

Abstract
Duchenne muscular dystrophy (DMD)–associated cardiac diseases are emerging as a major cause of morbidity and mortality in DMD patients, and many therapies for treatment of skeletal muscle failed to improve cardiac function. The reprogramming of patients’ somatic cells into pluripotent stem cells, combined with technologies for correcting the genetic defect, possesses great potential for the development of new treatments for genetic diseases. In this study, we obtained human cardiomyocytes from DMD patient–derived, induced pluripotent stem cells genetically corrected with a human artificial chromosome carrying the whole dystrophin genomic sequence. Stimulation by cytokines was combined with cell culturing on hydrogel with physiological stiffness, allowing an adhesion-dependent maturation and a proper dystrophin expression. The obtained cardiomyocytes showed remarkable sarcomeric organization of cardiac troponin T and α-actinin, expressed cardiac-specific markers, and displayed electrically induced calcium transients lasting less than 1 second. We demonstrated that the human artificial chromosome carrying the whole dystrophin genomic sequence is stably maintained throughout the cardiac differentiation process and that multiple promoters of the dystrophin gene are properly activated, driving expression of different isoforms. These dystrophic cardiomyocytes can be a valuable source for in vitro modeling of DMD-associated cardiac disease. Furthermore, the derivation of genetically corrected, patient-specific cardiomyocytes represents a step toward the development of innovative cell and gene therapy approaches for DMD.
Introduction
Duchenne muscular dystrophy (DMD) is one of the most common and severe inherited neuromuscular disorders, affecting 1 in 3,500 newborn males. DMD is caused by mutations in the dystrophin gene encoding a key structural protein of the dystrophin glycoprotein complex, which connects the contracting cytoskeletal machinery of skeletal and cardiac muscle fibers to the extracellular matrix scaffold.1 The absence of dystrophin in DMD patients causes a broad spectrum of physical consequences, eventually leading to a premature death.2 Approximately 20% of deaths are the result of cardiomyopathies and/or cardiac conduction abnormalities. The improvement in treatments of respiratory muscle disease allowed an increased life span of patients affected by DMD. Consequently, cardiomyopathies (which are present in ~90% of DMD patients) are emerging as a major cause of morbidity and mortality.3 In addition, many experimental therapies have mainly focused on skeletal muscle, aiming at the restoration of dystrophin expression in myofibers, and have failed to improve cardiac function.4
The derivation of DMD patient–specific cardiomyocytes (CMs) and the correction of their genetic defect could provide a valuable cell source for in vitro modeling and for studying DMD-related cardiac dysfunctions, in addition to potentially representing a significant advancement toward an effective therapy for DMD-associated cardiomyopathies.
The efficient reprogramming technology pioneered by Yamanaka and colleagues5 opened the perspective of deriving large numbers of disease-specific human cells in vitro. Human induced pluripotent stem (hiPS) cells are recently emerging as an ideal cell source for the generation of clinically relevant cardiac disease models.6 Several recent studies demonstrate how hiPS cell–derived CMs can be used for modeling the pathological phenotype of inherited cardiac disorders, such as the LEOPARD syndrome,7 type 1 and 2 long QT syndrome,8–10 catecholaminergic polymorphic ventricular tachycardia,11 arrhythmogenic right ventricular dysplasia/cardiomyopathy,12 and the dilated cardiomyopathy.13 However, to our knowledge, CMs from DMD patient–derived hiPS cells have never been obtained so far.
Disease-specific hiPS cell–derived CMs could represent a platform for studying in vitro the pathological dystrophic phenotype and for testing and validating the therapeutic approaches and their efficiency in restoring the normal phenotype.14 In addition, it has been recently demonstrated that hiPS cell–derived CMs can efficiently integrate in injured hearts of a guinea pig,15 providing a proof of principle for the application of these cells in regenerative medicine aiming at the treatment of cardiac dysfunctions.
In the perspective of a therapeutic application, the genetic defect of CMs derived from inherited disease–specific hiPS cells should be corrected before these cells are reengrafted in the patient.
DMD is among the most difficult genetic diseases to treat, and the dystrophin gene is the largest gene described in the human genome. Promising results have been obtained in rodent models of Duchenne cardiomyopathy using adeno-associated viruses carrying minimized synthetic dystrophin genes (mini- and microdystrophin).16 These approaches do not allow the insertion of a complete functional version of the dystrophin gene. On the other hand, exon-skipping approaches,17 which redirect the gene processing bypassing the mutation, can be applied only to defined ranges of patients, based on their specific mutations. Several studies suggest that utrophin may help in the preservation of heart function in young adult mdx mice and heterozygous mdx mice.18,19 However, similar to dystrophin, the large size of the utrophin gene also presents a significant challenge to gene delivery.16 Other approaches for the treatment of Duchenne cardiomyopathy include the forced expression of sarcoendoplasmic reticulum calcium-ATPase via adeno-associated virus gene transfer, aimed at restoring calcium homeostasis and improving cardiac contractility,20 without correction of the genetic defect.
