

Abstract
Computational protein design requires methods to accurately estimate free energy changes in protein stability or binding upon an amino acid mutation. From the different approaches available, molecular dynamics-based alchemical free energy calculations are unique in their accuracy and solid theoretical basis. The challenge in using these methods lies in the need to generate hybrid structures and topologies representing two physical states of a system. A custom made hybrid topology may prove useful for a particular mutation of interest, however, a high throughput mutation analysis calls for a more general approach. In this work, we present an automated procedure to generate hybrid structures and topologies for the amino acid mutations in all commonly used force fields. The described software is compatible with the Gromacs simulation package. The mutation libraries are readily supported for five force fields, namely Amber99SB, Amber99SB*-ILDN, OPLS-AA/L, Charmm22*, and Charmm36.
Keywords: free energy calculations, molecular dynamics, alchemy, thermostability, mutations
Introduction
In silico protein design has received a significant attention from the computational structural biology community in the last decade.1,2 One of the major goals in the field is the accurate estimation of the change in protein stability upon an amino acid mutation. A related challenge is predicting protein–protein or protein–DNA/RNA binding specificity subject to an amino acid or a nucleotide mutation. Computational approaches to address these questions differ in their predictive accuracy and computational cost.
The methods can be classified into two main branches.3 Statistical approaches build regression models trained on experimental free energy data,4,5 whereas simulation-based methods utilize energy functions which guide sampling in the protein conformational space. The computationally least expensive simulation methods rely on the statistically or empirically derived energy functions. These approaches are usually limited to sampling the side-chain rotamers, for example, FoldX,6 Robetta,7 but may also allow restrained backbone motions, for example, ddg_monomer protocol8 using the Rosetta potential.9 Another often used free energy calculation technique uses molecular dynamics (MD) or a distance constraint-based sampling10 combined with the solution of the Poisson–Boltzmann (or Generalized Born) equation.11–13
The computationally most expensive approaches rely on first principles. These methods are often termed alchemical, as they exploit unphysical pathways along a thermodynamic cycle, that is, atom morphing, creation, and annihilation. The methods use molecular mechanics force fields as an energy function; sampling of the correct thermodynamic ensemble is ensured by the thermostatted and barostatted dynamics. The strength of the alchemical approaches in capturing changes in the protein thermostability upon an amino acid mutation was demonstrated by Seeliger and de Groot.14 In the study, a set of 109 barnase mutations was analyzed and a remarkable agreement (correlation of 0.86) with experimentally obtained free energy values was observed. A similar setup was used to calculate change in the binding free energy of a DNA and a zinc-finger transcription factor on a DNA nucleotide mutation.15 Recently, MD-based alchemical free energy calculations have been successfully applied to predict stabilizing mutations in unbound ubiquitin, as well as in a complex with binding partners.16
Although being highly accurate and theoretically sound, the alchemical calculations also pose a number of technical challenges. As already mentioned, the computational cost of these methods is higher in comparison to the statistical and (semi)empirical approaches. Second, the alchemical methods utilize hybrid topologies of the molecules, the generation of which requires specialized software or tedious and error-prone manual topology generation.
In the current work, we present a method to automatically build hybrid protein structures and topologies for alchemical free energy calculations. The method is compatible with the force field organization and topology representation of Gromacs 4.517 and higher versions. Hybrid topology generation relies on the force field specific pregenerated mutation libraries. Currently, we demonstrate successful incorporation of five force fields, namely Amber99SB,18 Amber99SB*-ILDN,19,20 OPLS-AA/L,21,22 Charmm22*,23 and Charmm36.24 The provided toolkit enables generation of the mutation libraries in other force fields as well.
Methodology
Overall workflow
The workflow of the pmx-based hybrid structure and topology generation is summarized in Figure 1. The procedure starts with the hybrid structure generation (mutate.py): after providing an initial protein structure, the user may select one or more mutations to perform. As a result, a structure file is created containing a hybrid residue with the atoms of both, the wild type and mutated amino acids. The atom mappings for the hybrid structures are extracted from the pregenerated mutation libraries. The second step in the workflow requires the generation of a hybrid topology file. The mutation libraries contain descriptions of the hybrid amino acids in the format that is compatible with the standard Gromacs tool pdb2gmx. The topology generated at this stage does not yet have the force field parameters for the physical A and B states defined. In the third step, the parameters are added by means of the tool generate_hybrid_topology.py. The tool utilizes information from the mutation library and extracts the required bonded parameters from the force field files.
