

Conspectus
Cyanine derivatives, named from the Greek word kyanos meaning dark-blue, were discovered more than 150 years ago and remain one of the most widely used classes of organic dyes with contemporary applications in photography (panchromatic emulsions), information storage (CD-R and DVD-R media) and biochemistry (DNA and protein labeling) fields. Cyanine chromogens consist of a charged π-conjugated segment containing an odd number of sp2 carbon atoms with the chain capped at the extremities by two electronegative centers, typically nitrogen or oxygen atoms. Cyanines are characterized by a vanishing bond length alternation indicating nearly equal carbon–carbon bond lengths, as well as a very intense and sharp absorption band presenting a shoulder. This hallmark band undergoes a strong red shift when the chain is extended. This so-called vinyl shift is extremely large (ca. 100 nm for each pair of carbon atoms added in the π-conjugated path), making cyanines ideal building blocks for the design of devices with near-infrared applications. Numerous cyanines also exhibit emission bands with large quantum yields. These exceptional optical properties explain why both canonical cyanines and the corresponding fluoroborates (e.g., boron-dipyrromethene, BODIPY) remain the focus of an ever-growing body of experimental work.
In turn, this popularity has stimulated quantum mechanical investigations aiming, on the one hand, at probing the specific electronic nature of cyanine dyes and, on the other hand, at helping to design new dyes. However, the adiabatic approximation to time-dependent density functional theory, the most widespread ab initio model for electronically excited states, fails to accurately reproduce the absorption spectra of cyanine derivatives: it yields a systematic and large underestimation of the experimental wavelengths irrespective of the details of the computational protocol. In contrast, highly correlated wave function approaches provide accurate transition energies for model systems but are hardly applicable to real-life cyanines and BODIPY. This indicates that setting up a computationally tractable theoretical protocol that provides both robust and accurate optical spectra for cyanine-based dyes is a major challenge that has only been taken up lately.
In this Account, we compile the most recent advances in the field by considering both compact streptocyanines and large fluoroborates. For the former, we summarize the key results obtained with a large panel of theoretical approaches, allowing us not only to understand the origin of the cyanine challenge but also to pinpoint the schemes presenting the most promising accuracy/effort ratio. For the latter, we show via selected examples how theoretical models can be used to reproduce simultaneously experimental band shapes and transition energies, thus paving the way to an efficient in silico design of new compounds.
1. Introduction
Generally speaking, charged (cationic or anionic) cyanines represent one of the subclasses of the large family of streptopolymethine dyes.1,2 These have been used first as sensitizer for emulsion photography and then in advanced photonic materials,3,4 as well as photodynamic therapy agents or bioprobes for near-infrared (NIR) imaging.5,6 These dyes consist of a π-conjugated bridge composed of an odd number of sp2 carbon atoms linking electron-donating or -accepting groups. An unambiguous rationalization of the particular photophysical properties of polymethine dyes as controlled by the length and nature of the bridge (the so-called vinyl shift),7 the composition of the terminal moieties and their environment (solvent polarity, counterions, etc.),8,9 and other factors remains a matter of important debate. Until recently, polymethines were classified on the basis of their ground state electronic configurations.10 The cyanine configuration11 is defined by the charge being fully and symmetrically delocalized over the entire conjugated backbone (nearly equal bond length, and consequently negligible bond-length alternation, BLA). The relationship between this vanishing BLA and the vinyl shift can be qualitatively understood with simple models.12 The cyanine configuration results in sharp and intense absorption with a vibronic shoulder at higher energy. The dipolar configuration is characterized by a charge predominantly localized at one extremity (positive BLA) and a spectroscopic signature consisting of a broad and structureless charge transfer type transition that is blue-shifted compared with the cyanine one. As an alternative, a third electronic configuration that corresponds to a predominant charge-centered state called bis-dipole was experimentally and theoretically demonstrated by Maury and co-workers.13 These three limit configurations (i.e., the forms I–III) are represented in Scheme 1. Going from one form to another, meaning crossing the cyanine limit in either direction (I ↔ II) or passing from cyanine to bis-dipole (I ↔ III), remains a challenging task.7,8,13,14
Scheme 1. Schematic Representation of the Three Limit Structures of Cyanines.
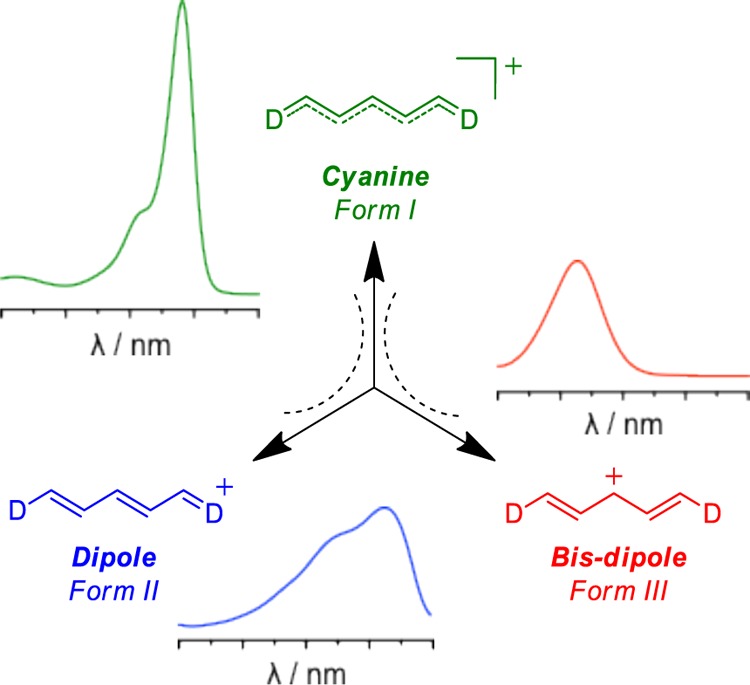
Adapted with permission from ref (13). Copyright 2014 American Chemical Society.
