

Abstract
mTOR is a rational target in renal cell carcinoma (RCC) because of its role in disease progression. However, the effects of temsirolimus, the only first-generation mTOR inhibitor approved by the FDA for first-line treatment of metastatic RCC, on tumor reduction and progression-free survival are minimal. Second-generation mTOR inhibitors have not been evaluated on RCC. We compared the effects of temsirolimus and MLN0128, a potent second-generation mTOR inhibitor, on RCC growth and metastasis using a realistic patient-derived tissue slice graft (TSG) model. TSGs were derived from three fresh primary RCC specimens by subrenal implantation of precision-cut tissue slices into immunodeficient mice that were randomized and treated with MLN0128, temsirolimus, or placebo. MLN0128 consistently suppressed primary RCC growth, monitored by magnetic resonance imaging (MRI), in three TSG cohorts for up to 2 months. Temsirolimus, in contrast, only transiently inhibited the growth of TSGs in one of two cohorts before resistance developed. In addition, MLN0128 reduced liver metastases, determined by human-specific quantitative polymerase chain reaction, in two TSG cohorts, whereas temsirolimus failed to have any significant impact. Moreover, MLN0128 decreased levels of key components of the two mTOR subpathways including TORC1 targets 4EBP1, p-S6K1, HIF1α and MTA1 and the TORC2 target c-Myc, consistent with dual inhibition. Our results demonstrated that MLN0128 is superior to temsirolimus in inhibiting primary RCC growth as well as metastases, lending strong support for further clinical development of dual mTOR inhibitors for RCC treatment.
Keywords: renal cell carcinoma, mTOR inhibitor, tumorgrafts
mTOR, as a component of two distinct protein complexes, TORC1 and TORC2, plays an important role in development and progression of renal cell carcinoma (RCC).1,2 Two mTOR inhibitors improved progression-free survival and quality of life and have been approved for metastatic RCC (mRCC) patients.3,4 These are temsirolimus for treatment-naïve patients with poor prognosis mRCC and everolimus for patients with mRCC who failed antiangiogenic therapy. These first-generation mTOR inhibitors were only modestly successful in clinical trials5 with efficacy limited by preferential inhibition of TORC14,5 and compensatory upregulation of the Akt pathway by TORC26,7; therefore, inactivation of both TORC1 and TORC2 may represent a more effective strategy.8 Indeed, second-generation mTOR inhibitors compete against ATP to bind the mTOR catalytic domain and effectively inhibit both TORC1 and TORC2, resulting in more significant antiproliferative effects compared to rapalogs.9 One such inhibitor, MLN0128, is currently in phase I clinical trials (http://clinicaltrials.gov/show/NCT01058707).
In preclinical studies of prostate cancer (PCa), treatment with MLN0128 not only significantly reduced PCa volume in mice but also completely blocked the progression of invasive PCa locally in the mouse prostate and profoundly inhibited the total number and size of distant metastases.10 In addition, MLN0128 inhibited the translation of four genes (YB1, MTA1, vimentin and CD44) that direct invasion and metastasis through 4EBP1 and S6k1 downstream of TORC1, and downregulated p-AKT, a known target of TORC2, confirming its dual inhibition of both subpathways.10
In this work, we evaluated the effects of MLN0128 on RCC growth and metastasis using a patient-derived tissue slice graft (TSG) model recently developed in our laboratory.11 We monitored tumor growth noninvasively by magnetic resonance imaging (MRI) and compared tumor growth rates in mice treated with MLN0128 or temsirolimus vs. placebo. In addition, liver metastases in TSG-bearing mice were quantified and the effects of MLN0128 and temsirolimus were compared. Finally, the mechanisms of MLN0128 and temsirolimus activity were examined by determination of the levels of key components of the TORC1 and TORC2 sub-pathways and target genes.
Material and Methods
Tissue acquisition
Tissues were freshly obtained from three patients undergoing nephrectomy at Stanford between September 2011 and May 2012 under an Institutional Review Board-approved protocol. Frozen sections were used to histologically confirm the diagnosis of clear cell RCC. Clinicopathological features of the cases are summarized in Table 1.
Table 1.
Pathological features and engraftment rates of RCC cases in this study
| Case | Sex | Age | Pathologic stage | Nodal and metastatic stage | Furhman grade | Additional pathologic features | TSG generation and engraftment rate (engrafted/total) | Comments |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 42 | T3a | NxMx | III | First, 55% (12/22) | VHL mutation1 | |
| 2 | F | 59 | T3a | NxM1 (bone and brain) | IV | Rhabdoid | First, 72% (21/29) | Cytoreductive nephrectomy after neoadjuvant sunitinib2 |
| 3 | F | 64 | T3a | NxMx | III | Sixth3, 83% (10/12) | Postoperative metastases4 |
The patient has a known mutation of the Von-Hippel-Lindau (VHL) gene that is validated in this study.
