-8993(96)00266-1. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Boulanger LM. Role of immune molecules in the establishment and plasticity of glutamatergic synapses. Eur J Neurosci. 2010;32:207–217. doi: 10.1111/j.1460-9568.2010.07342.x. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Davenport CM, Tyler CM, Cheng TT, Spencer MB, Boulanger LM. MHC class I modulates NMDA receptor function and AMPA receptor trafficking. Proc Natl Acad Sci U S A. 2010;107:22278–22283. doi: 10.1073/pnas.0914064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay PA, McAllister AK. Novel roles for immune molecules in neural development: implications for neurodevelopmental disorders. Frontiers in synaptic neuroscience. 2010;2:136. doi: 10.3389/fnsyn.2010.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gejman PV, Sanders AR, Kendler KS. Genetics of schizophrenia: new findings and challenges. Annu Rev Genomics Hum Genet. 2011;12:121–144. doi: 10.1146/annurev-genom-082410-101459. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Genetics of autism spectrum disorders. Trends Cogn Sci. 2011;15:409–416. doi: 10.1016/j.tics.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn MW, Elmer BM, Garay PA, Liu XB, Needleman LA, El-Sabeawy F, McAllister AK. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci. 2011;14:442–451. doi: 10.1038/nn.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether J, Hansen R, Kharrazi M, Ashwood P, Van de Water J. Increased midgestational IFN-gamma, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol Autism. 2011;2:13. doi: 10.1186/2040-2392-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith CA, Rogers DP. The case for autoimmunity in the etiology of schizophrenia. Pharmacotherapy. 2008;28:730–741. doi: 10.1592/phco.28.6.730. [DOI] [PubMed] [Google Scholar]
- Guerini FR, Bolognesi E, Chiappedi M, De Silvestri A, Ghezzo A, Zanette M, Rusconi B, Manca S, Sotgiu S, Agliardi C, Clerici M. HLA polymorphisms in Italian children with autism spectrum disorders: results of a family based linkage study. J Neuroimmunol. 2011;230:135–142. doi: 10.1016/j.jneuroim.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Guerini FR, Bolognesi E, Manca S, Sotgiu S, Zanzottera M, Agliardi C, Usai S, Clerici M. Family-based transmission analysis of HLA genetic markers in Sardinian children with autistic spectrum disorders. Hum Immunol. 2009;70:184–190. doi: 10.1016/j.humimm.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Guerini FR, Manca S, Sotgiu S, Tremolada S, Zanzottera M, Agliardi C, Zanetta L, Saresella M, Mancuso R, De Silvestri A, Fois ML, Arru G, Ferrante P. A family based linkage analysis of HLA and 5-HTTLPR gene polymorphisms in Sardinian children with autism spectrum disorder. Hum Immunol. 2006;67:108–117. doi: 10.1016/j.humimm.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Haga S, Aizawa T, Ishii T, Ikeda K. Complement gene expression in mouse microglia and astrocytes in culture: comparisons with mouse peritoneal macrophages. Neurosci Lett. 1996;216:191–194. doi: 10.1016/0304-3940(96)13040-8. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Annals of neurology. 2012;71:444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, Lotspeich L, Croen LA, Ozonoff S, Lajonchere C, Grether JK, Risch N. Genetic Heritability and Shared Environmental Factors Among Twin Pairs With Autism. Arch Gen Psychiatry. 2011 doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamshere ML, Walters JT, Smith R, Richards AL, Green E, Grozeva D, Jones I, Forty L, Jones L, Gordon-Smith K, Riley B, O'Neill T, Kendler KS, Sklar P, Purcell S, Kranz J, Morris D, Gill M, Holmans P, Craddock N, Corvin A, Owen MJ, O'Donovan MC. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25:604–615. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA, Travers P, Walport M, Shlomchik M. Immunobiology. Garland Science; Oxford, UK: 2004. [Google Scholar]