As an alternative to cyanines, boron-dipyrromethene (i.e., BODIPY) and other fluoroborates considered to be cis-constrained cyanines (see Scheme 2) are recognized as the most effective and versatile family of organic fluorophores developed to date. This is explained by their extremely rich chemistry and their remarkable photophysical properties inclucing a sharp fluorescence emission band with large quantum yield.15−17 Applications of these derivatives are already found in bioimaging, photodynamic therapy, advanced optics and optoelectronics, and photovoltaics.18,19
Scheme 2. Representation of (1) the BODIPY Structure (center), (2) the Formal Relationship between BODIPY and Streptocyanines (top), and (3) Synthesized Variants of the BODIPY.

Clockwise starting from top right: extension of the π-path, stiffening of the lateral arms, addition of electron-donating groups, typical aza-BODIPY, substituted NBO compound, boranil derivative, and fused-dimer structure.
For rationalizing experimental measurements, as well as for predicting optical properties of to-be-synthesized derivatives, quantum mechanical tools are extremely popular. In the framework of optical spectra and electronically excited states, the toolbox of theoretical chemists is now vast,20 comprising both wave function methods, for example, the single reference EOM-CC (equation-of-motion coupled-cluster), CIS(D) (configuration interaction singles with a perturbative correction for doubles), ADC (algebraic-diagrammatic construction propagator approach), and the multireference CAS (complete active space) schemes as well as density-based approaches. In the latter category, TD-DFT (time-dependent density functional theory) is certainly the most popular method due to its remarkably small computational cost and to the availability of analytical derivatives allowing for easy exploration of the excited-state potential energy surfaces.21 In this Account, we aim to provide an overview of the applications of these approaches for cyanine and BODIPY derivatives, two broad families of chromophores that consistently grieve computational chemists. With particular emphasis upon the simultaneous calculation of transition energies and the accurate restoration of absorption and emission band topologies (that require the calculation of excited-state vibrations), we herein point out the strengths and weaknesses of adiabatic TD-DFT and the opportunities offered by CIS(D) as well as by GW/BSE (Greens function/Bethe–Salpeter equation) that allow for addressing these weaknesses.
2. Model Chains and Cyanines
For the increasingly long streptocyanine dyes (CNx in Scheme 3), numerous methodologies have been tested with some key results listed in Table 1; the interested reader can find expanded data in the Supporting Information. It is important to note that all data reported in Table 1 and in the Supporting Information have been obtained in the vertical approximation, that is, using a frozen ground-state geometry and completely neglecting vibronic couplings. While this approximation allows one to use a large range of theoretical schemes, the obtained theoretical figures cannot be rigorously compared with experiment, notably because CNx derivatives undergo strong deformations after absorption, which make the constant geometry assumption an inadequate hypothesis.22 The first ab initio calculations on the CNx derivatives appeared in 2001 with the works of Fabian23 and Schreiber and co-workers.24 Already at that time, it turned out that the adiabatic approximation to TD-DFT relying on the B3LYP hybrid exchange-correlation functional could not provide accurate transition energies (TD-DFT values are overshot by ca. 0.5–1.0 eV, a deviation well beyond TD-DFT’s expected accuracy for low-lying excited-states)25 nor restore the vinyl shift (underestimated by a factor of ca. 2).23,24 An examination of Table S-2 in the Supporting Information demonstrates that these large deviations are not related to the selection of specific atomic basis sets or ground-state geometries. Indeed, the typical variations noted when changing these two parameters are 1 order of magnitude smaller than the reported TD-DFT error. Likewise, substitution of the chain ends with methyl groups (NMe2 instead of NH2 in Scheme 3) does not impact the conclusions.22,24,26,27 The early CAS-PT2 calculations of Schreiber et al. revealed no significant multireference character for the states of interest, for example, the main determinant represents 88% of the first excited-state in CN7;24 hence the TD-DFT discrepancies could not be ascribed to the monodeterminantal nature of the theory. In the same vein, one cannot explain this error by the well-known charge-transfer (CT) failure of TD-DFT,28 because (i) TD-DFT overestimates the transition energies of CNx whereas it would underestimate the energies of CT states,23,24,29 (ii) the use of range-separated hybrids, for example, CAM-B3LYP, does not provide any significant improvement,26 (iii) the examination of the frontier orbitals involved in the transition indicates an important overlap between the relevant occupied and virtual orbitals,30 and (iv) optimally tuned functionals that minimize the delocalization error do not cure the problem.31 The fact that the obtained transition energies are rather insensitive to the selection of a specific (pure or hybrid) exchange-correlation functional is one of the theoretical signatures of cyanine derivatives. This can be illustrated by comparing the results obtained for CN9 with two extreme functionals in the Minnesota family, namely M06-L (no exact exchange) and M06-HF (100% exact exchange): a small shift of 0.26 eV is noted (see the Supporting Information).30 A second theoretical signature of cyanines is that the Tamm–Dancoff approximation (TDA), generally having a marginal impact on estimates of TD-DFT, induces a strong increase of the transition energies by ca. 0.5 eV,22,31 that is, even larger errors than the original TD-DFT produces. In fact, the cyanine failure of TD-DFT can be attributed to the difficulty of capturing the differential electron-correlation effects between the ground and excited states.31−34 To the best of our knowledge, this was first noted by Grimme and Neese,32 who proposed the use of their double hybrid exchange-correlation functional, B2PLYP, as a fix. While this approach including a correction for doubly excited-states indeed brings significant improvements, the vinyl shift remains underestimated, and the absolute error remains far from negligible.32 To shed a qualitative light on these differential electron-correlation effects, Masunov evaluated the difference of total density between the first excited and ground states in a medium sized cyanine.35 He showed that there are strong depletions (accumulations) of density on even (odd) atomic centers, the absolute variations being about three times the one observed in the corresponding polyene. In other words, despite the strong overlap between the frontier orbitals involved in the transition, a particularly strong reorganization of the electron density takes place in cyanines. This is likely the reason why Minnesota functionals providing a more accurate treatment of electron correlation, for example, M08-SO, are among the best performing global hybrids for the CNx series,30 though the errors remain large (see Table 1). More thorough theoretical analyses of the origin of the cyanine problem were recently performed by the groups of Autschbach and Ziegler.31,34 They found that time-dependent approaches (both TD-DFT and TDA) afford too large singlet energies but too small triplet energies, the singlet–triplet separation being consequently strongly overestimated (by ca. 1 eV), an error almost independent of the selected functional.31 They related this inaccurate singlet–triplet gap to an inadequate representation of an exchange integral between π and π* orbitals. On the contrary, both the singlet energies and the singlet–triplet separations obtained with time-independent approaches, for example, ΔSCF, are significantly modified when varying the exact exchange percentage. It was noticed that BHHLYP (50% of exact exchange) emerges as a good compromise in that ΔSCF protocol (see Table 1),34 a result subsequently confirmed by Filatov and Huix-Rotllant with a similar approach.33
Scheme 3. Model Cyanine Chains of Increasing Chain Length.