The patient presented with metastatic disease at time of diagnosis and underwent brain and bone metastasectomy, followed by two cycles of neoadjuvant sunitinib before cytoreductive nephrectomy and the implantation of the primary tumor tissue into mice. The patient developed liver metastases within a year after surgery.
Tumor tissues from this patient were serially passaged in mice and the tumor graft line used in this study was the sixth generation of TSG.
Postoperative development of new lung and pancreatic metastases within 6 months after surgery.
Precision-cutting and subrenal implantation of tissue Slices
Precision-cutting and subrenal implantation of tissue slices were performed as previously described.11 All animal work was done in accordance with institutional regulations for laboratory animal studies. RAG2−/−γC−/− mice,12 6–8 weeks of age, were engrafted with RCC tissue slices.
Immunohistochemistry
Immunohistochemistry was performed as previously described.13 The sources and dilutions of the antibodies used in this study are listed in Supporting Information Table 1.
VHL sequencing
DNA was extracted from TSGs preserved in Allprotect tissue reagent (Qiagen, Germantown, MD) using an AllPrep DNA/RNA/Protein Mini Kit (Qiagen) according to the manufacturer’s directions. The three exons of VHL were selected for polymerase chain reaction (PCR) amplification and direct sequencing. PCR primers for VHL exons 1–3 are listed in Supporting Information Table 2.
MRI of TSGs
A Discovery MR901 7.0 T MRI system (Agilent, Santa Clara, CA) was used at the Stanford University Small Animal Imaging Facility for in vivo imaging. A custom T2-weighted sequence was developed for abdominal imaging of the grafts in situ on the mouse kidney. Blinded 3D volumetric modeling was performed with OsiriX 4.1 (Pixmeo, Bernex, Switzerland). Specific growth rate (SGR) was calculated as SGR = ln(V2/V1)/(t2 − t1) with (t2 − t1) as time (in days) between the two MRI measurements V2 and V1 (in cm3).14
Drug administration
Temsirolimus (Sigma-Aldrich, St. Louis, MO) was administered by intraperitoneal injection, once a week, 10 mg/kg/week, as shown to be an effective dose against multiple types of cancers including RCC in mouse models.15,16 It was stocked at 50 mg/ml in 100% EtOH and diluted in 5% Tween-80 and 5% polyethylene glycol 400 to the final concentration on the day of injection. MLN0128 (ChemieTek, Indianapolis, IN) was administered by oral gavage daily, 1 mg/kg/day. It was formulated in 5% polyvinylpropylene, 15% NMP (N-methylpyrrolidone) and 80% water. This formulation served as placebo, which was administered daily by oral gavage. The pharmacokinetic characteristics of MLN0128 in tumor xenograft models have been described previously.17 The dose and schedule used in this study were based on the previously published pharmacokinetic data and other previous studies demonstrating maximal mTOR inhibition while minimizing off-target effects.10,18–20 Tissues were collected within 24 hr after the last treatment.
Quantitative real-time PCR
Tissues were preserved in Allprotect tissue reagent (Qiagen) at −20°C before RNA extraction using an AllPrep DNA/RNA/Protein Mini Kit (Qiagen). The quality of RNA was determined using an Agilent 2100 Bioanalyzer (Agilent). Quantitative real-time PCR (qRT-PCR) was performed as previously described.21 Primer sequences for human-specific and universal GAPDH are listed in Supporting Information Table 2.
Immunoblot analysis
TSGs were homogenized in Tissue Protein Extraction Reagent (Pierce, Appleton, WI) in the presence of proteinase/phosphatase inhibitor cocktail (Millipore, Billerica, MA). Protein concentration was determined using the Bradford assay (Bio-Rad, Hercules, CA). Protein separation and detection were performed as previously described.21 Antibodies used in this study are listed in Supporting Information Table 1. Signal intensities were quantified with the Image J software (NIH).
Statistical analysis
Student’s t-test was used for two-arm experiments and ANOVA for three-arm experiments. A p < 0.05 was considered significant. Statistical tests were performed with Excel Stats.