- Jia P, Wang L, Fanous AH, Chen X, Kendler KS, Zhao Z. A bias-reducing pathway enrichment analysis of genome-wide association data confirmed association of the MHC region with schizophrenia. J Med Genet. 2012;49:96–103. doi: 10.1136/jmedgenet-2011-100397. [DOI] [PubMed] [Google Scholar]
- Johnson WG, Buyske S, Mars AE, Sreenath M, Stenroos ES, Williams TA, Stein R, Lambert GH. HLA-DR4 as a risk allele for autism acting in mothers of probands possibly during pregnancy. Arch Pediatr Adolesc Med. 2009;163:542–546. doi: 10.1001/archpediatrics.2009.74. [DOI] [PubMed] [Google Scholar]
- Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neurosci Biobehav Rev. 2008;32:1519–1532. doi: 10.1016/j.neubiorev.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipnis J, Derecki NC, Yang C, Scrable H. Immunity and cognition: what do age-related dementia, HIV-dementia and 'chemo-brain' have in common? Trends Immunol. 2008;29:455–463. doi: 10.1016/j.it.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Krzewski K, Strominger JL. The killer's kiss: the many functions of NK cell immunological synapses. Curr Opin Cell Biol. 2008;20:597–605. doi: 10.1016/j.ceb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LC, Zachary AA, Leffell MS, Newschaffer CJ, Matteson KJ, Tyler JD, Zimmerman AW. HLA-DR4 in families with autism. Pediatr Neurol. 2006;35:303–307. doi: 10.1016/j.pediatrneurol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Lee RH, Mills EA, Schwartz N, Bell MR, Deeg KE, Ruthazer ES, Marsh-Armstrong N, Aizenman CD. Neurodevelopmental effects of chronic exposure to elevated levels of pro-inflammatory cytokines in a developing visual system. Neural development. 2010;5:2. doi: 10.1186/1749-8104-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008;18:545–551. doi: 10.1016/j.tcb.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Sweeten TL, McMahon WM, Fujinami RS. Autistic disorder and viral infections. J Neurovirol. 2005;11:1–10. doi: 10.1080/13550280590900553. [DOI] [PubMed] [Google Scholar]
- Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luber-Narod J, Rogers J. Immune system associated antigens expressed by cells of the human central nervous system. Neurosci Lett. 1988;94:17–22. doi: 10.1016/0304-3940(88)90263-7. [DOI] [PubMed] [Google Scholar]
- Machold RP, Ploegh HL. Intermediates in the assembly and degradation of class I major histocompatibility complex (MHC) molecules probed with free heavy chain-specific monoclonal antibodies. The Journal of experimental medicine. 1996;184:2251–2259. doi: 10.1084/jem.184.6.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain, Behavior, and Immunity. 2012 doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzardo AM, Henkhaus R, Dhillon S, Butler MG. Plasma cytokine levels in children with autistic disorder and unrelated siblings. Int J Dev Neurosci. 2011 doi: 10.1016/j.ijdevneu.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, van de Water J. Breaking boundaries in neural-immune interactions. Neuron. 2009;64:9–12. doi: 10.1016/j.neuron.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Huang YH, Datwani A, Shatz CJ. H2-K(b) and H2-D(b) regulate cerebellar long-term depression and limit motor learning. Proc Natl Acad Sci U S A. 2009;106:6784–6789. doi: 10.1073/pnas.0902018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res. 2011;69:26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P, Altaye M, Wills-Karp M. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol. 2006;172:198–205. doi: 10.1016/j.jneuroim.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Mostafa GA, Shehab AA. The link of C4B null allele to autism and to a family history of autoimmunity in Egyptian autistic children. Journal of neuroimmunology. 2010;223:115–119. doi: 10.1016/j.jneuroim.2010.03.025. [DOI] [PubMed] [Google Scholar]