A highly promising gene delivery tool for the correction of the DMD gene is represented by human artificial chromosomes (HACs). HAC is an artificially created exogenous minichromosome having the ability to replicate and segregate autonomously in target human cells and to be stably maintained at episomal level, without integration into the host genome. In addition, HACs have the capacity to carry large genomic loci with all their regulatory elements.21
An HAC vector carrying, for the first time, the whole dystrophin genomic locus including the associated regulatory elements (DYS-HAC) was developed by Hoshiya et al.22 Furthermore, Kazuki et al.23 have demonstrated the complete correction of hiPS cells derived from a DMD patient, using the DYS-HAC, pioneering an innovative and promising therapeutic approach for the treatment of DMD and DMD-associated cardiac diseases.
The first preclinical proof of safety and efficacy of DYS-HAC–mediated therapy has been provided by Tedesco et al.24 In this study, the authors showed a significantly ameliorated phenotype in the mdx dystrophic mouse model after the transplantation of mdx mesoangioblasts genetically corrected with the DYS-HAC. In addition, the same group recently reported the differentiation in mesoangioblast-like stem/progenitor cells from DMD patient–derived hiPS cells, carrying the genetic correction with the DYS-HAC.25
In this article, we aim to differentiate DMD patient–derived, genetically corrected hiPS cells into CMs with a mature phenotype and to assess the maintenance of the DYS-HAC during the differentiation of hiPS cells. In particular, we aim at investigating the correct activation, at HAC level, of the complex mechanisms regulating dystrophin expression, such as multiple promoter activities, which should be finely regulated in a development- and tissue-specific way.
An ad hoc cardiac differentiation procedure, combining the delivery of cytokines with mechanical stimulation, by culturing cells on hydrogel with physiological stiffness, has been designed to allow full CM maturation. DYS-HAC–mediated dystrophin expression restoration has been assessed at different stages of the differentiation process.
The dystrophic CMs established in this work could potentially represent a valuable cell source to be used for in vitro modeling of DMD-associated cardiomyopathies; and, at the same time, the genetically corrected dystrophic CMs possess a promising therapeutic potential for the treatment of DMD.
Results
Differentiation of hiPS cells toward the cardiac lineage
To obtain DMD-specific CMs, their genetic correction, and a positive control of dystrophin expression, the following hiPS cell lines differentiated toward the cardiac lineage were: (i) hiPS cells derived from a DMD patient with deletion of exons 4–43 of the muscle isoform Dp427m (DMD hiPS cells); (ii) DMD patient–specific hiPS cells genetically corrected by an HAC carrying the full-length genomic dystrophin sequence (DYS-HAC hiPS cells); and (iii) hiPS cells derived from a healthy individual (healthy hiPS cells), carrying a normal genotype with a wild-type copy of the dystrophin gene.
First, DMD, DYS-HAC, and healthy hiPS cell lines were cultured and expanded in their undifferentiated state for up to 10 passages. During expansion, hiPS cell colonies maintained the expression of pluripotency markers such as Oct4, Sox2, c-Myc, Tra-1-60, and Tra-1-81, as evaluated by immunofluorescence (see Supplementary Figure S1a). In addition, DYS-HAC hiPS cells were monitored for the expression of the enhanced green fluorescent protein, a marker contained in the DYS-HAC (see Supplementary Figure S1b). The expanded colonies were then used for embryoid bodies (EBs) generation.
DMD, DYS-HAC, and healthy hiPS cell–derived EBs were differentiated toward the cardiac lineage through an ad hoc optimized procedure. Overall outline of this procedure is shown in Figure 1a.
Figure 1.

Differentiation of hiPS cells. (a) Schematic representation of the differentiation procedure. (b) Undifferentiated colonies of DYS-HAC hiPS cells cultured on murine embryonic fibroblast feeder cells. (c) EBs generated from DYS-HAC hiPS cell colonies on day 4 of the differentiation procedure. (d) Cultured cells 2 days after EB adhesion. Epifluorescence images show the expression of EGFP in each condition, indicating the presence of the DYS-HAC. BMP4, bone morphogenetic protein 4; DKK1, DICKKOPF-1; DYS-HAC, human artificial chromosome carrying the whole dystrophin genomic sequence; EBs, embryoid bodies; EGFP, enhanced green fluorescent protein; hiPS cells, human induced pluripotent stem cells; VEGF, vascular endothelial growth factor; bFGF, basic fibroblast growth factor.
Remarkably, the enhanced green fluorescent protein gene, driven by a CAG promoter contained in the DYS-HAC,23 was expressed during all the stages of the differentiation procedure, in DYS-HAC hiPS cells (Figure 1b), in DYS-HAC hiPS cell–derived EBs (Figure 1c), and in cultured cells after EBs adhesion (Figure 1d), indicating the stable maintenance of the DYS-HAC.