Table 1. Vertical Transition Energies to the Lowest Excited-State of Model Cyanines (See Scheme 3) Calculated with Various Theoretical Approachesa.
| method | CN3 | CN5 | CN7 | CN9 | CN11 | ref |
|---|---|---|---|---|---|---|
| Multireference Wave Function Approaches | ||||||
| DMC | 7.38 | 5.03 | 3.83 | 3.09 | 2.62 | (22) |
| CAS-PT2 | 7.73 | 4.85 | 3.55 | 2.70 | 2.16 | (24) |
| CAS-PT2 | 6.99 | 4.46 | 3.30 | 2.59 | 2.10 | (22) |
| CAS-PT2 (IPEA shift) | 7.19 | 4.69 | 3.52 | 2.81 | 2.46 | (22) |
| Single-Reference Wave Function Approaches | ||||||
| ADC(2) | 4.64 | 3.46 | 2.78 | 2.32 | (33) | |
| CIS(D) | 4.85 | 3.65 | 2.95 | 2.48 | (31) | |
| CC2 | 7.26 | 4.97 | 3.79 | 3.10 | 2.64 | (22) |
| CC3 | 7.16 | 4.84 | 3.65 | 2.96 | 2.53 | (22) |
| GW/BSE | 4.80 | 3.63 | 2.96 | 2.48 | (36) | |
| Density Functional Theory Approaches | ||||||
| TD-B2PLYP | 7.17 | 5.12 | 3.87 | 3.23 | 2.80 | (32) |
| TD-B3LYP | 7.60 | 5.28 | 4.12 | 3.44 | 2.99 | (23) |
| TD-CAM-B3LYP | 7.61 | 5.28 | 4.13 | 3.44 | 2.98 | (26) |
| TD-LC-PBE*b | 5.26 | 4.09 | 3.40 | 2.94 | (31) | |
| TD-LC-PBE0*b | 5.37 | 4.19 | 3.49 | 3.02 | (31) | |
| TD-M06-2X | 5.23 | 4.09 | 3.41 | 2.95 | (30) | |
| TD-M08-SO | 5.16 | 4.04 | 3.37 | 2.91 | (30) | |
| Δ SCF-BHHLYP | 7.33 | 4.81 | 3.55 | 2.79 | 2.29 | (34) |
| Δ SCF-BHHLYPc | 4.87 | 3.72 | 3.06 | 2.62 | (33) | |
| SF-BHHLYPd | 5.09 | 3.95 | 3.27 | 2.82 | (33) | |
All values are in eV. For methods implying the selection of an active space, the values obtained with the optimal space as selected in the original works are reported. In this table, as well as in the text, TD refers to the adiabatic approximation of TD-DFT, except when explicitly noted. See the Supporting Information for details about the basis sets and geometries used. Expanded tables with computational details are available in the Supporting Information as well.
Optimally tuned approaches.
SSR variations of the ΔSCF model.
Spin-flip approach.
Having identified and explained the limitations of TD-DFT, let us now turn toward alternative approaches. By comparison of the three CAS-PT2 results listed in Table 1, it is obvious that the details of the calculations (size of the active space, application of an IPEA shift) are of prime importance especially for short chains.22,24 As stated above, these CAS-PT2 calculations did not revealed strong multireference character, suggesting that one can therefore trust the most correlated single-reference approach, CC3, which provides values of 4.84 and 3.65 eV for CN5 and CN7, respectively. These figures are between those of the two most refined multireference schemes, namely, DMC (5.03 and 3.83 eV) and CAS-PT2 with a large active space and an IPEA shift (4.69 and 3.52 eV). Interestingly, all methods that explicitly account for double excitations, namely, CC2, CIS(D), and ADC(2) yield quite accurate trends that are close to the CC3 reference. CC2 slightly overestimates the CC3 results (by ca. 0.10–0.15 eV),22 whereas ADC(2) leads to the opposite error (underestimation by ca. 0.15–0.20 eV).33 As noted by Send et al. and consistently with the above analysis, the correlation energy strongly depends on double excitations for all cyanines, and the contribution of the triple excitations is larger than in other π-conjugated derivatives.22 In practice, CIS(D) that is systematically within 0.05 eV of the CC3 reference appears as very effective due to its relatively small computational cost.31 The same holds for the GW/BSE approach, which explicitly and self-consistently accounts for the correlation between the electron and the hole.36 Given its limited computational cost (ca. twice that of TD-DFT with the same scaling) and its ease of use, GW/BSE probably stands as one of the best compromises among ab initio approaches for cyanines.
Real-life cyanines, used for practical applications, present a conjugated skeleton similar to the one of the CNx derivatives but with much bulkier end groups. Of course, for those dyes, the large TD-DFT deviations noted for the transition energies of the model systems persist.13,23,27,29,37 As stated in the Introduction, these derivatives develop a hallmark shoulder that was first modeled by Champagne and co-workers.29 Using a vibronic approach and the BHHLYP functional, they could reproduce the experimental absorption band shape of a typical compound (see Figure 1). This result hinted that although TD-DFT fails to provide accurate transition energies, this approach is able to correctly describe the ground and excited state geometries and vibrational frequencies. The same group applied a similar methodology to quantify the impact of stacking on vibrationally resolved spectra.38 Lately, we have investigated a homologous series of conjugated dyes in such a way as to smoothly explore a range of transitions going from well-localized behavior to classical cyanine.13 We showed that the hallmark shoulder in cyanine could be mainly ascribed to an asymmetric deformation mode implying the bonds of the conjugated pathway, whereas in the corresponding bis-dipolar dyes (see Scheme 1), the band shapes were related to symmetric vibrations, implying changes in the single/double bond character of the CC linkages.