Results
TSGs derived from RCC tissues maintained histological and genetic fidelity
TSG cohorts were generated from three patient-derived specimens by implanting precision-cut slices of fresh RCC tissues under the renal capsule of immunodeficient mice. The pathological features and engraftment rates of RCC cases in this study are listed in Table 1. Cohort 1 TSGs were derived from a treatment-naïve patient undergoing nephrectomy. The donor for cohort 2 TSGs had undergone neoadjuvant sunitinib, providing an opportunity to investigate whether RCC previously treated with a tyrosine kinase inhibitor remained responsive to mTOR inhibitors. TSGs that had been passaged five times in mice were used for cohort 3 because these TSGs had demonstrated consistent metastatic potential. The donor of cohort 3 TSGs was treatment-naïve at the time of nephrectomy, and developed postoperative metastases. All three TSG cohorts showed similar histological phenotypes to the parent tumors (Fig. 1). Specifically, TSG cohorts 1 and 2 showed high intensity of membranous CAIX staining similar to parent tumors (Figs. 1a–1d), whereas both parent tumor and its derivative TSGs for case 3 showed modest CAIX staining (Figs. 1e and 1f). In addition, all three parent tumors and TSGs showed patchy CD10 staining (Figs. 1g–1l). Moreover, all TSGs and parent tumors were negative for CD117 (Figs. 1m–1r) and CK7 except for weak patchy CK7 staining observed for both parent tumor and TSGs of case 1 (Figs. 1s–1x). Overall, the staining patterns observed for CAIX, CD10, CD117 and CD7 were as expected for clear cell RCC. Finally, DNA sequencing revealed the same VHL mutation (473 T->C in exon 3) in parent tumor and TSGs for case 1 (Figs. 1y and 1z). These results demonstrated that TSGs maintained histological and genetic characteristics of the parent tumors.
Figure 1.
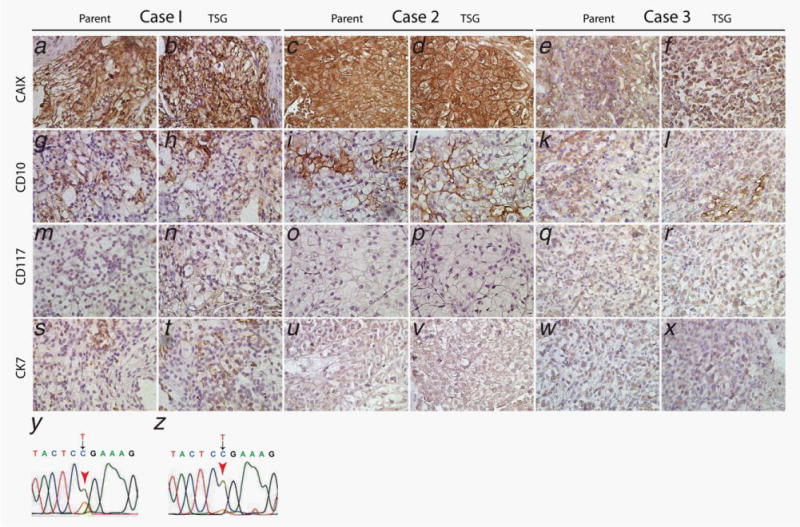
TSGs preserved histological features and biomarker expression of corresponding parental tumors. All three cases and their TSGs displayed intermediate to strong expression of CAIX (a–f) and patchy staining of CD10 (g–l). All three cases and their TSGs were negative for CD117 (m–r). Case 1 and its TSGs showed weak staining for CK7 (s and t). Cases 2 and 3 and their TSGs were negative for CK7 (u–x). Case 1 and its TSGs harbored the same mutation in VHL gene exon 3 (y and z). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
MLN0128 is a potent inhibitor of RCC growth
Four weeks after implantation, TSG-bearing mice were randomized to control and treated arms based on TSG volume measured by MRI and 3D volumetric modeling (Figs. 2a and 2b), so that TSG volume distribution in each arm was similar. SGR was also taken into account for TSG cohorts 1 and 3 with multiple pretreatment MRI measurements. MLN0128 significantly reduced tumor growth in all three cohorts (Fig. 2c). Specifically, for TSG cohort 1, mice treated with MLN0128 for 2 months had a negative mean post-treatment SGR of −2.98%/day vs. 1.46%/day for placebo, indicating significant shrinkage of the tumors by MLN0128 (p = 0.0005). For TSG cohort 2, mean SGR in MLN0128-treated mice was 0.008%/day during the 2-month treatment period compared to 3.7%/day in placebo arm (p < 0.001), demonstrating a nearly complete inhibition of tumor growth by MLN0128. For TSG cohort 3, MLN0128 decreased mean SGR by 50% during a 10-day treatment period (5.8 vs. 13.8% in placebo, p = 0.03). These results demonstrated that MLN0128 is a potent inhibitor of RCC growth.
Figure 2.