Figure 1.
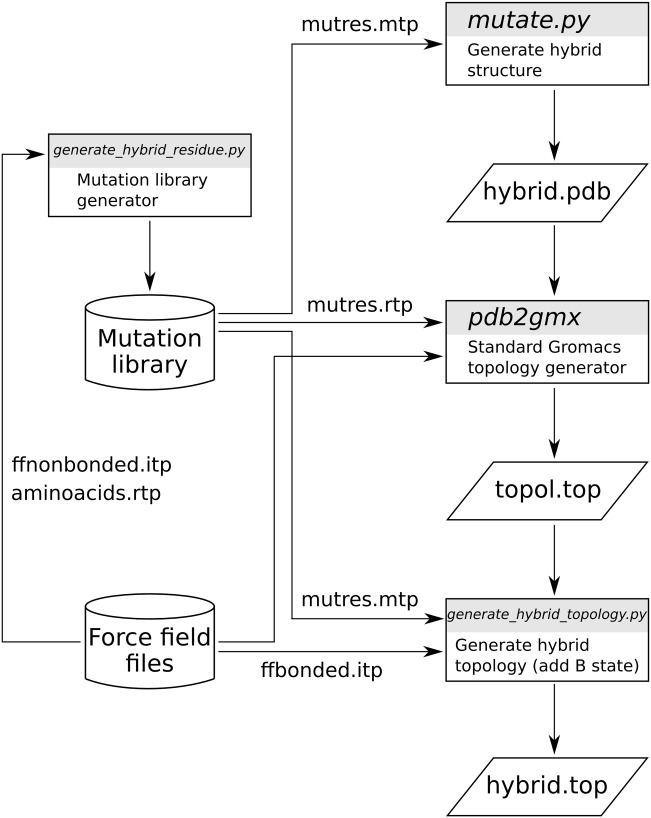
An overall workflow of the pmx hybrid topology and structure generation. The force field specific mutation libraries are created by generate_hybrid_residue.py using the amino acid topologies, bonded, and nonbonded parameters as defined in the force field of interest. By default pmx provides mutation libraries for five commonly used force fields. A hybrid mutated structure is generated by mutate.py. The Gromacs standard tool pdb2gmx uses the hybrid structure to create its topology, which subsequently is processed by generate_hybrid_topology.py by adding the required parameters for the A and B states.
The aforementioned steps require a force field specific pregenerated mutation library. Although we have generated libraries for five commonly used force fields, we also provide the tool (generate_hybrid_residue.py) to incorporate new force fields. To provide a deeper insight into the pmx-workflow, we continue with the description of the mutation library generator and follow with an outline of the structure and topology construction.
Mutation library
To generate an entry in a mutation library, generate_hybrid_residue.py requires as input two structures of the amino acids, force field nonbonded parameter and amino acid topology files. The structures need to be superimposed on the backbone atoms. After reading in the residue structures and topologies into the pmx data structures, the tool aligns the side chains by setting them to the same rotameric states. The actual atom mapping between the residues follows one of two routes: pairing predefined atoms or mapping as many atoms as possible in the corresponding branches of the residues being morphed.
The predefined atom pairing is used for residues containing aromatic rings: to avoid breaking the rings, these residues are morphed into one another considering backbone and the atoms up to Cγ (including Cγ). For the highly similar aromatic residues, the ring atoms are mapped to be morphed as well, for example, phenylalanine to tyrosine and different histidine protonation forms among each other. If an aromatic residue is morphed into a nonaromatic amino acid, the atoms up to Cβ are mapped. For glycine mutations, only the backbone atoms are mapped. Atom mapping for the rest of the residues is performed by, first, pairing atoms up to Cβ. Second, the branches of the side chains are traversed and atom morphs are identified by a distance criterion, that is, atoms separated by less than 0.55 Å are matched. The latter strategy works due to the previous superposition of the backbone atoms and side-chain alignment. For the atoms that are not paired to other atoms dummies are assigned. The 0.55 Å distance criterion prevents matching distant atoms, but also allows taking into consideration minor superpositioning inaccuracies.