Figure 1.

Comparison between TD-DFT (dotted lines) and experimental (full lines) band shapes for a typical cyanine dye. Note that an offset of 0.5 eV has been applied on the theoretical curve so that the TD-DFT 0–0 energy matches its experimental counterpart. Adapted with permission from ref (29). Copyright 2006 Elsevier.
3. BODIPY and Related Derivatives
For BODIPY and other fluoroborates, one can split the simulations into two categories, those performed within the vertical approximation (frozen ground-state geometry and neglect of vibrational effects, see above) and those going beyond that simple scheme by exploring and characterizing the potential energy surface of the excited state.
With the former strategy, it is technically possible to perform reference wave function calculations, at least for compact derivatives, but only a few studies are available.39−41 As for streptocyanines, these works, for example, the coupled-cluster analysis in ref (41), revealed that although BODIPY derivatives do not possess a strong multiconfigurational character, they have first singlet excited states presenting a substantial (and rather constant for all structures) contribution from the double excitations. In turns, this indicates that conventional TD-DFT might lead to an overestimation of the transition energies. However, several TD-DFT studies performed with popular hybrid functionals, such as B3LYP or PBE0, have led to vertical transition wavelengths nicely matching the experimental longest wavelength of maximal absorption (λmax) for both aza-BODIPY42,43 and BODIPY.44,45 Indeed in all these studies, rather small mean absolute deviations between theory and experiment have been obtained (0.05–0.28 eV, depending on the work), but the determination coefficients relating calculated and measured data are also small (R2 in the 0.45–0.79 range) indicating a limited reproduction of the auxochromic trends. Similar conclusions have been reached for the calculation of vertical fluorescence energies of both aza-BODIPY46 and BODIPY.47 The apparent accuracy can be explained by an error cancelation phenomenon between (i) the neglect of vibrational (and often solvation) effects and (ii) the inherent inaccuracy of TD-DFT for describing transition energies to states involving a significant double character.
Because gas phase vertical transition energies cannot be measured in practice, the second strategy has, despite its computational cost, been recently advocated.48−52 In these studies, theoretical 0–0 energies (that can be directly compared with measured absorption–fluorescence crossing point) or band shapes are used to provide meaningful comparisons with experiments. It has also been shown that solvation effects are quite difficult to capture with theoretical tools for BODIPY derivatives; consequently, advanced solvation models are often needed.40,53 Figure 2 displays the 0–0 energies obtained with a refined TD-DFT model carried out on a panel of 58 BODIPY derivatives, collected and grouped from several works. As can be seen, TD-DFT overshoots the transition energies by an average of 0.37 eV but yields very consistent transition energies (R2 = 0.97). These results are in sharp contrast with the data obtained within the cruder vertical approximation (as above). This trend is also consistent with the results obtained with a very similar protocol by Boens and co-workers for “frustrated” BODIPY derivatives.54 Since 0–0 calculations imply the determination of the excited-state vibrational frequencies, they become rapidly impossible with highly accurate approaches. Because of this, we have recently suggested to correct the TD-DFT 0–0 energies by using data obtained with theoretical approaches suited for cyanine derivatives, such as GW/BSE55 or SOS-CIS(D).51,52 In those calculations, the geometrical, vibrational, and solvation effects are obtained with TD-DFT, while the use of the more accurate approach is limited to vertical gas-phase calculations, making the protocol computationally tractable even for rather large (ca. 150 atoms) fluorophores. The results obtained with such a SOS-CIS(D)//TD-DFT hybrid method are also displayed in Figure 2, which shows that the theoretical estimates are now both accurate (MAE of 0.08 eV) and consistent (R2 = 0.97). We note that such a mixed protocol has been tested in more general frameworks,56,57 but appears particularly efficient for cyanine-like compounds.
Figure 2.
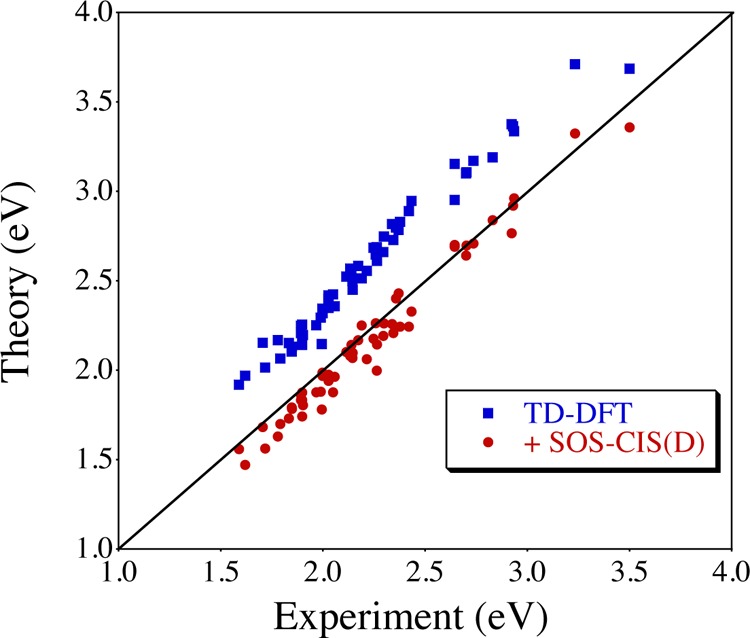
Comparison between TD-DFT and experimental 0–0 energies (eV) for a set of 58 BODIPY derivatives built from the data of refs (49, 51, and 52). All TD-DFT calculations have been performed at the SS-PCM-TD-M06-2X/6-311+G(2d,p)//PCM-M06-2X/6-31G(d) level. The impact of an SOS-CIS(D) vertical correction is also illustrated. The central line indicates a perfect match between theory and experiment.
