

Abstract
A 1-year-old male Indian rhesus macaque presented with a bilateral blindness. Ocular examination, gross and histopathological evaluation, and immunohistochemistry were performed. The major findings were retinal telangiectasia, accumulation of exudate in the intraretinal and subretinal space, and retinal detachment. Coat-like retinopathy was diagnosed, and it has not been previously reported in veterinary medicine.
Keywords: Coat's disease, retinal telangiectasis and exudate, non-human primate
Introduction
A normally born, 1-year-old male Indian rhesus macaque (macaca mulatta) presented with a bilateral blindness. Upon ocular examination, the intraocular pressure was 11 mm Hg in the right eye and 20 mm Hg in the left eye. The left eye showed phthisis bulbi with aqueous flare and retinal detachment. The right eye showed buphthalmos with severe miosis. Fluorescein staining was negative in both eyes. Routine serology showed that the animal had serum antibody titers of 1:640 to West Nile virus (WNV) by hemagglutination-inhibition assay. A decision to humanely euthanasia the animal was made due to the lack of responsiveness to antibiotic treatment and deteriorating physical condition. This animal received prior approval from the institutional animal care and use committee (IACUC) of the TNPRC in Covington, LA. This study was conducted within the guidelines for ethical use of animals in United States Public Health Service policy as outlined in the Guide for the Care and Use of Laboratory Animals.
Grossly the animal had poor body condition with little body fat and moderately skeletal muscle atrophy. The left eye was small with a cloudy anterior chamber, corneal opacity, and a lens cataract. (Fig. 1) The vitreous fluid was yellow-brown containing the detached, coiled retina. The right eye was mildly enlarged with similar, but milder lesions to those noted in the left eye. All other organs were grossly unremarkable.
Fig. 1.
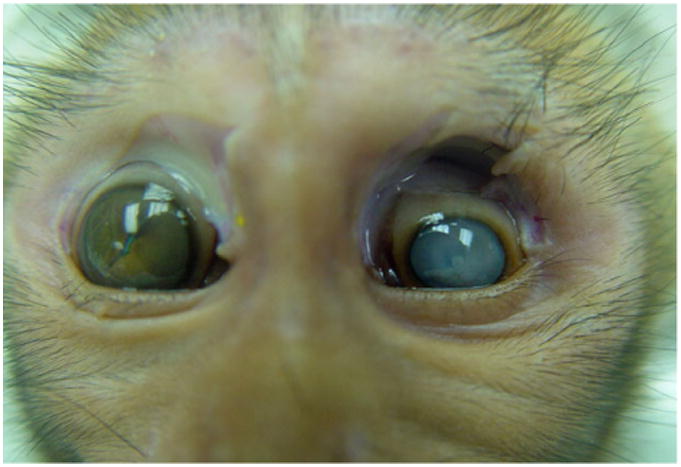
One-year-old male Indian rhesus macaque (macaca mulatta). Eyes. Shrunken left eye with cloudy anterior chamber, corneal opacity, and a lens cataract. The right eye was characterized by buphthalmos with a cataract and an irregular iris.
Histopathologically, the irregular, coiled retina of the left eye was completely detached from the retinal pigmented epithelia. The subretinal space was filled with an exudate composed of fibrin, hemosiderin-laden macrophages, and rare cholesterol clefts surrounded by multinucleate giant cells. (Fig. 2) Multifocally, the inner plexiform layer was expanded by edema, fibrin, and numerous vacuoles. The capillaries and mid-sized vessels were irregular, moderately to severely dilated, and lined by a thin layer of endothelial cells (telangiectasia). These blood vessels often had moderate to severe perivascular edema and/or mild lymphoplasmacytic perivasculitis. (Fig. 3) Some of the retinal vessels showed considerably endothelial fenestration and interendothelial cell separation. Marked telangiectasia with the loss of basement membrane integrity produced the abnormality of vascular permeability and formation of saccular dilation to microaneurysms. (Fig. 4) Occasionally, lymphoplasmacytic vasculitis was also noted. The cells with vacuolar degeneration (lipid-laden macrophages) in the retina and brown pigmentation (hemosiderin-laden macrophages) in the subretinal space were positive for the macrophage marker CD68 by immunohistochemistry (Fig. 5). Other lesions in the left eye included multifocal keratitis, cataract, lens rupture, broad anterior and posterior synechia, and anterior uveitis. Similar, but milder, lesions were noted in the right eye. All other organs were histologically unremarkable.