To generate the full mutation library, the generate_hybrid_residue.py procedure is performed for every amino acid pair. The collected output is a complete mutation database (mutres.mtp), which contains information required by pmx in the following steps. The database describes atom mappings, coordinates of the hybrid residues, rules to morph the dihedrals that are found in the aminoacids.rtp file and rotatable side-chain bonds with the according atom dependencies. Atom masses are extracted from the atomtypes.atp database. The mutres.rtp file contains entries of the hybrid residue topologies that are compatible with the input expected by the pdb2gmx tool.
For the Charmm family of force fields, the grid-based energy correction map (CMAP)25 correction entries are included in the mutres.rtp file. However, the current Gromacs implementation17 does not include the CMAP's contribution into the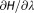 during an alchemical transition. Nevertheless, alchemical transitions with the enabled CMAP correction are correct for all the amino acids, except glycine and proline, because the
during an alchemical transition. Nevertheless, alchemical transitions with the enabled CMAP correction are correct for all the amino acids, except glycine and proline, because the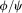 dihedral angle correction grids are identical for all the residues, apart from Gly and Pro. The summary of the generated mutation libraries is provided in the Table1. Proline mutations are not supported in any of the force fields, as such a mutation would require bond breaking.
dihedral angle correction grids are identical for all the residues, apart from Gly and Pro. The summary of the generated mutation libraries is provided in the Table1. Proline mutations are not supported in any of the force fields, as such a mutation would require bond breaking.
Table 1.
Summary of the mutation libraries and available mutations for the force fields included in pmx.
| Force field | FF name in pmx | Mutations without CMAP | Mutations with CMAP |
|---|---|---|---|
| Amber99SB | amber99sbmut | All except Pro | – |
| Amber99SB*-ILDN | amber99sb-star-ildn-mut | All except Pro | – |
| OPLS-AA/L | oplsaamut | All except Pro | – |
| Charmm22* | charmm22starmut | All except Pro | All except Pro and Gly |
| Charmm36 | charmm36mut | All except Pro | All except Pro and Gly |
Hybrid structure
Hybrid structures are created by the tool mutate.py, which requires as input the structure of a protein and the name of the force field to be used. The path to the mutation libraries is set by the Gromacs environmental variable GMXLIB. The residues to be mutated can be selected interactively after executing mutate.py or via an external text file.
Once the mutation is defined, the corresponding hybrid residue entry is extracted from the mutres.mtp database. As the coordinates for the hybrid residues are already pregenerated, the selected hybrid amino acid from the mutation library is superimposed onto the protein wild type residue based on the backbone and Cβ atoms (if glycine is involved only the backbone atoms are used). Subsequently, the side-chain of the hybrid residue is set to the same rotameric state as the wild type amino acid. Finally, the hybrid residue atoms corresponding to the wild type atoms are set to the wild type atom positions, thus ensuring completely unchanged geometry of the protein in its native state even with the hybrid residue included. The last step in the procedure may introduce a minor distortion in the B state, however, the correct geometry is restored in a few energy minimization steps prior to starting the simulations.
In case a terminal residue is to be mutated, it needs to be capped. Generation of hybrid residues for uncapped terminal residues is not currently supported by pmx.
Hybrid topology
The topology for the structure generated by the mutate.py can be created using the native Gromacs tool pdb2gmx. The tool utilizes the mutation library file mutres.rtp for a specific force field. At this stage, the generated topology contains no explicitly specified bonded parameters for either A or B state. It is the function of the generate_hybrid_topology.py program to extract the force field parameters and appropriately define them in the topology file for both physical states. In this way, generate_hybrid_topology.py mimics some functionality of the Gromacs preprocessor grompp.