Similar to the standard cyanine, unsubstituted BODIPY (center of Scheme 2) presents absorption and emission bands that are nearly perfect mirror images, with intense peaks accompanied by characteristic shoulders displaced by ca. 15 nm from and approximatively half as intense as the main peak.58 Vibronic TD-DFT calculations reproduce the presence and relative position of this shoulder, though the height of the emission shoulder is underrated.49 Figure 3 presents a comparison between experimental and theoretical band topologies for two larger fluoroborates.49 It is obvious that the proposed theoretical protocol very accurately restores both the band positions and the band shapes: the first dye (in red in Figure 3) presents two peaks of nearly equal intensity followed by a shoulder with a ca. 50% height, whereas for the second compound (in black in Figure 3), the second peak is the most intense with a more intense shoulder than for the first dye. All these key features can be reproduced by a TD-DFT approach with SOS-CIS(D) corrected transition energies, highlighting the performances of such a hybrid approach. For both dyes, the presence of multiple maxima in the absorption band can be mainly attributed to a vibrational mode at ca. 1550 cm–1 that corresponds to in-plane wagging of the CH and distortion of the aromatic cycles.49
Figure 3.
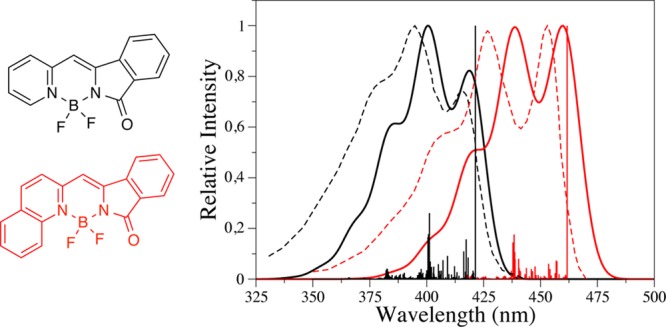
Comparison between TD-DFT and experimental absorption band shapes for two BODIPY derivatives. The positions of the theoretical band were set by accounting for SOS-CIS(D) corrections. Dotted lines represent experiment adapted with permission from ref (100). Copyright 2008 American Chemical Society. Full lines represent theory (both convoluted and stick spectra), adapted from ref (49) with permission from the Royal Society of Chemistry.
4. Further Challenges
Despite the undeniable improvements offered by recent computational protocol with respect to straightfoward vertical TD-DFT approaches, important physical aspects that may strongly affect photoinduced phenomena are still missing in the description. Whereas solvent effects are taken into account implicitly in the ground- and excited-state wave functions, a proper treatment of dynamics, explicit solvent molecules, counterions,8 or even aggregation of the dyes in various networks59 is potentially required and would necessitate further methodological developments. Likewise, if cyanine or BODIPY chromophores are combined with late transition metals,60 spin–orbit couplings should be included in the description. These are the next important challenges that computational approaches of the photophysical behavior of cyanines or related chromophores will probably face.
5. Conclusions and Outlook
In this Account, we have surveyed theoretical calculations aimed at reproducing the optical spectra of cyanines and their derivatives. Experimentally, cyanines are characterized by well-identified structural and spectroscopic signatures: a vanishing bond length alternation and a sharp absorption peak accompanied by a shoulder that are red-shifted by ca. 100 nm for each pair of carbon atoms added in the conjugated path. The theoretical simulations of the vertical transition energies performed with time-dependent density functional theory may deliver contrasted results: the values appeared strongly overshot for model streptocyanines but reasonably in line with experiment for large BODIPY derivatives. This latter outcome is related to an error compensation between, on the one hand, the vertical approximation and, on the other hand, the inherent inadequacy of TD-DFT for cyanines. Therefore, knowing how to identify a cyanine excited state with theory is a crucial prerequisite for obtaining a good answer for a good reason. There are indeed two straightforward theoretical ways to pinpoint a cyanine transition: (i) the adiabatic TD-DFT transitions energies are almost independent of the selected exchange-correlation functional, and (ii) these energies are strongly upshifted when the TDA approximation is applied. When a cyanine excited state is revealed, standard TD-DFT methods that do not capture the differential electron correlation between the ground and excited states systematically yield overly large transition energies but rather accurate geometries and vibrational signatures. To improve the former, it is mandatory to take into account the contributions of doubly excited states; all methods including such contributions, for example, CC2, ADC(2), CIS(D), and GW/BSE, provide very satisfying results, whereas the use of multireference schemes is generally not required. As an improved approach, it has been proposed to combine TD-DFT structures and vibrations to vertical calculations performed with one of the above “double” approaches. Such a computationally appealing protocol allows for physically meaningful predictions of both the 0–0 energies and the band shapes of a large panel of BODIPY derivatives with a remarkable accuracy. An example of such protocol is detailed in the Supporting Information. Even if the easy-handling approach consisting of the determination of the vertical transition energies from GS structures still persists, one may expect that the herein-proposed strategy will soon become the norm when the goal is reproducing the experimental features. Future challenges in this field of computationally reproducing optical properties concerns the proper treatment of (i) dynamical aspects (temperature, solvent, etc.) that often play decisive roles in the overall spectra, (ii) aggregation processes, and (iii) spin–orbit couplings that cannot be eluded when late-transition metals are combined with cyanines.60
Acknowledgments
The authors are indebted to their coauthors on the topic for fruitful discussions (alphabetical order): C. Adamo, X. Blase, P. Boulanger, A. Charaf-Eddin, S. Chibani, I. Ciofini, I. Duchemin, A. D. Laurent, O. Maury, B. Mennucci, and D. G. Truhlar. D.J. thanks K. J. Chen for her careful reading of the manuscript. D.J. acknowledges the European Research Council (ERC) and the Région des Pays de la Loire for financial support in the framework of Starting Grant (Marches -278845) and a recrutement sur poste stratégique, respectively.
Biographies
Boris Le Guennic was born in Pabu (France) in 1976 and received his Ph.D. in Chemistry from the University of Rennes in 2002. He then successively moved to the Universities of Erlangen, Buffalo, and Bonn for postdoctoral stays in the groups of Jochen Autschbach and Markus Reiher. In 2005, he was appointed as CNRS researcher at ENS de Lyon (France). He is currently CNRS researcher at University of Rennes where his research is devoted mainly to the modeling of magnetic and optical properties in lanthanide complexes and organic chromophores.