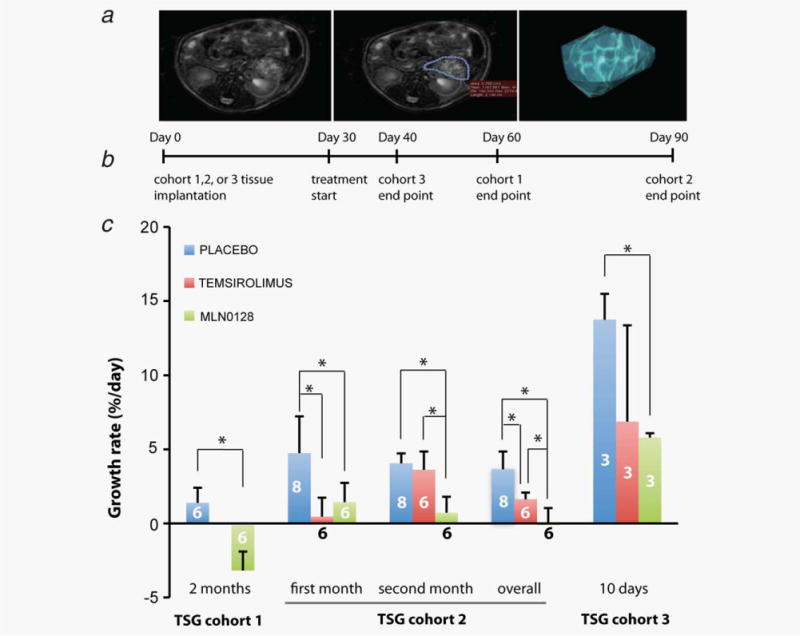
MLN0128 was superior to temsirolimus in tumor growth inhibition. (a) TSG volume was measured by MRI and 3D volumetric modeling. (b) Diagram of study timeline. (c) SGR of TSGs in mice treated with either MLN0128, temsirolimus or placebo in three cohorts. * indicates statistical significance (p < 0.05). The numbers on the columns are the numbers of mice in each arm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
MLN0128 is superior to temsirolimus in RCC growth inhibition
We compared the efficacy of MLN0128 to temsirolimus in growth inhibition of TSG cohorts 2 and 3. Specifically, we assessed tumor volume and calculated SGR at 1 and 2 months after treatment in TSG cohort 2. After the first month of treatment, both MLN0128 and temsirolimus significantly decreased SGR compared to placebo (Fig. 2c). The difference between MLN0128 and temsirolimus was not significant. Between 1 and 2 months of treatment, temsirolimus did not significantly reduce SGR compared to placebo, suggesting the development of resistance to temsirolimus. However, MLN0128 continued to significantly reduce SGR compared to both placebo (p = 0.002) and temsirolimus (p = 0.005) (Fig. 2c). Overall, for the entire period of treatment (SGR between the MRI pretreatment and at 2 months), both temsirolimus (p = 0.009) and MLN0128 (p < 0.0001) showed significant inhibition of tumor growth, with MLN0128 more potent than temsirolimus (p = 0.044). For TSG cohort 3, after 10 days of treatment, MLN0128 significantly reduced SGR (p = 0.02), whereas the reduction by temsirolimus was not significant (p > 0.05) (Fig. 2c). Overall, these results suggest that MLN0128 is superior to temsirolimus in inhibiting RCC tumor growth.
MLN0128, but not temsirolimus, suppresses growth of liver metastasis of RCC
Consistent with the clinical follow-up of case 3 who developed lung and pancreatic metastases within 6 months after surgery, we observed gross liver and lung metastases expressing Ku70, a human-specific nuclear antigen (Fig. 3a), in mice carrying TSGs from this patient. Lung (not shown) and liver metastases were positive for CAIX (Fig. 3b) and negative for CD10 (Fig. 3c), consistent with the observation that most of the parent tumor and primary TSGs are positive for CAIX (Figs. 1e and 1f) and negative for CD10 (Figs. 1k and 1l). These results suggest that the metastases maintained the immunophenotype of the parent tumor and primary TSG.
Figure 3.
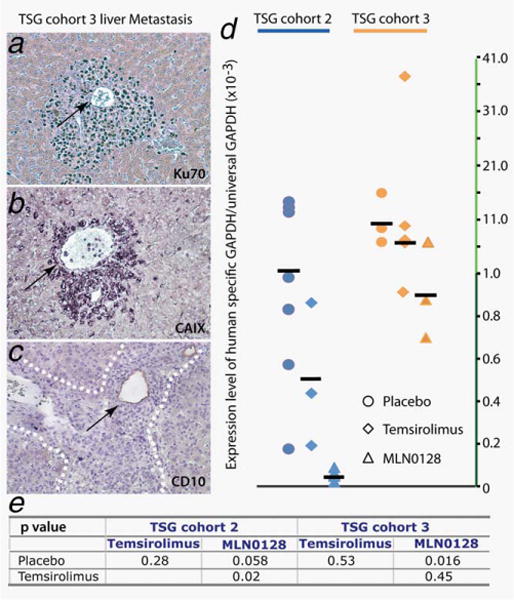
MLN0128 was superior to temsirolimus in inhibiting metastases. mRCC cells were observed surrounding blood vessels (arrows in a–c) in livers of mice carrying TSG cohort 3. They expressed Ku70 (a), a human-specific nuclear antigen, CAIX (b) and were negative for CD10 (c). MLN0128 but not temsirolimus reduced the mean ratio of mRCC cells vs. total cells, measured as the ratio of human-specific GAPDH to total GAPDH by qRT-PCR (d). The effects were statistically significant as shown by p-values of Student’s t-test (e). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
We quantified the ratio of mRCC cells to total cells using qRT-PCR. mRCC cells were detected with human-specific GAPDH primers and normalized to total GAPDH detected using primers recognizing both human and mouse GAPDH. MLN0128 significantly reduced the ratio of mRCC cells in the liver in TSG cohort 3 compared to placebo (Fig. 3d; p = 0.02). Consistent with the fact that case 2 developed liver metastases within a year after surgery, mRCC cells were also detected in the livers of cohort 2 mice by qRT-PCR, and the reduction of the ratio of mRCC to total cells by MLN0128 was near significant in this cohort (p = 0.058). In contrast, temsirolimus did not significantly reduce the ratio of mRCC to total cells in the liver although it was lower compared to placebo in both cases (Fig. 3d). For TSG cohort 2, mice treated with MLN0128 showed significantly lower levels of mRCC cells in the liver compared to those treated with temsirolimus (p = 0.02). As a negative control, we confirmed that human GAPDH was not detected in mice without TSGs. In addition, no human GAPDH was present in livers of mice bearing TSG cohort 1, indicating the absence of metastatic disease. These results suggest that MLN0128, but not temsirolimus, suppresses growth of liver metastases of RCC.