The script reads in the mutation library data (mutres.mtp) and the bonded force field parameters (ffbonded.itp). First, the atom types, masses, and partial charges for the B state are assigned. By default, the dummy atom masses are set to 1.0 a.u., however, this feature may be disabled for a dummy to retain the mass of an atom of the corresponding physical state. Afterwards, the bond and angle parameters for the A and B states are specified. For every bond/angle in the topology, the atom type-based search in ffbonded.itp is performed and the parameters for the A and B states are extracted. If a dummy atom is present in one of the states, the bond/angle parameters of the other state are retained. The nonbonded parameters, bonds and angles follow a 1 to 1 mapping between the states A and B, that is, for one atom type, bond or angle in the state A there is only one atom/bond/angle in the state B. Therefore, these parameters can be directly morphed between the states.
For the dihedral angles, the 1 to 1 mapping may not necessarily hold, as Gromacs allows defining several periodic functions for one dihedral. Therefore, a general solution to the dihedral angle parameter morphing is to define seperate entries for switching the dihedral potential to zero and turning it back on when going from one physical state to another. In other words, for dihedrals, a transition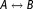 is split into A
is split into A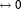 and
and . If a dummy atom is involved in a dihedral, the on/off switching is not required and direct morphing is used. The amino acid specific dihedrals and some impropers, in case of the OPLS-AA/L force field, that are explicitly predefined in the bonded parameter database, are always switched on/off independent of whether a dummy atom is forming the dihedral.
. If a dummy atom is involved in a dihedral, the on/off switching is not required and direct morphing is used. The amino acid specific dihedrals and some impropers, in case of the OPLS-AA/L force field, that are explicitly predefined in the bonded parameter database, are always switched on/off independent of whether a dummy atom is forming the dihedral.
For the Charmm force fields, the CMAP entries are not assigned a B-state, because the alchemical CMAP parameter morph is not supported by Gromacs. Instead the CMAP entry generated by pdb2gmx is retained in the final hybrid topology.
pmx data structures
Although the current work concentrates on the protein hybrid stucture and topology generation, application of the pmx package may be much broader. pmx comprises a number of classes and modules that can be utilized to set up MD simulations, manipulate structure, topology, atom index files, and build analysis tools. Although a more detailed description of the main classes and modules in the package can be found on the pmx website, here we illustrate the two essential data structures for handling macromolecular structures and topologies.
The full structure of a biomolecule is stored in an instance of the Model class, which contains the lists of chains, residues, and atoms (Fig. 2). An instance of the Chain class contains the lists of residues and atoms, while an instance of the Molecule class stores the list of atoms only. The description of the atoms, for example, name, id, coordinates so forth, is provided by the attributes of an Atom class instance.
Figure 2.
A schematic illustration of the two main pmx data structures used in the hybrid structure and topology generation. The top row describes the classes containing protein structure information, whereas the bottom rows describe the topological information. A detailed description can be found in the main text.
Objects of a molecular topology are created from the Topology class. The class inherits from the TopolBase lists with the topology information parsed from the contents of a Gromacs .itp or .top file. An instance of the TopolBase class also instantiates Molecule and Atom classes with the attributes read from the user provided topology file. A Topology class instance contains the lists of the bonded and nonbonded force field parameters that are parsed and stored in the attributes of the BondedParser and NBParser classes, respectively.
Installation
The latest pmx version can be downloaded from the git repository from http://code.google.com/p/pmx/. The installation is performed via the standard Python setup.py utility. pmx requires Python 2.6 or 2.7 and the NumPy library (http://www.numpy.org/). The environmental variable PYTHONPATH needs to point to the installation path, whereas GMXLIB has to be set to the mutation force field directory.
Validation
The validity of the hybrid structure and topology porting into different force fields was assessed by calculating the free energy changes over a thermodynamic cycle. Provided the correct implementation of the mutation libraries and the pmx tools, the calculated change of the free energy over a complete cycle must match the theoretical value of 0 kJ/mol.