Denis Jacquemin was born in Dinant (Belgium) in 1974 and received his Ph.D. in Chemistry from the University of Namur in 1998, before moving to the University of Florida for his postdoctoral stay. In 2003, he was appointed as Research Associate of the FNRS in Namur (Belgium). He is currently full Professor at the Université de Nantes (France) and junior member of the Institut Universitaire de France. He obtained an ERC starting grant (2012–2016) and is the recipient of the Watoc’s Dirac Medal (2014). His research is focused on modeling the electronically excited states in dyes and photochromes.
Supporting Information Available
Expanded tables and detailed computational protocol. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Fabian J.; Nakazumi H.; Matsuoka M. Near-Infrared Absorbing Dyes. Chem. Rev. 1992, 92, 1197–1226. [Google Scholar]
- Mishra A.; Behera R. K.; Behera P. K.; Mishra B. K.; Behera G. B. Cyanines during the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2011. [DOI] [PubMed] [Google Scholar]
- Hales J. M.; Matichak J.; Barlow S.; Ohira S.; Yesudas K.; Brédas J.-L.; Perry J. W.; Marder S. R. Design of Polymethine Dyes with Large Third-Order Optical Nonlinearities and Loss Figures of Merit. Science 2010, 327, 1485–1488. [DOI] [PubMed] [Google Scholar]
- Bellier Q.; Makarov N. S.; Bouit P.-A.; Rigaut S.; Kamada K.; Feneyrou P.; Berginc G.; Maury O.; Perry J. W.; Andraud C. Excited State Absorption: A Key Phenomenon for the Improvement of Biphotonic Based Optical Limiting at Telecommunication Wavelengths. Phys. Chem. Chem. Phys. 2012, 14, 15299–15307. [DOI] [PubMed] [Google Scholar]
- Wang X.; Sun J.; Zhang W.; Ma X.; Lv J.; Tang B. A near-Infrared Ratiometric Fluorescent Probe for Rapid and Highly Sensitive Imaging of Endogenous Hydrogen Sulfide in Living Cells. Chem. Sci. 2013, 4, 2551–2556. [Google Scholar]
- Hu C.; Sun W.; Cao J.; Gao P.; Wang J.; Fan J.; Song F.; Sun S.; Peng X. A Ratiometric Near-Infrared Fluorescent Probe for Hydrazine and Its in Vivo Applications. Org. Lett. 2013, 15, 4022–4025. [DOI] [PubMed] [Google Scholar]
- Tolbert L. M.; Zhao X. Beyond the Cyanine Limit: Peierls Distortion and Symmetry Collapse in a Polymethine Dye. J. Am. Chem. Soc. 1997, 119, 3253–3258. [Google Scholar]
- Bouit P. A.; Aronica C.; Toupet L.; Le Guennic B.; Andraud C.; Maury O. Continuous Symmetry Breaking Induced by Ion Pairing Effect in Heptamethine Cyanine Dyes: Beyond the Cyanine Limit. J. Am. Chem. Soc. 2010, 132, 4328–4335. [DOI] [PubMed] [Google Scholar]
- Hu H.; Przhonska O. V.; Terenziani F.; Painelli A.; Fishman D.; Ensley T. R.; Reichert M.; Webster S.; Bricks J. L.; Kachkovski A. D.; Hagan D. J.; van Stryland E. W. Two-Photon Absorption Spectra of a near-Infrared 2-Azaazulene Polymethine Dye: Solvation and Ground-State Symmetry Breaking. Phys. Chem. Chem. Phys. 2013, 15, 7666–7678. [DOI] [PubMed] [Google Scholar]
- Terenziani F.; Przhonska O. V.; Webster S.; Padilha L. A.; Slominsky Y. L.; Davydenko I. G.; Gerasov A. O.; Kovtun Y. P.; Shandura M. P.; Kachkovski A. D.; Hagan D. J.; van Stryland E. W.; Painelli A. Essential-State Model for Polymethine Dyes: Symmetry Breaking and Optical Spectra. J. Phys. Chem. Lett. 2010, 1, 1800–1804. [Google Scholar]
- Dähne S.; Radeglia R. Revision der Lewis-Calvin-Regel zur Charakterisierung Vinyloger Polyen- und Polymethinähnlicher Verbindungen. Tetrahedron 1971, 27, 3673–3693. [Google Scholar]
- Autschbach J. Why the Particle-in-a-Box Model Works Well for Cyanine Dyes but Not for Conjugated Polyenes. J. Chem. Educ. 2007, 84, 1840–1845. [Google Scholar]
- Pascal S.; Haefele A.; Monnereau C.; Charaf-Eddin A.; Jacquemin D.; Le Guennic B.; Andraud C.; Maury O. Expanding the Polymethine Paradigm: Evidence for the Contribution of a Bis-Dipolar Electronic Structure. J. Phys. Chem. A 2014, 118, 4038–4047. [DOI] [PubMed] [Google Scholar]
- Würthner F.; Archetti G.; Schmidt R.; Kuball H.-G. Solvent Effect on Color, Band Shape, and Charge-Density Distribution for Merocyanine Dyes Close to the Cyanine Limit. Angew. Chem., Int. Ed. 2008, 47, 4529–4532. [DOI] [PubMed] [Google Scholar]
- Loudet A.; Burgess K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [DOI] [PubMed] [Google Scholar]
- Ulrich G.; Ziessel R.; Harriman A. The Chemistry of Fluorescent Bodipy Dyes: Versatility Unsurpassed. Angew. Chem., Int. Ed. 2008, 47, 1184–1201. [DOI] [PubMed] [Google Scholar]
- Nepomnyashchii A. B.; Bard A. J. Eletrochemistry and Electrogenerated Chemiluminescence of BODIPY Dyes. Acc. Chem. Res. 2012, 45, 1844–1853. [DOI] [PubMed] [Google Scholar]
- Bouit P. A.; Kamada K.; Feneyrou P.; Berginc G.; Toupet L.; Maury O.; Andraud C. Two-Photon Absorption-Related Properties of Functionalized BODIPY Dyes in the Infrared Range up to Telecommunication Wavelengths. Adv. Mater. 2009, 21, 1151–1154. [Google Scholar]