- Needleman LA, Liu XB, El-Sabeawy F, Jones EG, McAllister AK. MHC class I molecules are present both pre-and postsynaptically in the visual cortex during postnatal development and in adulthood. Proc Natl Acad Sci U S A. 2010;107:16999–17004. doi: 10.1073/pnas.1006087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nature reviews. Immunology. 2011;11:823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J Exp Med. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordahl CW, Lange N, Li DD, Barnett LA, Lee A, Buonocore MH, Simon TJ, Rogers S, Ozonoff S, Amaral DG. Brain enlargement is associated with regression in preschool-age boys with autism spectrum disorders. Proc Natl Acad Sci U S A. 2011;108:20195–20200. doi: 10.1073/pnas.1107560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odell D, Maciulis A, Cutler A, Warren L, McMahon WM, Coon H, Stubbs G, Henley K, Torres A. Confirmation of the association of the C4B null allelle in autism. Human immunology. 2005;66:140–145. doi: 10.1016/j.humimm.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun. 2011 doi: 10.1016/j.bbi.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onore C, Enstrom A, Krakowiak P, Hertz-Picciotto I, Hansen R, Van de Water J, Ashwood P. Decreased cellular IL-23 but not IL-17 production in children with autism spectrum disorders. J Neuroimmunol. 2009;216:126–129. doi: 10.1016/j.jneuroim.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH. Modeling autistic features in animals. Pediatr Res. 2011;69:34R–40R. doi: 10.1203/PDR.0b013e318212b80f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter LS, Scott SA, Barron E, Chui HC. MHC class II-positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res. 1992;33:549–558. doi: 10.1002/jnr.490330407. [DOI] [PubMed] [Google Scholar]
- Peudenier S, Hery C, Ng KH, Tardieu M. HIV receptors within the brain: a study of CD4 and MHC-II on human neurons, astrocytes and microglial cells. Res Virol. 1991;142:145–149. doi: 10.1016/0923-2516(91)90051-4. [DOI] [PubMed] [Google Scholar]
- Plioplys AV, Greaves A, Kazemi K, Silverman E. Lymphocyte function in autism and Rett syndrome. Neuropsychobiology. 1994;29:12–16. doi: 10.1159/000119056. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt AC, Rodgers RJ, Clapcote SJ. The role of neurexins in schizophrenia and autistic spectrum disorder. Neuropharmacology. 2012;62:1519–1526. doi: 10.1016/j.neuropharm.2011.01.024. [DOI] [PubMed] [Google Scholar]
- Rezaie P, Male D. Colonisation of the developing human brain and spinal cord by microglia: a review. Microscopy research and technique. 1999;45:359–382. doi: 10.1002/(SICI)1097-0029(19990615)45:6<359::AID-JEMT4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Ribic A, Zhang M, Schlumbohm C, Matz-Rensing K, Uchanska-Ziegler B, Flugge G, Zhang W, Walter L, Fuchs E. Neuronal MHC class I molecules are involved in excitatory synaptic transmission at the hippocampal mossy fiber synapses of marmoset monkeys. Cellular and molecular neurobiology. 2010;30:827–839. doi: 10.1007/s10571-010-9510-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, Law PA. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. 2009;163:907–914. doi: 10.1001/archpediatrics.2009.98. [DOI] [PubMed] [Google Scholar]
- Santos SG, Powis SJ, Arosa FA. Misfolding of major histocompatibility complex class I molecules in activated T cells allows cis-interactions with receptors and signaling molecules and is associated with tyrosine phosphorylation. J Biol Chem. 2004;279:53062–53070. doi: 10.1074/jbc.M408794200. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Stevens B. Synapse elimination during development and disease: immune molecules take centre stage. Biochem Soc Trans. 2010;38:476–481. doi: 10.1042/BST0380476. [DOI] [PubMed] [Google Scholar]
- Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron. 2009;64:40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JF, Tancredi DJ, Hertz-Picciotto I. Independent and dependent contributions of advanced maternal and paternal ages to autism risk. Autism research : official journal of the International Society for Autism Research. 2010;3:30–39. doi: 10.1002/aur.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe'er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23:116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh VK, Warren R, Averett R, Ghaziuddin M. Circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatr Neurol. 1997;17:88–90. doi: 10.1016/s0887-8994(97)00045-3. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WC. Complement regulatory proteins and autoimmunity. Autoimmunity. 2006;39:403–410. doi: 10.1080/08916930600739647. [DOI] [PubMed] [Google Scholar]