Molecular mechanisms of MLN0128 action in RCC
To determine the mechanisms of MLN0128 action, we compared the protein expression of mTOR pathway components including p-4EBP1, p-S6K1 and p-AKT by quantitative immunoblot (95% confidence intervals listed in Table 2). The expression of proteins that are known to play a role in cancer progression and are regulated by the two mTOR complexes, TORC1 and TORC2, was also examined. These include TORC1 targets, HIF1α22 and MTA1,10 and TORC2-modulated c-Myc.23 For TSG cohort 1, quantitation of protein expression by immunoblot using Image J revealed a significant downregulation of p-4EBP1, HIF1α, MTA1 and c-Myc by MLN0128 compared to placebo (Fig. 4a). However, there was no significant downregulation of (S473)p-AKT. For TSG cohort 3, we observed a superior ability of MLN0128 compared to placebo and temsirolimus to downregulate p-4EBP1, p-S6K1 and HIF1α (Fig. 4b). There was no significant effect of MLN0128 on (S473)p-AKT expression compared to placebo. In contrast, temsirolimus did not significantly affect p-4EBP1, p-S6K1 or HIF1α (Fig. 4b). In addition, temsirolimus significantly upregulated (S473)p-AKT compared to placebo. Immunohistochemistry revealed nuclear and cytoplasmic staining of p-4EBP1 in both patient 1 and 3 primary tumors (Fig. 4c). Furthermore, MLN0128-treated TSGs in both cohorts displayed decreased expression of p-4EBP1 compared to placebo (Fig. 4c). These results demonstrated that, unlike temsirolimus, MLN0128 consistently downregulated components of the TORC1 subpathway and its regulated genes, and differentially regulated TORC2 target genes compared to temsirolimus (Fig. 4d).
Table 2.
95% confidence intervals for quantitative immunoblot analysis of mTOR pathway components
| TSG cohot 1 | TSG cohort 3 | ||||
|---|---|---|---|---|---|
| Protein | Arm | 95% Confidence interval | Protein | Arm | 95% Confidence interval |
| p-4.FRP1 | Placebo | (1.46, 1.97) | Placebo | (0.87, 1.08) | |
| MLN0128 | (2.13, 2.58) | P-4EBP1 | Temsirolimus | (0.81, 0.95) | |
| HIF1-alpha | Placebo | (1.48, 1.81) | MLN0128 | (1.69, 2.22) | |
| MLN0128 | (1.83, 2.21) | Placebo | (1.19, 1.54) | ||
| MTA1 | Placebo | (1.72, 1.95) | HIF1-alpha | Temsirolimus | (1.34, 1.54) |
| MLN0128 | (2.02, 2.44) | MLN0128 | (1.55, 1.88) | ||
| c-MYC | Placebo | (0.96, 1.21) | Placebo | (0.71, 1.03) | |
| MLN0128 | (1.30, 1.51) | P-S6K1 | Temsirolimus | (0.68, 0.93) | |
| MLN0128 | (1.08, 1.23) | ||||
| Placebo | (1.17, 1.71) | ||||
| p-AKT | Temsirolimus | (0.84, 1.12) | |||
| MLN0128 | (1.05, 1.36) | ||||
Figure 4.