- Rousseau T.; Cravino A.; Bura T.; Ulrich G.; Ziessel R.; Roncali J. Multi-donor Molecular Bulk Heterojunction Solar Cells: Improving Conversion Efficiency by Synergistic Dye Combinations. J. Mater. Chem. 2009, 19, 2298–2300. [Google Scholar]
- González L.; Escudero D.; Serrano-Andrès L. Progress and Challenges in the Calculation of Electronic Excited States. ChemPhysChem 2012, 13, 28–51. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Jacquemin D. The Calculations of Excited-State Properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [DOI] [PubMed] [Google Scholar]
- Send R.; Valsson O.; Filippi C. Electronic Excitations of Simple Cyanine Dyes: Reconciling Density Functional and Wave Function Methods. J. Chem. Theory Comput. 2011, 7, 444–455. [DOI] [PubMed] [Google Scholar]
- Fabian J. Electronic Excitation of Sulfur-Organic Compounds - Performance of Time-Dependent Density Functional Theory. Theor. Chem. Acc. 2001, 106, 199–217. [Google Scholar]
- Schreiber M.; Bub V.; Fülscher M. P. The Electronic Spectra of Symmetric Cyanine Dyes: A CASPT2 Study. Phys. Chem. Chem. Phys. 2001, 3, 3906–3912. [Google Scholar]
- Laurent A. D.; Jacquemin D. TD-DFT Benchmarks: A Review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar]
- Jacquemin D.; Perpète E. A.; Scalmani G.; Frisch M. J.; Kobayashi R.; Adamo C. An Assessment of the Efficiency of Long-Range Corrected Functionals for Some Properties of Large Compounds. J. Chem. Phys. 2007, 126, 144105. [DOI] [PubMed] [Google Scholar]
- Fabian J. TDDFT-Calculations of Vis/NIR Absorbing Compounds. Dyes Pigm. 2010, 84, 36–53. [Google Scholar]
- Dreuw A.; Head-Gordon M. Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: the Zincbacteriochlorin-Bacteriochlorin and Bacteriochlorophyll-Spheroidene Complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. [DOI] [PubMed] [Google Scholar]
- Champagne B.; Guillaume M.; Zutterman F. TDDFT Investigation of the Optical Properties of Cyanine Dyes. Chem. Phys. Lett. 2006, 425, 105–109. [Google Scholar]
- Jacquemin D.; Zhao Y.; Valero R.; Adamo C.; Ciofini I.; Truhlar D. G. Verdict: Time-Dependent Density Functional Theory “Not Guilty” of Large Errors for Cyanines. J. Chem. Theory Comput. 2012, 8, 1255–1259. [DOI] [PubMed] [Google Scholar]
- Moore B.; Autschbach J. Longest-Wavelength Electronic Excitations of Linear Cyanines: The Role of Electron Delocalization and of Approximations in Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2013, 9, 4991–5003. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Neese F. Double-Hybrid Density Functional Theory for Excited Electronic States of Molecules. J. Chem. Phys. 2007, 127, 154116. [DOI] [PubMed] [Google Scholar]
- Filatov M.; Huix-Rotllant M. Assessment of Density Functional Theory Based Δ SCF and Linear Response Methods for Longest Wavelength Excited States of Extended π-Conjugated Molecular Systems. J. Chem. Phys. 2014, 141, 024112. [DOI] [PubMed] [Google Scholar]
- Zhekova H.; Krykunov M.; Autschbach J.; Ziegler T. Applications of Time Dependent and Time Independent Density Functional Theory to the First π to π* Transition in Cyanine Dyes. J. Chem. Theory Comput. 2014, 10, 3299–3307. [DOI] [PubMed] [Google Scholar]
- Masunov A. E. Theoretical Spectroscopy of Carbocyanine Dyes Made Accurate by Frozen Density Correction to Excitation Energies Obtained by TD-DFT. Int. J. Quantum Chem. 2010, 110, 3095–3100. [Google Scholar]
- Boulanger P.; Jacquemin D.; Duchemin I.; Blase X. Fast and Accurate Electronic Excitations in Cyanines with the Many-Body Bethe-Salpeter Approach. J. Chem. Theory Comput. 2014, 10, 1212–1218. [DOI] [PubMed] [Google Scholar]
- Meguellati K.; Ladame S.; Spichty M. A Conceptually Improved TD-DFT Approach for Predicting the Maximum Absorption Wavelength of Cyanine Dyes. Dyes Pigm. 2011, 90, 114–118. [Google Scholar]
- Guthmuller J.; Zutterman F.; Champagne B. Prediction of Vibronic Coupling and Absorption Spectra of Dimers from Time-Dependent Density Functional Theory: The Case of a Stacked Streptocyanine. J. Chem. Theory Comput. 2008, 4, 2094–2100. [DOI] [PubMed] [Google Scholar]
- Cakmak Y.; Kolemen S.; Duman S.; Dede Y.; Dolen Y.; Kilic B.; Kostereli Z.; Yildirim L. T.; Dogan A. L.; Guc D.; Akkaya E. U. Designing Excited States: Theory-Guided Access to Efficient Photosensitizers for Photodynamic Action. Angew. Chem., Int. Ed. 2011, 50, 11937–11941. [DOI] [PubMed] [Google Scholar]
- Briggs E. A.; Besley N. A.; Robinson D. QM/MM Excited State Molecular Dynamics and Fluorescence Spectroscopy of BODIPY. J. Phys. Chem. A 2013, 117, 2644–2650. [DOI] [PubMed] [Google Scholar]
- Valiev R.; Sinelnikov A.; Aksenova Y.; Kuznetsova R.; Berezin M.; Semeikin A.; Cherepanov V. The Computational and Experimental Investigations of Photophysical and Spectroscopic Properties of BF2 Dipyrromethene Complexes. Spectrochim. Acta A 2014, 117, 323–329. [DOI] [PubMed] [Google Scholar]