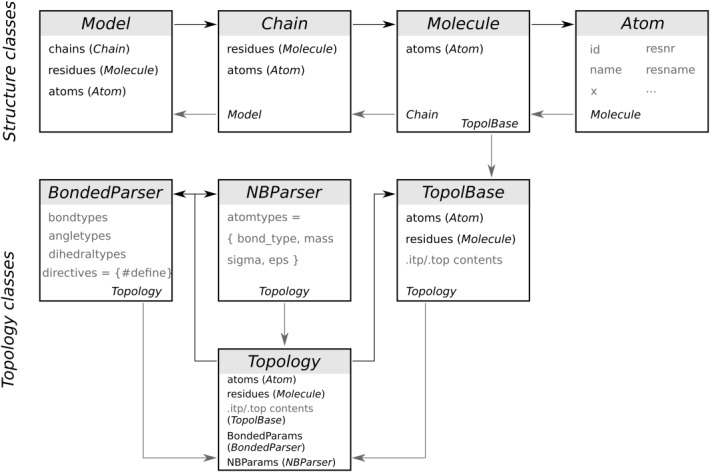
To construct the thermodynamic cycles, we utilized the double system in a single simulation box setup, as illustrated in Figure 3A. In the example, the system contains two capped tripeptides: Gly-V2F-Gly and Gly-F2V-Gly. The peptides were positioned ∼3 nm apart. Upon an alchemical transition individual peptides were morphed (Val to Phe and Phe to Val), as a result, the physical states A and B remained unchanged.
Figure 3.
Thermodynamic cycles used for validation. A) The double system in a single simulation box setup. Two tripeptides with the hybrid structures, V2F and F2V, are placed in the same box. During an alchemical transition, valine is morphed into phenylalanine and phenylalanine into valine. The overall state of the system remains unchanged, therefore, the calculated ΔΔG must be equal to 0 kJ/mol. B: results of the ΔΔG calculation over a closed thermodynamic cycle for nine amino acid mutation pairs for five force fields.
In total, nine mutation pairs were simulated (Fig. 3B). To calculate the free energy differences, we performed 10 independent equilibrium simulations for every mutation, 10 ns each, for the states A and B separately. Position restraints were applied on the hybrid residue Cα atoms throughout the course of the equilibrium runs to prevent peptide interaction. Concoord10 was used to generate starting peptide conformations. The system was solvated with TIP3P water.26 For the Charmm family of force fields the TIP3P water model with Van der Waals parameters on hydrogen atoms was used. The simulations were performed at 300 K temperature with the velocity rescaling thermostat.27 The pressure was kept at 1 bar by means of the Parrinello–Rahman barostat.28 Long range electrostatic interations were treated with the Particle Mesh Ewald approach.29 The simulation box was kept neutral by adding 0.15 M sodium and chloride ions. From every equilibrium trajectory, after discarding the first 2 ns, 100 fast nonequilibrium molecular dynamic runs were spawned morphing the system from one physical state to another in 50 ps. A soft-core potential30 was enabled for the alchemical transition simulations.
The free energy difference calculation was based on the Crooks Fluctuation Theorem31,32 using the maximum likelihood estimator.33 For every mutation, 10 free energy values were calculated (one for every 10 ns equilibrium simulation pair). The statistical error for each ΔΔG value was estimated using the bootstrapping approach. Subsequently, the error was added and subtracted from the estimated free energy, thus yielding two extreme ΔΔG values. The standard deviation over 20 such values for every mutation was reported as an overall uncertainty, effectively reflecting on both, the statistical error and the initial condition influence.
Results
A practical example
To give a more practical insight into the pmx-based hybrid structure and topology generation, we will provide an example for a mutation of valine into phenylalanine in a Gly-Val-Gly tripeptide in the Charmm22* force field.