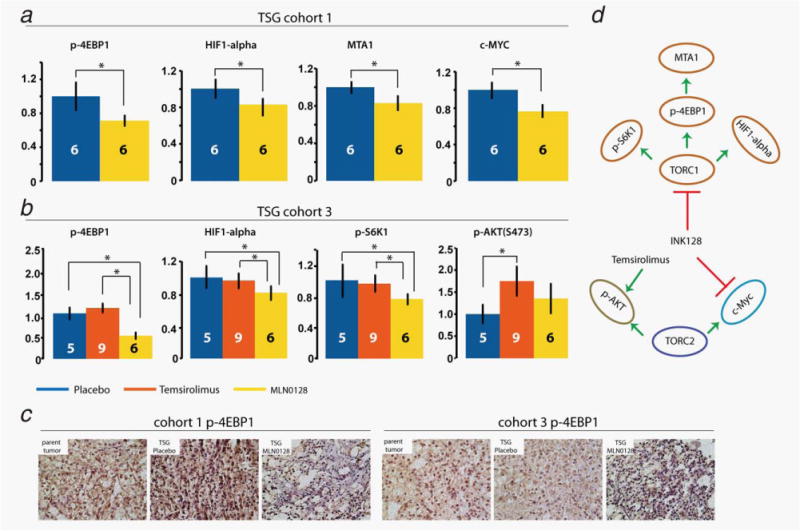
MLN0128 downregulated key components of the two mTOR subpathways, TORC1 and TORC2. Quantitative immunoblot demonstrated downregulation of p-4EBP1, HIF1α, MTA1 and c-Myc in TSG cohort 1 by MLN0128 (a). In TSG cohort 3 (b), MLN0128 was superior to placebo and temsirolimus in downregulating p-4EBP1, p-S6K1 and HIF1α. In addition, temsirolimus significantly upregulated (S473)p-AKT compared to placebo. * indicates statistical significance (p < 0.05). (c) Expression of p-4EBP1 in parent tumors and derivative TSGs for cohorts 2 and 3, treated with placebo or MLN0128, as determined by immunohistochemistry. (d) Diagram of MLN0128 actions in RCC. The numbers on the columns are the numbers of mice in each arm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
In our study, we compared the effects of temsirolimus and MLN0128, a potent second-generation mTOR inhibitor, on RCC growth and metastasis using a realistic patient-derived TSG model.11 The TSG model of RCC faithfully recapitulates tumor pathology, gene expression, genetic mutation and drug response.11 The high engraftment rate and metastatic potential of this authentic model, in conjunction with the ability to generate large first-generation animal cohorts and to quantitate tumor volume at the orthotopic site by MRI, proffer significant advantages compared to other preclinical platforms. In particular, the establishment of large animal cohorts from individual specimens will facilitate comparative effectiveness studies of the many novel agents under development for RCC such as MLN0128. One of the limitations of the TSG model is that it depends on the availability of patient specimens. In addition, tissue needs to be implanted immediately after acquisition from surgical specimens. It may be feasible to implant cryopreserved tissue slices but our preliminary results suggest that the take rate is lower than when using fresh tissue slices.
The finding that MLN0128 shows potent therapeutic activity in a realistic preclinical model of human RCC is noteworthy. Since 2005, seven targeted agents have been approved by regulatory authorities for various uses in advanced RCC or mRCC patients.24 However, the response rates (<50%) and increased length of progression-free survival (≤6 months) for these agents are far from satisfactory.24 In particular, the objective response rate in mRCC for temsirolimus is low, and patients rapidly develop resistance.25 In our study, MLN0128 was not only a potent inhibitor of primary RCC growth at the orthotopic site but also effectively repressed liver metastases of RCC. MLN0128 was active against all three clear cell RCC cases tested in this study, albeit the number was small. In contrast, temsirolimus either lost its anticancer activity over time as in TSG cohort 2 or was not effective overall as in TSG cohort 3. In addition, MLN0128 significantly reduced the burden of liver metastases in both TSG cohorts 2 and 3, whereas no effect was observed for temsirolimus. Currently, MLN0128 is being evaluated in a Phase I trial to determine the safety, tolerability, dose-limiting toxicities and pharmacokinetics in subjects with advanced cancer. Our results warrant further clinical development of MLN0128 and perhaps other dual inhibitors of mTOR for RCC treatment.