- Spiliotis ET, Pentcheva T, Edidin M. Probing for membrane domains in the endoplasmic reticulum: retention and degradation of unassembled MHC class I molecules. Mol Biol Cell. 2002;13:1566–1581. doi: 10.1091/mbc.01-07-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de Frutos R, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Yoon JL, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St Clair D, Goldstein DB, Stefansson K, Collier DA. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D. The contribution of TNFalpha to synaptic plasticity and nervous system function. Adv Exp Med Biol. 2011;691:541–557. doi: 10.1007/978-1-4419-6612-4_57. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stetler RA, Gan Y, Zhang W, Liou AK, Gao Y, Cao G, Chen J. Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog Neurobiol. 2010;92:184–211. doi: 10.1016/j.pneurobio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stubbs EG. Autistic symptoms in a child with congenital cytomegalovirus infection. Journal of autism and childhood schizophrenia. 1978;8:37–43. doi: 10.1007/BF01550276. [DOI] [PubMed] [Google Scholar]
- Stubbs EG, Magenis RE. HLA and autism. Journal of autism and developmental disorders. 1980;10:15–19. doi: 10.1007/BF02408429. [DOI] [PubMed] [Google Scholar]
- Stubbs EG, Ritvo ER, Mason-Brothers A. Autism and shared parental HLA antigens. J Am Acad Child Psychiatry. 1985;24:182–185. doi: 10.1016/s0002-7138(09)60445-3. [DOI] [PubMed] [Google Scholar]
- Stubbs G. Shared parental HLA antigens and autism. Lancet. 1981;2:534. doi: 10.1016/s0140-6736(81)90926-0. [DOI] [PubMed] [Google Scholar]
- Sweeten TL, Posey DJ, McDougle CJ. Brief report: autistic disorder in three children with cytomegalovirus infection. J Autism Dev Disord. 2004;34:583–586. doi: 10.1007/s10803-004-2552-y. [DOI] [PubMed] [Google Scholar]
- Syken J, Grandpre T, Kanold PO, Shatz CJ. PirB restricts ocular-dominance plasticity in visual cortex. Science. 2006;313:1795–1800. doi: 10.1126/science.1128232. [DOI] [PubMed] [Google Scholar]
- Szatmari P. Is Autism, at Least in Part, a Disorder of Fetal Programming? Arch Gen Psychiatry. 2011 doi: 10.1001/archgenpsychiatry.2011.99. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D'Antuono M, Cafe C, Giovedi S, Bue MC, D'Arcangelo G, Onofri F, Benfenati F. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem. 2000;75:634–643. doi: 10.1046/j.1471-4159.2000.0750634.x. [DOI] [PubMed] [Google Scholar]
- Thomas A, Gasque P, Vaudry D, Gonzalez B, Fontaine M. Expression of a complete and functional complement system by human neuronal cells in vitro. Int Immunol. 2000;12:1015–1023. doi: 10.1093/intimm/12.7.1015. [DOI] [PubMed] [Google Scholar]
- Torres AR, Maciulis A, Stubbs EG, Cutler A, Odell D. The transmission disequilibrium test suggests that HLA-DR4 and DR13 are linked to autism spectrum disorder. Hum Immunol. 2002;63:311–316. doi: 10.1016/s0198-8859(02)00374-9. [DOI] [PubMed] [Google Scholar]
- Torres AR, Sweeten TL, Cutler A, Bedke BJ, Fillmore M, Stubbs EG, Odell D. The association and linkage of the HLA-A2 class I allele with autism. Hum Immunol. 2006;67:346–351. doi: 10.1016/j.humimm.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Traynor TR, Majde JA, Bohnet SG, Krueger JM. Intratracheal double-stranded RNA plus interferon-gamma: a model for analysis of the acute phase response to respiratory viral infections. Life Sci. 2004;74:2563–2576. doi: 10.1016/j.lfs.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Turner T, Pihur V, Chakravarti A. Quantifying and modeling birth order effects in autism. PLoS ONE. 2011;6:e26418. doi: 10.1371/journal.pone.0026418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochemistry research international. 2011;2011:618127. doi: 10.1155/2011/618127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of neurology. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Kim SU, McGeer PL. Expression of complement C4 and C9 genes by human astrocytes. Brain Res. 1998;809:31–38. doi: 10.1016/s0006-8993(98)00811-7. [DOI] [PubMed] [Google Scholar]