Since pmx is designed to be compatible with Gromacs, it is convenient to start with a protein structure already preprocessed by Gromacs. Running pdb2gmx in the first place ensures the atom ordering and naming in accordance to the Gromacs and selected force field conventions. Note that we only need the structure file generated in this step, but not the topology:
pdb2gmx -f val.pdb -o val_pdb2gmx.pdb -ff charmm22starmut
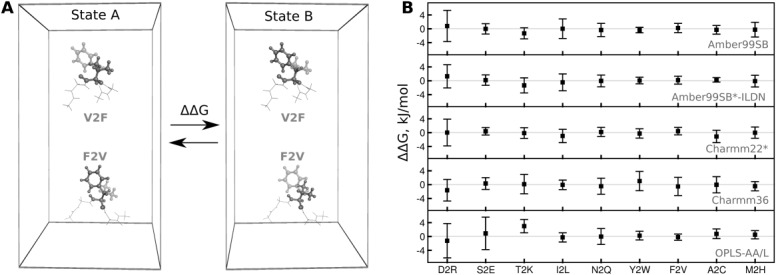
The mutation and hybrid structure generation is performed by the mutate.py program. The preprocessed structure of the tripeptide (Fig. 4A) is supplied as an input. When prompted, amino acid no. 2 (i.e., valine) is selected to be mutated into Phe:
python mutate.py -f val_pdb2gmx.pdb -o V2F.pdb -ff charmm22starmut
Figure 4.
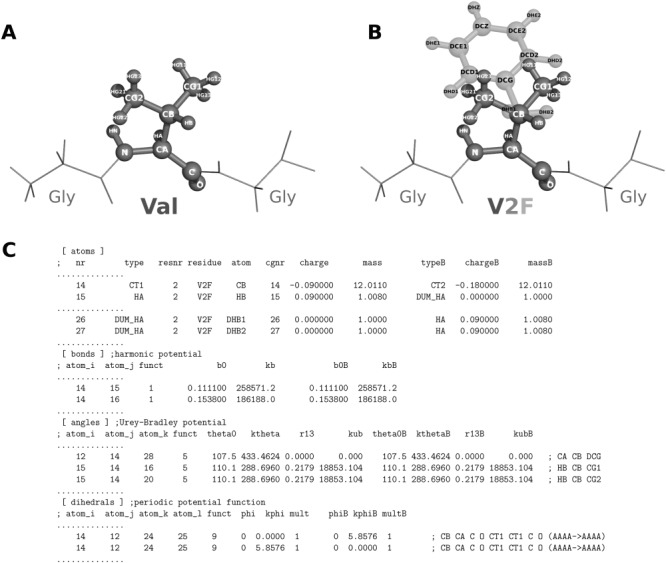
Illustration of the input/output structures and an output topology of the pmx scripts. A) A capped Gly-Val-Gly tripeptide as a starting structure for the mutation. B) Structure of the tripeptide with the hybrid V2F amino acid. Valine (dark gray) is in the physical state A, whereas phenylalanine (light gray) is in the state B. C) An excerpt from the hybrid V2F topology.
The generated hybrid residue contains atoms of both amino acids: valine and phenylalanine (Fig. 4B). The atoms that are not present in valine (state A), but exist in phenylalanine (state B) are marked as dummies. In the next step, the hybrid structure is supplied to pdb2gmx to generate the topology file:
pdb2gmx -f V2F.pdb -o V2F_pdb2gmx.pdb -p topol.top -ff charmm22starmut
The hybrid topology is created by the script generate_hybrid_topology.py:
python generate_hybrid_topology.py -p topol.top -o hybrid.top -ff charmm22starmut
The output of a successful topology generation should resemble the excerpts shown in Figure 4C. The given topology example illustrates that the Cβ atom of valine changes charge when morphed into phenylalanine and the Hβ atom becomes a dummy at the B state. The phenylalanines Hβ1 and Hβ2 are present in the state B, but become dummies in state A. Similarly, the parameters for bonds and angles are paired to be morphed between the states. The excerpt from the dihedral section illustrates that the dihedrals are switched off (force constant 0.0 kJ mol−1) and afterwards switched back on.
Validation thermodynamic cycles
Thermodynamic cycles were constructed to validate the pmx-based hybrid residue structure and topology generation (Fig. 3A). The estimated double free energy differences are provided in Figure 3B. The calculated ΔΔG values fall closely to the theoretical 0 kJ/mol mark for all the force fields. This result confirms the consistency of hybrid topology generation and proper mutation library implementation within the pmx framework.