- Quartarolo A. D.; Russo N.; Sicilia E. Structures and Electronic Absorption Spectra of a Recently Synthesised Class of Photodynamic Therapy Agents. Chem.—Eur. J. 2006, 12, 6797–6803. [DOI] [PubMed] [Google Scholar]
- Le Guennic B.; Maury O.; Jacquemin D. Aza-Boron-Dipyrromethene Dyes: TD-DFT Benchmarks, Spectral Analysis and Design of Original Near-IR Structures. Phys. Chem. Chem. Phys. 2012, 14, 157–164. [DOI] [PubMed] [Google Scholar]
- Lazarides T.; McCormick T. M.; Wilson K. C.; Lee S.; McCamant D. W.; Eisenberg R. Sensitizing the Sensitizer: The Synthesis and Photophysical Study of Bodipy-Pt(II)(diimine)(dithiolate) Conjugates. J. Am. Chem. Soc. 2011, 133, 350–364. [DOI] [PubMed] [Google Scholar]
- Awuah S. G.; Polreis J.; Biradar V.; You Y. Singlet Oxygen Generation by Novel NIR BODIPY Dyes. Org. Lett. 2011, 13, 3884–3887. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yu H.; Xiao Y. Replacing Phenyl Ring with Thiophene: An Approach to Longer Wavelength Aza-dipyrromethene Boron Difluoride (Aza-BODIPY) Dyes. J. Org. Chem. 2012, 77, 669–673. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhao J.; Guo H.; Xiz L. Geometry Relaxation-Induced Large Stokes Shift in Red-Emitting Borondipyrromethenes (BODIPY) and Applications in Fluorescent Thiol Probes. J. Org. Chem. 2012, 77, 2192–2206. [DOI] [PubMed] [Google Scholar]
- Chibani S.; Le Guennic B.; Charaf-Eddin A.; Maury O.; Andraud C.; Jacquemin D. On the Computation of Adaiabtic Energies in Aza-Boron-Dipyrromethene Dyes. J. Chem. Theory Comput. 2012, 8, 3303–3313. [DOI] [PubMed] [Google Scholar]
- Chibani S.; Le Guennic B.; Charaf-Eddin A.; Laurent A. D.; Jacquemin D. Revisiting the Optical Signatures of BODIPY with Ab Initio Tools. Chem. Sci. 2013, 4, 1950–1963. [Google Scholar]
- Zakrzewska A.; Zalesny R.; Kolehmainen E.; Osmialowski B.; Jedrzejewska B.; Agren H.; Pietrzak M. Substituent Effects on the Photophysical Properties of Fluorescent 2-Benzoylmethylenequinoline Difluoroboranes: A Combined Experimental and Quantum Chemical Study. Dyes Pigm. 2013, 99, 957–965. [Google Scholar]
- Charaf-Eddin A.; Le Guennic B.; Jacquemin D. Excited-States of BODIPY-Cyanines: Ultimate TD-DFT Challenges?. RSC Adv. 2014, 4, 49449–49456. [Google Scholar]
- Chibani S.; Laurent A. D.; Le Guennic B.; Jacquemin D. Improving the Accuracy of Excited State Simulations of BODIPY and aza-BODIPY Dyes with a Joint SOS-CIS(D) and TD-DFT Approach. J. Chem. Theory Comput. 2014, 10, 4574–4582. [DOI] [PubMed] [Google Scholar]
- Jacquemin D.; Chibani S.; Le Guennic B.; Mennucci B. Solvent Effects on Cyanine Derivatives: A PCM Investigation. J. Phys. Chem. A 2014, 118, 5343–5348. [DOI] [PubMed] [Google Scholar]
- Boens N.; Leen V.; Dehaen W.; Wang L.; Robeyns K.; Qin W.; Tang X.; Beljonne D.; Tonnele C.; Paredes J. M.; Ruedas-Rama M. J.; Orte A.; Crovetto L.; Talavera E. M.; Alvarez-Pez J. M. Visible Absorption and Fluorescence Spectroscopy of Conformationally Constrained, Annulated BODIPY Dyes. J. Phys. Chem. A 2012, 116, 9621–9631. [DOI] [PubMed] [Google Scholar]
- Boulanger P.; Chibani S.; Le Guennic B.; Duchemin I.; Blase X.; Jacquemin D. Combining the Bethe-Salpeter Formalism with Time-Dependent DFT Excited-State Forces to Describe Optical Signatures: NBO Fluoroborates as Working Examples. J. Chem. Theory Comput. 2014, 10, 4548–4556. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. Assessment of TD-DFT Methods and of Various Spin Scaled CISnD and CC2 Versions for the Treatment of Low-Lying Valence Excitations of Large Organic Dyes. J. Chem. Phys. 2010, 132, 184103. [Google Scholar]
- Winter N. O. C.; Graf N. K.; Leutwyler S.; Hattig C. Benchmarks for 0–0 Transitions of Aromatic Organic Molecules: DFT/B3LYP, ADC(2), CC2, SOS-CC2 and SCS-CC2 Compared to High-Resolution Gas-Phase Data. Phys. Chem. Chem. Phys. 2013, 15, 6623–6630. [DOI] [PubMed] [Google Scholar]
- Schmitt A.; Hinkeldey B.; Wild M.; Jung G. Synthesis of the Core Compound of the Bodipy Dye Class: 4,4’-Bora-(3a,4a)-Diaza-s-Indacene. J. Fluoresc. 2009, 19, 755–758. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Xiao Y.; Li D.; Fu M.; Qian X. Novel Fluorescent Fluorine–Boron Complexes: Synthesis, Crystal Structure, Photoluminescence, and Electrochemistry Properties. J. Org. Chem. 2008, 73, 1571–1574. [DOI] [PubMed] [Google Scholar]
- Nüesch F.; Moser J. E.; Shklover V.; Grätzel M. Merocyanine Aggregation in Mesoporous Networks. J. Am. Chem. Soc. 1996, 118, 5420–5431. [Google Scholar]
- Majumdar P.; Yuan X.; Li S.; Le Guennic B.; Ma J.; Zhang C.; Jacquemin D.; Zhao J. Cyclometalated Ir(III) Complexes with Styryl-BODIPY Ligands Showing Near IR Absorption/Emission: Preparation, Study of Photophysical Properties and Application As Photodynamic/Luminescence Imaging Materials. J. Mater. Chem. B 2014, 2, 2838–2854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.