Fig. 2.
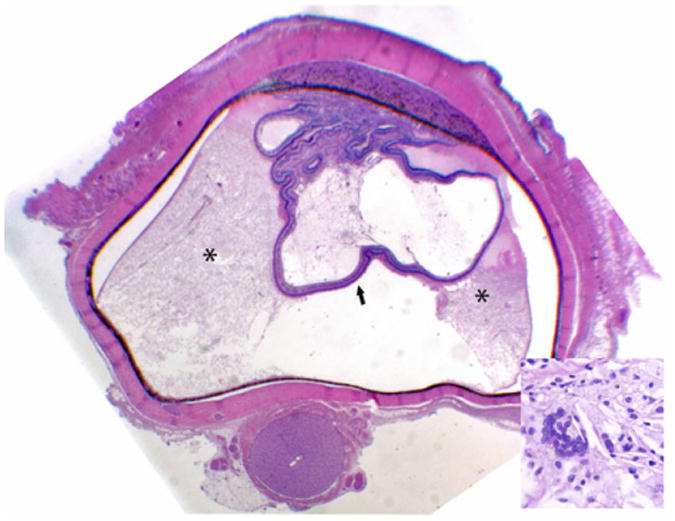
Subgross photography of the left eye. The retina is completely detached (arrows) and the subretinal space is filled with an exudate (*). Anterior and posterior synechia, and keratitis are also present. Insert. Cholesterol clefts with multinucleate giant cells in the subretinal exudate. HE. 400×.
Fig. 3.
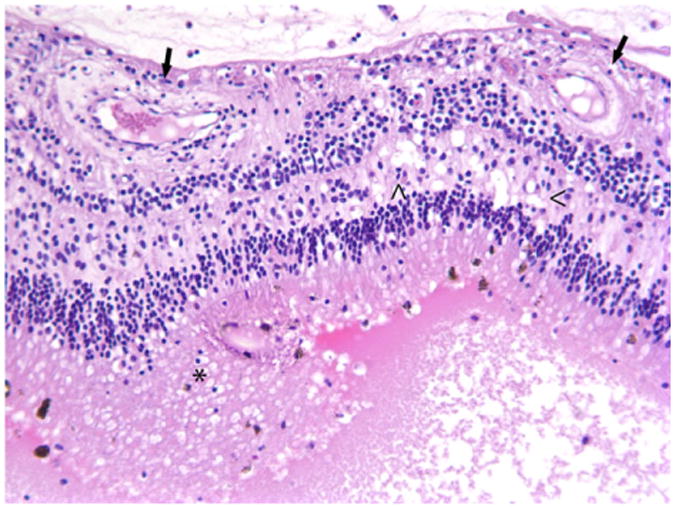
Histology of the retina of the left eye. Retinal blood vessels (arrows) were dilated with perivascular edema. Vacuolization predominantly in the inner plexiform layer (<) of the retina was observed, and the subretinal space was filled with large an eosinophilic exudate containing many lipid and hemosiderin-laden macrophages (*). HE. 200×.
Fig. 4.
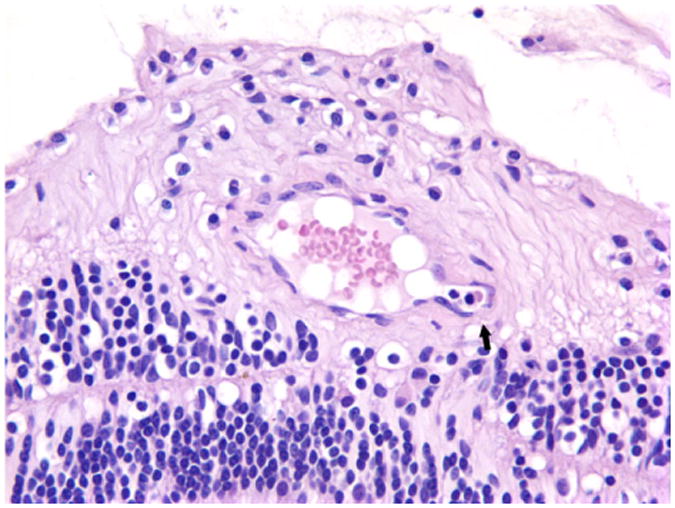
Histology of the retina of the left eye. The retinal vessel showed greatly endothelial fenestration and interendothelial cell separation with the loss of basement membrane integrity and formation of microaneurysm (arrow). HE. 400×.