- Warren RP, Burger RA, Odell D, Torres AR, Warren WL. Decreased plasma concentrations of the C4B complement protein in autism. Arch Pediatr Adolesc Med. 1994;148:180–183. doi: 10.1001/archpedi.1994.02170020066011. [DOI] [PubMed] [Google Scholar]
- Warren RP, Cole P, Odell JD, Pingree CB, Warren WL, White E, Yonk J, Singh VK. Detection of maternal antibodies in infantile autism. J Am Acad Child Adolesc Psychiatry. 1990;29:873–877. doi: 10.1097/00004583-199011000-00005. [DOI] [PubMed] [Google Scholar]
- Warren RP, Odell JD, Warren WL, Burger RA, Maciulis A, Daniels WW, Torres AR. Strong association of the third hypervariable region of HLA-DR beta 1 with autism. J Neuroimmunol. 1996;67:97–102. doi: 10.1016/0165-5728(96)00052-5. [DOI] [PubMed] [Google Scholar]
- Warren RP, Singh VK, Cole P, Odell JD, Pingree CB, Warren WL, DeWitt CW, McCullough M. Possible association of the extended MHC haplotype B44-SC30-DR4 with autism. Immunogenetics. 1992;36:203–207. doi: 10.1007/BF00215048. [DOI] [PubMed] [Google Scholar]
- Warren RP, Singh VK, Cole P, Odell JD, Pingree CB, Warren WL, White E. Increased frequency of the null allele at the complement C4b locus in autism. Clinical and experimental immunology. 1991;83:438–440. doi: 10.1111/j.1365-2249.1991.tb05657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzba-Bobrowicz T, Kosno-Kruszewska E, Lewandowska E, Lechowicz W, Schmidt-Sidor B. Major histocompatibility complex class II (MHC II) expression during development of human fetal brain and haemopoietic organs. Adv Exp Med Biol. 2001;495:93–101. doi: 10.1007/978-1-4615-0685-0_13. [DOI] [PubMed] [Google Scholar]
- Williams DB, Barber BH, Flavell RA, Allen H. Role of beta 2-microglobulin in the intracellular transport and surface expression of murine class I histocompatibility molecules. J Immunol. 1989;142:2796–2806. [PubMed] [Google Scholar]
- Wingate M, Mulvihill B, Kirby R, Pettygrove S, Cunniff C, Meaney F, Schulz E, Miller L, Robinson C, Quintana G, Kaiser M, Lee L, Landa R, Newschaffer C, Constantino J, Fitzgerald R, Zahorodny W, Daniels J, Giarelli E, Pinto-Martin J, Levy S, Nicholas J, Charles JZ, J., Maenner M, Durkin M, Rice C, Baio J, Van Naarden Braun K, Phillips K, Doernberg N, Yeargin-Allsopp M. Prevalence of autism spectrum disorders--Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. Morbidity and mortality weekly report. Surveillance summaries. 2012;61:1–19. [PubMed] [Google Scholar]
- Xu HP, Chen H, Ding Q, Xie ZH, Chen L, Diao L, Wang P, Gan L, Crair MC, Tian N. The immune protein CD3zeta is required for normal development of neural circuits in the retina. Neuron. 2010;65:503–515. doi: 10.1016/j.neuron.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y, Fujimoto C, Nakajima E, Isagai T, Matsuishi T. Possible association between congenital cytomegalovirus infection and autistic disorder. J Autism Dev Disord. 2003;33:455–459. doi: 10.1023/a:1025023131029. [DOI] [PubMed] [Google Scholar]
- Yenari MA. Heat shock proteins and neuroprotection. Advances in experimental medicine and biology. 2002;513:281–299. doi: 10.1007/978-1-4615-0123-7_10. [DOI] [PubMed] [Google Scholar]
- Zerbo O, Iosif AM, Delwiche L, Walker C, Hertz-Picciotto I. Month of conception and risk of autism. Epidemiology. 2011;22:469–475. doi: 10.1097/EDE.0b013e31821d0b53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer's disease. J Neurochem. 2008;106:2080–2092. doi: 10.1111/j.1471-4159.2008.05558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]