The charge changing mutations (D2R, S2E, and T2K) exhibit larger fluctuations around the expected outcome value in comparison to the charge conserving mutations (I2L, N2Q, Y2W, F2V, A2C, and M2H). The ΔΔG value in case of the charge inversion involving aspartate to arginine mutation fluctuates the most.
Discussion
As illustrated by the closed thermodynamic cycle validation, pmx successfully generated hybrid structures and topologies for different force fields. This test also demonstrates that the structures and topologies are created consistently in the directions A2B and B2A, that is, A is described identically in both A2B and B2A topologies and the same holds for B. It is the internal consistency that allows cancellation of the dummy effects during the transitions in the single system in a double box setup, subsequently leading to a closed thermodynamic cycle.
The performed validation checks provide no information on the accuracy of the alchemical calculations in terms of agreement to the experimental measurements. The results from the closed thermodynamic cycles, however, give a rough estimate of a statistical uncertainty that could be expected for a certain type of mutation in a force field of interest. For example, charge change involving mutations exhibit larger fluctuations, which may partially be attributed to the finite-size artefacts related to the treatment of electrostatic interactions.34
In the validation step, glycine involving mutations were intentionally discarded due to the specifics of the glycine residue. During an alchemical transition from glycine, the side chain of a new amino acid may appear in a high energy region of the Ramachandran plot. In turn, this would yield poor convergence of the free energy calculation, which is not instructive for the current purpose of the method validation. Another glycine related issue is that in the current Gromacs implementation17 morphing of the CMAP correction is not possible. Therefore, for a glycine involving mutation in a Charmm family force field, CMAP needs to be switched off. Although glycine mutations are not presented in this study, they are included in the pmx mutation libraries. However, careful case-specific testing is advised for such mutations.
Proline involving mutations are not currently supported, since there are several difficulties concerning the proline alchemical morphing that would require additional investigation. First, the required bond creation/breakage would prevent using the constraints on all bonds. If constraints are not used, the creation of a bond may still appear to be slowly converging in terms of free energy. Another caveat lies in trapping proline in a rarely populated stereoisomeric form when creating a bond. An approximate solution to proline mutation could be leaving all the bonds intact and turning off the angles and dihedrals that couple the N and Cα atoms via the pyrrolidine ring. However, all these options require further investigation.
pmx enables force field specific mutation library generation via the utilities of generate_hybrid_residue.py. The tool was designed to be compatible with the Amber, Charmm, and OPLS force field families. Therefore, incorporation of a new related force field is straightforward. However, due to the specifics of every force field, additional modification to the software may be required for the addition of other force fields. For example, definitions of the bonded parameters in the GROMOS35 force field family within the Gromacs framework differs from the other force fields. Thus, creating a GROMOS port would naturally become more involved. Another useful application of the library generation framework would be the creation of a mutation database involving nonstandard amino acids. This would enable computational evaluation of the post-translational modification effects in terms of free energies. Although the generation of new mutation libraries is possible, it requires advanced pmx usage. For the hybrid structure and topology generation using the provided force fields (Table1), the user simply needs to follow the steps outlined in the practical example of the Results section.
The objectives for the future pmx development include the aforementioned nonstandard amino acid mutation library generation. Another direction involves the nucleic base mutation update to the new pmx framework from an earlier implementation.15 A more technical prospect is to ensure pmx compatibility with Python 3.0 and higher versions.