Fig. 5.
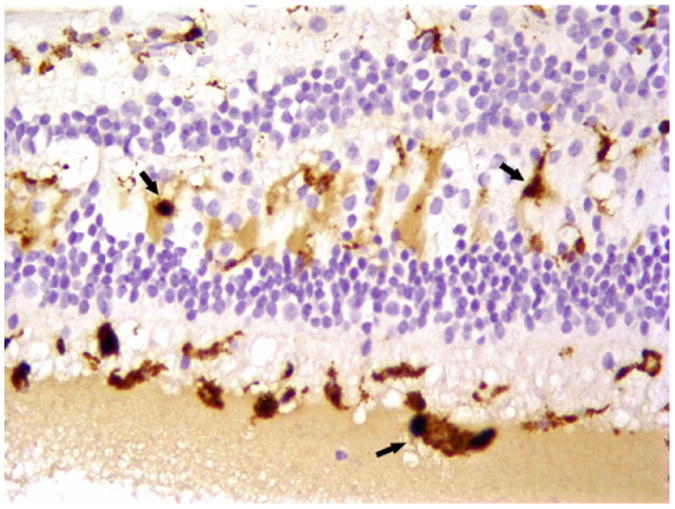
Immunohistochemistry of the left eye. Immunohistochemistry revealed that the vacuolated cells observed in the H&E of the retina were CD68-positive macrophages (arrows). Macrophages were also found in the infiltrates in the subretinal space. Magnification 200×.
First described by Gorge Coats in 1908, Coats disease is an idiopathic congenital ocular disease mainly characterized by retinal telangiectasia and exudative retinopathy. Coats disease occurs more commonly in children and has a clear male predominance and usually unilateral manifestation [17]. However, bilateral Coats-like disease has also been reported in young and adult patients [1, 14]. The most common clinical signs of Coats disease are decreased visual acuity, strabismus, and leukocoria. The clinical signs can be very variable ranging from barely visible vascular changes to extensive telangiectasias and exudation. The subretina and intraretina often contains yellow lipid-rich exudates with retinal vascular abnormalities. The exudation can lead to a visible, partially, or totally serous retinal detachment. Fluorescein angiography usually can demonstrate retinal telangiectasia, aneurysms, venous beading, and avascularity [19].
Coats disease has been classified into five stages from retinal telangiectasia only in stage 1 to the advanced end-stage disease in stage 5 in humans. [18] Based on the clinical and histopathological findings, the Coats-like retinopathy in the current case was comparable to the condition of stage 5 disease, which is clinically blind and non-painful eye with a total retinal detachment, cataract, and phthisis bulbi. [18] The hallmark pathological lesions in this animal were retinal telangiectasia, retinal detachment, and exudation in the intraretinal and subretinal space. The exudate contained a large amount of thick yellow lipoproteinaceous fluid admixing with lipid-laden macrophages and cholesterol clefts. Theses lesions have been described in nearly all human patients [17]. In humans with Coats disease, leakage of lipoproteinaceous fluid from telangiectasic retinal vessels was slowly progressive with increasing exudative retinal detachment [11, 17]. Excessive leakage into adjacent retina causes recognizable intraretinal and subretinal cholesterol exudates, hemorrhage, edema, lymphocytic infiltration, and deposition of lipid and fibrin. These exudates induce retinal degeneration and infiltration of phagocytic, lipid-laden ‘ghost’ cells [12]. Our immunohistochemistry of CD68 revealed that these inflammatory cells within the retina and subretinal space were macrophages, which was consistent with the results in human Coats disease [6, 9].
The etiology of Coats disease remains unknown. Coats disease, especially bilateral presentations, has been observed in patients with systemic disorders or hereditary diseases such as Alport syndrome, Turner's syndrome, and facioscapulohumeral muscular dystrophy (FSHD) [12, 14]. Facioscapulohumeral dystrophy has been associated with bilateral advanced Coats disease [7, 14, 20]. However, only 0.7% to 1.7% of patients with Coats disease have FSHD [20]. FSHD is an autosomal dominant myopathy caused by a deletion of D4Z4 macrosatellite repeats in the subtelomeric region of chromosome 4q35. The genetic testing of FSHD in the current case was unsuccessful, due to that the extraction of required size (>10 kbp) DNA fragments for PCR and sequence was failed from the available paraffin-embedded tissues in our laboratory. While the animal had a significant titer against West Nile virus (WNV), the ocular lesions were not consistent with WNV infection. Furthermore, immunohistochemistry for WNV and other flaviviruses was negative on eyes, brain, and spinal cord as was RT-PCR for WNV performed on vitreous fluid (data not shown).