The high efficacy of MLN0128 that we observed is likely due to the fact that it inhibits both mTORC1 and mTORC2 by competing for the ATP-binding site on mTOR26 in opposition to the allosteric mTOR inhibitors, namely the rapalogs (rapamycin, temsirolimus and everolimus) that only inhibit TORC1.27 Alternatively but not exclusively, MLN0128 inhibits TORC1 to a greater degree than the rapalogs in that it inhibits both subpathways of TORC1 (p-4EBP1 and p-S6K1), whereas rapalogs mostly inhibit p-S6K1. Hsieh et al. reported that MLN0128 efficiently repressed both TORC1 subpathways (p-4EBP1 and p-S6K1) in PCa.10 In addition, MLN0128 decreased levels of p-AKT by inhibiting TORC2, but the effect was modest,10 probably because of the regulation of AKT phosphorylation through other pathways.28 In our RCC TSG model, the two TORC1 subpathways were significantly downregulated by MLN0128 compared to both placebo and temsirolimus, suggesting that better control of TORC1 contributed to efficient inhibition of TSG growth and the superiority to temsirolimus. The lack of downregulation of TORC1 subpathways by temsirolimus is consistent with the lack of tumor growth inhibition by temsirolimus in TSG cohort 3. Upregulation of p-AKT by temsirolimus, probably by a feedback loop between S6K1 and PI3K through the insulin receptor substrate 1 (IRS-1),6,29–31 may also explain the superiority of MLN0128 in TSG cohort 3. Unlike in PCa,10 we did not observe downregulation of p-AKT by MLN0128 in TSG cohorts 1 or 3, suggesting different mechanisms of action in different tumor types. Even in the same tumor type, i.e., clear cell RCC, we observed differential regulation of gene expression by MLN0128. For example, MLN0128 significantly downregulated c-Myc in TSG cohort 1 but not 3. This is not surprising considering the molecular heterogeneity of human RCC.32–34
Our results were largely consistent with two recent studies evaluating antitumor efficacy of MLN0128 in xenograft models of breast cancer and leukemia.18,20 In both studies, MLN0128 outperformed rapalogs in suppressing tumor growth at a much lower concentration.18,20 Moreover, these studies attributed this improved potency of MLN0128 to its ability to effectively block both mTORC1-and mTORC2-mediated signaling and prevent feedback to AKT. Specifically, MLN0128 decreased the levels of p-4EBP1, pS6K1 and p-AKT, whereas rapalogs increased AKT activity and failed to reduce p-4EBP1.18,20 One difference is that Gokmen-Polar et al. showed that both MLN0128 and rapamycin blocked VEGF-induced lung metastasis in VEGF-driven breast cancer xenograft models, whereas our study demonstrated that MLN0128, but not temsirolimus, inhibited liver metastasis in RCC TSG-bearing mice. This disparate finding could be due to the differences in model systems, rapalogs used and/or cancer types examined. Indeed, Hsieh et al. revealed the ability of MLN0128, but not rapamycin, in suppressing lymph node metastasis in a PTEN knockout PCa model.10 Given the complex regulatory role of mTOR signaling in the development of metastasis, further studies are necessary to determine underlying mechanisms of these different findings.
In summary, our results demonstrate that MLN0128 is a promising anticancer agent for human RCC and that TSGs can serve as a representative preclinical model for discovery of effective therapies for RCC.
Supplementary Material
What’s new?
Second-generation mTOR inhibitors, which target both the TORC1 and the TORC2 mTOR subpathways, could be more effective against renal cell carcinoma (RCC) than first-generation inhibitors, which target only TORC1. This study lends support to that idea, revealing that the novel second-generation mTOR inhibitor MLN0128 consistently suppresses primary RCC growth in a patient-derived tissue slice graft (TSG) model, whereas the FDA-approved first-generation inhibitor temsirolimus only transiently inhibited TSGs. The results suggest that dual mTOR inhibitors may have superior activity compared with first-generation drugs and should be evaluated in human trials for RCC.
Acknowledgments
The authors thank Dr. Benjamin Chung for help with tissue acquisition and providing clinical data and Dr. Jesse McKenney for pathological consultation.
Grant sponsors: Association Française d’Urologie, the Ferdinand Eisenberger Grant of the German Society of Urology ID SaM1/FE-11, Instrumentarium Science Foundation, Finland and the Finnish Medical Foundation
Abbreviations
- mRCC
metastatic renal cell carcinoma
- MRI
magnetic resonance imaging
- PCa
prostate cancer
- PCR
polymerase chain reaction
- qRT-PCR
quantitative real-time PCR
- RCC
renal cell carcinoma
- SGR
specific growth rate
- TSG
tissue slice graft Additional
Footnotes
Supporting Information may be found in the online version of this article
Conflict of interest: Nothing to report
References
- 1.Pantuck AJ, Seligson DB, Klatte T, et al. Prognostic relevance of the mTOR pathway in renal cell carcinoma: implications for molecular patient selection for targeted therapy. Cancer. 2007;109:2257–67. doi: 10.1002/cncr.22677. [DOI] [PubMed] [Google Scholar]