Conclusions
The pmx software enables automated hybrid amino acid structure and topology generation for all common biomolecular force fields. These allow high-quality large-scale automated alchemical free energy calculations due to amino acid mutations. Input/output of the program is compatible with the latest Gromacs versions. Utilities to include new mutation libraries for other force fields are included in the package. pmx can be downloaded from http://code.google.com/p/pmx/
References
- Pantazes R, Grisewood M, Maranas C. Curr. Opin. Struct. Biol. 2011;21:467. doi: 10.1016/j.sbi.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Fleishman S. Curr. Opin. Struct. Biol. 2013;23:903. doi: 10.1016/j.sbi.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Khan S, Vihinen M. Hum. Mutat. 2010;31:675. doi: 10.1002/humu.21242. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Casadio R. Nucl. Acids Res. 2005;33:W306. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Rossi I, Casadio R. BMC Bioinf. 2008;9:S6. doi: 10.1186/1471-2105-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerois R, Nielsen J, Serrano L. J. Mol. Biol. 2002;320:369. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Kortemme T, Baker D. Proc. Natl. Acad. Sci. USA. 2002;99:14116. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg E, Leaver-Fay A, Baker D. Proteins Struct. Funct. Bioinf. 2011;79:830. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Baker D. Annu. Rev. Biochem. 2008;77:363. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- De Groot B, Van Aalten D, Scheek R, Amadei A, Vriend G, Berendsen H. Proteins Struct. Funct. Bioinf. 1997;29:240. doi: 10.1002/(sici)1097-0134(199710)29:2<240::aid-prot11>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Srinivasan J, Cheatham T, Cieplak P, Kollman P, Case D. J. Am. Chem. Soc. 1998;120:9401. [Google Scholar]
- Kollman P, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W. Acc. Chem. Res. 2000;33:889. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Benedix A, Becker C, de Groot B, Caflisch A, Böckmann R. Nat. Methods. 2009;6:3. doi: 10.1038/nmeth0109-3. [DOI] [PubMed] [Google Scholar]
- Seeliger D, de Groot B. Biophys. J. 2010;98:2309. doi: 10.1016/j.bpj.2010.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger D, Buelens F, Goette M, de Groot B, Grubmüller H. Nucl. Acids Res. 2011;39:8281. doi: 10.1093/nar/gkr531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielssens S, Peters J, Ban D, Pratihar S, Seeliger D, Sharma M, Giller K, Sabo T, Becker S, Lee D, Griesinger C, de Groot B. Angew. Chem. Int. Ed. 2014;126:10535. doi: 10.1002/anie.201403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts M, Smith J, Kasson P, van der Spoel D, Hess B, Lindahl E. Bioinformatics. 2013;29:845. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins Struct. Funct. Bioinf. 2006;65:712. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Hummer G. J. Phys. Chem. B. 2009;113:9004. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Proteins Struct. Funct. Bioinf. 2010;78:1950. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W, Maxwell D, Tirado-Rives J. J. Am. Chem. Soc. 1996;118:11225. [Google Scholar]
- Kaminski G, Friesner R, Tirado-Rives J, Jorgensen W. J. Phys. Chem. B. 2001;105:6474. [Google Scholar]
- Piana S, Lindorff-Larsen K, Shaw D. Biophys. J. 2011;100:L47. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R, Zhu X, Shim J, Lopes P, Mittal J, Feig M, MacKerell A., Jr J. Chem. Theory Comput. 2012;8:3257. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A, Feig M, Brooks C. J. Comput. Chem. 2004;25:1400. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926. [Google Scholar]
- Bussi G, Donadio D, Parrinello M. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M, Rahman A. J. Appl. Phys. 1981;52:7182. [Google Scholar]
- Essmann U, Perera L, Berkowitz M, Darden T, Lee H, Pedersen L. J. Chem. Phys. 1995;103:8577. [Google Scholar]
- Gapsys V, Seeliger D, de Groot B. J. Chem. Theory Comput. 2012;8:2373. doi: 10.1021/ct300220p. [DOI] [PubMed] [Google Scholar]
- Crooks G. J. Stat. Phys. 1998;90:1481. [Google Scholar]
- Crooks G. Phys. Rev. E. 1999;60:2721. doi: 10.1103/physreve.60.2721. [DOI] [PubMed] [Google Scholar]
- Shirts M, Bair E, Hooker G, Pande V. Phys. Rev. Lett. 2003;91:140601. doi: 10.1103/PhysRevLett.91.140601. [DOI] [PubMed] [Google Scholar]
- Rocklin G, Mobley D, Dill K, Hünenberger P. J. Chem. Phys. 2013;139:184103. doi: 10.1063/1.4826261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, Scott WRP, Tironi IG. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Zürich, Switzerland: Hochschulverlag AG an der ETH Zürich; 1996. [Google Scholar]