It is very important for clinicians to differentiate bilateral Coats disease from other abnormalities that have quite similar exudative retinopathy. Retinoblastoma (RB) is one of the most important conditions that are often confused clinically with Coats disease. [16] The distinction between these two diseases is especially difficult when the tumor diffusely spreads or is exophytic, which consists of tumor cells growing under the neuro-sensory retina, resulting in a non-rhegmatogenous retinal detachment. [15] However, histopathological differences are evident. Not only are neoplastic cells present in RB, but also the vascular changes in two diseases are usually different. Like observed in the current case, Coats disease often has an irregular caliber dilation with small aneurysmal or saccular capillaries. In contrast, the blood vessels in RB are more uniformly dilated and associated with a retinal mass. In Coats disease, the subretinal fluid is yellow or yellow-green and often contains glistening cholesterol crystals, but the subretinal material in exophytic retinoblastoma appears as white or gray indolent material, often forming clumps of tumor cells. [19] Retinitis pigmentosa (RP) is an inherited retinal degeneration characterized by abnormalities of the photoreceptors (rods and cones) or the retinal pigment epithelium of the retina. The earliest observed changes in RP are arteriolar narrowing, fine dust-like intraretinal pigmentation, and loss of pigment from the pigment epithelium. [4] As photoreceptor deterioration progresses, severe retinal vessel attenuation and waxy pallor of the optic nerve become apparent in individuals with advanced RP. Rare patients with advanced RP can have exudative vasculopathy, often called Coats-like disease. [8, 10] Retinal exudation is associated with many other pediatric conditions including juvenile diabetes [13] and laser therapy [3]. Diabetic retinopathy is an ocular manifestation affecting up to 80% of patients with juvenile diabetes. The retinal exudation is due to the hyperglycemia-induced intramural pericyte death and retinal blood vessels becoming more permeable. [5] The current case had no signs of juvenile diabetes. The levels of blood glucose in the current case were within the normal range (77 and 81 mg/dl). Immunohistochemistry demonstrated a strong expression of insulin in the islets of Langerhans in this animal (data not shown). This animal, also, had no history of laser therapy. Rare cases of bilateral Coats retinopathy have been reported in a 2-year girl with severe bone marrow hypoplasia [10] and Reiter's disease [2]. However, complete blood counts, histology, and laboratory tests suggested that the current cases were not consistent with RB, RP, juvenile diabetes, bone marrow hypoplasia, and Reiter's disease.
The diagnosis of the current case is challenging because of a lack of comparative clinical and histopathological characteristics in the published literature in veterinary medicine. The hallmark lesions in this animal were retinal telangiectasia, retinal detachment, and exudation in the intraretinal and subretinal space, which are consistent with Coats-like disease in humans.
Acknowledgments
The authors thank Dr. Hans E. Grossniklaus in the Section of Ocular Oncology and Pathology, Emory Eye Center, Atlanta, GA, for consultation, Dr. Robert B. Tesh at the University of Texas for the flaviviral hemagglutination-inhibition assay, and Dr. Yugendar Bommineni at the Veterinary Laboratory of New Mexico State University for the flaviviral RT-PCR. This work was supported by the National Center for Research Resources and the Office of Research Infrastructure Programs (ORIP) of the National Institutes of Health through Grant Number OD011104.