- 2.Robb VA, Karbowniczek M, Klein-Szanto AJ, et al. Activation of the mTOR signaling pathway in renal clear cell carcinoma. J Urol. 2007;177:346–52. doi: 10.1016/j.juro.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 3.Pal SK, Quinn DI. Differentiating mTOR inhibitors in renal cell carcinoma. Cancer Treat Rev. 2013;39:709–19. doi: 10.1016/j.ctrv.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barthelemy P, Hoch B, Chevreau C, et al. mTOR inhibitors in advanced renal cell carcinomas: from biology to clinical practice. Crit Rev Oncol Hematol. 2013;88:42–56. doi: 10.1016/j.critrevonc.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Voss MH, Molina AM, Motzer RJ. mTOR inhibitors in advanced renal cell carcinoma. Hematol Oncol Clin North Am. 2011;25:835–52. doi: 10.1016/j.hoc.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10:992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- 8.Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29:3733–44. doi: 10.1038/onc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janes MR, Limon JJ, So L, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–13. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsieh AC, Liu Y, Edlind MP, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thong AE, Zhao H, Ingels A, et al. Tissue slice grafts of human renal cell carcinoma: an authentic preclinical model with high engraftment rate and metastatic potential. Urol Oncol. 2013 doi: 10.1016/j.urolonc.2013.05.008. pii: S1078-1439(13)00208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Rijn RS, Simonetti ER, Hagenbeek A, et al. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2−/− gammac−/− double-mutant mice. Blood. 2003;102:2522–31. doi: 10.1182/blood-2002-10-3241. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Nolley R, Chen Z, et al. Tissue slice grafts: an in vivo model of human prostate androgen signaling. Am J Pathol. 2010;177:229–39. doi: 10.2353/ajpath.2010.090821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehrara E, Forssell-Aronsson E, Ahlman H, et al. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res. 2007;67:3970–5. doi: 10.1158/0008-5472.CAN-06-3822. [DOI] [PubMed] [Google Scholar]
- 15.Nishikawa T, Takaoka M, Ohara T, et al. Antiproliferative effect of a novel mTOR inhibitor temsirolimus contributes to the prolonged survival of orthotopic esophageal cancer-bearing mice. Cancer Biol Ther. 2013;14:230–6. doi: 10.4161/cbt.23294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makhov PB, Golovine K, Kutikov A, et al. Modulation of Akt/mTOR signaling overcomes sunitinib resistance in renal and prostate cancer cells. Mol Cancer Ther. 2012;11:1510–17. doi: 10.1158/1535-7163.MCT-11-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel CG, Patel M, Chakravarty A, et al. Clinical pharmacokinetics (PK) and translational PK-pharmacodynamic (PD) modeling and simulation to predict antitumor response of various dosing schedules to guide the selection of a recommended phase II dose (RP2D) and schedule for the investigational agent MLN0128. J Clin Oncol. 2013;31(Suppl) abstr 2567. [Google Scholar]
- 18.Janes MR, Vu C, Mallya S, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2013;27:586–94. doi: 10.1038/leu.2012.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pourdehnad M, Truitt ML, Siddiqi IN, et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci USA. 2013;110:11988–93. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gokmen-Polar Y, Liu Y, Toroni RA, et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat. 2012;136:673–82. doi: 10.1007/s10549-012-2298-8. [DOI] [PubMed] [Google Scholar]
- 21.Zhao H, Nolley R, Chen Z, et al. Inhibition of monoamine oxidase A promotes secretory differentiation in basal prostatic epithelial cells. Differentiation. 2008;76:820–30. doi: 10.1111/j.1432-0436.2007.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toschi A, Lee E, Gadir N, et al. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem. 2008;283:34495–9. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–46. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mihaly Z, Sztupinszki Z, Surowiak P, et al. A comprehensive overview of targeted therapy in metastatic renal cell carcinoma. Curr Cancer Drug Targets. 2012;12:857–72. doi: 10.2174/156800912802429265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 26.Schenone S, Brullo C, Musumeci F, et al. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18:2995–3014. doi: 10.2174/092986711796391651. [DOI] [PubMed] [Google Scholar]
- 27.Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 28.Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005;24:7401–9. doi: 10.1038/sj.onc.1209099. [DOI] [PubMed] [Google Scholar]
- 29.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22:169–76. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Zhao M, Hao M, et al. Dual inhibition of PI3K and mTOR mitigates feedback-loop pAkt activation to improve tamoxifen response in breast cancer cells. Mol Cancer Res. 2013;11:1269–78. doi: 10.1158/1541-7786.MCR-13-0212. [DOI] [PubMed] [Google Scholar]
- 31.Wan X, Harkavy B, Shen N, et al. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–40. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 32.Brannon AR, Haake SM, Hacker KE, et al. Meta-analysis of clear cell renal cell carcinoma gene expression defines a variant subgroup and identifies gender influences on tumor biology. Eur Urol. 2012;61:258–68. doi: 10.1016/j.eururo.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brannon AR, Reddy A, Seiler M, et al. Molecular stratification of clear cell renal cell carcinoma by consensus clustering reveals distinct subtypes and survival patterns. Genes Cancer. 2010;1:152–63. doi: 10.1177/1947601909359929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staller P. Genetic heterogeneity and chromatin modifiers in renal clear cell carcinoma. Future Oncol. 2010;6:897–900. doi: 10.2217/fon.10.50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.