References
- 1.Alexandridou A, Stavrou P. Bilateral Coats' disease: long-term follow up. Acta Ophthalmol Scand. 2002;80:98–100. doi: 10.1034/j.1600-0420.2002.800120.x. [DOI] [PubMed] [Google Scholar]
- 2.Belcon MC, Bensen WG, Zahoruk RM. Bilateral retinal detachment in a case of Reiter's disease. Postgrad Med J. 1984;60:47–8. doi: 10.1136/pgmj.60.699.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehmann D, Greve M. Intravitreal bevacizumab for exudative retinal detachment post laser therapy for retinopathy of prematurity. Can J Ophthalmol. 2014;49:228–31. doi: 10.1016/j.jcjo.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 5.Fenwick EK, Pesudovs K, Rees G, Dirani M, Kawasaki R, Wong TY, Lamoureux EL. The impact of diabetic retinopathy: understanding the patient's perspective. Br J Ophthalmol. 2011;95:774–82. doi: 10.1136/bjo.2010.191312. [DOI] [PubMed] [Google Scholar]
- 6.Fernandes BF, Odashiro AN, Maloney S, Zajdenweber ME, Lopes AG, Burnier MN., Jr Clinical-histopathological correlation in a case of Coats' disease. Diagn Pathol. 2006;1:24. doi: 10.1186/1746-1596-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganesh A, Kaliki S, Shields CL. Coats-like retinopathy in an infant with preclinical facioscapulohumeral dystrophy. J AAPOS. 2012;16:204–6. doi: 10.1016/j.jaapos.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Khan JA, Ide CH, Strickland MP. Coats'-type retinitis pigmentosa. Surv Ophthalmol. 1988;32:317–32. doi: 10.1016/0039-6257(88)90094-x. [DOI] [PubMed] [Google Scholar]
- 9.Lim WK, Nussenblatt RB, Chan CC. Immunopathologic features of inflammatory coats disease. Arch Ophthalmol. 2005;123:279–81. doi: 10.1001/archopht.123.2.279. [DOI] [PubMed] [Google Scholar]
- 10.McCluskey P, Kearns M, Taylor F, Sarks J, Horvath J, Tiller D. Coats' type retinitis pigmentosa and subretinal neovascularisation in a patient with renal failure. Lancet. 1989;2:1401. doi: 10.1016/s0140-6736(89)92017-5. [DOI] [PubMed] [Google Scholar]
- 11.Patel HK, Augsburger JJ, Eagle RC., Jr Unusual presentation of advanced Coats' disease. J Pediatr Ophthalmol Strabismus. 1995;32:120–2. doi: 10.3928/0191-3913-19950301-14. [DOI] [PubMed] [Google Scholar]
- 12.Reichstein DA, Recchia FM. Coats disease and exudative retinopathy. Int Ophthalmol Clin. 2011;51:93–112. doi: 10.1097/IIO.0b013e318200de51. [DOI] [PubMed] [Google Scholar]
- 13.Salman AG. Intravitreal bevacizumab for pediatric exudative retinal diseases. Saudi J Ophthalmol. 2011;25:193–7. doi: 10.1016/j.sjopt.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shields CL, Zahler J, Falk N, Furuta M, Eagle RC, Jr, Espinosa LE, Fischer PR, Shields JA. Neovascular glaucoma from advanced Coats disease as the initial manifestation of facioscapulohumeral dystrophy in a 2-year-old child. Arch Ophthalmol. 2007;125:840–2. doi: 10.1001/archopht.125.6.840. [DOI] [PubMed] [Google Scholar]
- 15.Shields JA, Parsons HM, Shields CL, Shah P. Lesions simulating retinoblastoma. J Pediatr Ophthalmol Strabismus. 1991;28:338–40. doi: 10.3928/0191-3913-19911101-12. [DOI] [PubMed] [Google Scholar]
- 16.Shields JA, Shields CL. Differentiation of coats' disease and retinoblastoma. J Pediatr Ophthalmol Strabismus. 2001;38:262–6. doi: 10.3928/0191-3913-20010901-05. quiz 302–263. [DOI] [PubMed] [Google Scholar]
- 17.Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol. 2001;131:561–71. doi: 10.1016/s0002-9394(00)00883-7. [DOI] [PubMed] [Google Scholar]
- 18.Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol. 2001;131:572–83. doi: 10.1016/s0002-9394(01)00896-0. [DOI] [PubMed] [Google Scholar]
- 19.Shields JA, Shields CL. Review: coats disease: the 2001 LuEsther T. Mertz lecture. Retina. 2002;22:80–91. doi: 10.1097/00006982-200202000-00014. [DOI] [PubMed] [Google Scholar]
- 20.Statland JM, Sacconi S, Farmakidis C, Donlin-Smith CM, Chung M, Tawil R. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology. 2013;80:1247–50. doi: 10.1212/WNL.0b013e3182897116. [DOI] [PMC free article] [PubMed] [Google Scholar]