During the first 16 days of the differentiation procedure, EBs were cultured in suspension and subjected to a staged protocol, adapted from the one developed by Kattman et al.,26 based on the addition of specific cytokines—known to play a key role in cardiogenesis during embryonic development—to the culture medium. This 16-day differentiation procedure resulted in a relevant percentage of spontaneously contracting EBs (see Supplementary Videos S1 and S2), ranging from a minimum of 14% for DMD hiPS cell–derived EBs to a maximum of 44% for healthy hiPS cell–derived EBs (Table 1).
Table 1. Percentage of spontaneously contracting EBs and cTnT-positive cells obtained for each cell line used.
| hiPS cell lines | Contracting EBs obtained (% ± SD): day16 | cTnT-positive cells (% ± SD): day 24 |
|---|---|---|
| DMD hiPS cells | 14 ± 4 | 7,6 ± 0,7 |
| DYS-HAC hiPS cells | 19 ± 5 | 8,2 ± 1,5 |
| Healthy hiPS cells | 44 ± 2 | 9,8 ± 4,6 |
cTnT, cardiac troponin T; DMD, Duchenne muscular dystrophy; DYS-HAC, human artificial chromosome carrying the whole dystrophin genomic sequence; EBs, embryoid bodies; hiPS cells, human induced pluripotent stem cells; SD, standard deviation calculated on data obtained from 5 independent analyses.
Our recent data demonstrated that a proper cardiac differentiation requires cell–substrate interactions to promote functional and structural maturation of hiPS cell–derived CMs.36 For this reason, contracting EBs were cultured in suspension up to day 20 and then seeded on hydrogel substrate with a physiological stiffness for additional 4 days. Following adhesion, a remarkable maturation of CMs was observed in terms of both cytoskeletal architecture and cardiac markers expression (Figure 2). CMs obtained on day 24 were characterized by a remarkable sarcomeric organization, as revealed by immunofluorescence of α-actinin, cardiac troponin T (cTnT), and F-actin, and gap junction formation, as revealed by immunofluorescence of connexin 43 (Figure 2a). The percentage of cTnT-positive CMs obtained on the overall population was ~10% (Table 1). The same percentage increased to 44 ± 2% when only contracting EBs were selected.
Figure 2.

Characterization of hiPS cell–derived CMs. (a) Immunofluorescence of α-actinin; cardiac troponin T (cTnT) and F-actin; connexin 43 (Cnx43); sarcoendoplasmic reticulum calcium-ATPase (SERCA2a); and GATA4 in adhered CMs on day 24 of the differentiation procedure. Nuclei are counterstained with DAPI. (b) RT-PCR shows the expression of NKX2.5, cTnT, and MLC2v in EBs obtained from DMD, healthy, and DYS-HAC hiPS cells cultured in suspension (on day 20) and on the EB-derived adhered cells on day 24 of the differentiation procedure. (c) Typical calcium transients displayed by hiPS cell–derived CMs on day 24 of the differentiation procedure. The histogram reports quantitative evaluation of calcium reuptake rate by the half-life of calcium decay and the calcium release phase as the time to peak. Data are presented as SD. CMs, cardiomyocytes; DAPI, 4′,6-diamidino-2-phenylindole; DMD, Duchenne muscular dystrophy; DYS-HAC, human artificial chromosome carrying the whole dystrophin genomic sequence; hiPS cells, human induced pluripotent stem cells; MLC2v, ventricular myosin light chain; RT-PCR, reverse transcriptase–polymerase chain reaction.
Expression of sarcomeric cardiac-specific proteins, in particular cTnT and ventricular myosin light chain, in adhered CMs, was also confirmed by reverse transcriptase–polymerase chain reaction (RT-PCR) (Figure 2b). For DMD hiPS cell–derived CMs, expression of ventricular myosin light chain (MLC2v), a marker of terminally differentiated ventricular CMs, was observed only after adhesion. Mesoderm- and cardiac-specific transcription factors GATA4 and NKX2.5 were also expressed in all conditions tested, as revealed by RT-PCR (Figure 2b) and immunofluorescence (Figure 2a), respectively.
CMs cultured in adhesion displayed both spontaneous and electrically induced calcium transients lasting less than 1 second, typical of calcium cycling during contraction (Figure 2c), together with a diffuse intracellular distribution of cardiac-specific sarcoendoplasmic reticulum calcium-ATPase (Figure 2a), a key element of the calcium handling machinery needed for calcium reuptake after contraction.
Taken together, these results show that functionally differentiated CMs were derived from DMD, DYS-HAC, and healthy hiPS cells. The observed differences in terms of percentage of spontaneously contracting EBs and cTnT-positive CMs on the overall population (Table 1) can be due to the intrinsic variability related to the use of different hiPS cell lines and the efficiency of the cardiogenic protocol itself.
HAC-driven expression of dystrophin sequences originally deleted in the DMD patient
We then focused on the genetically corrected CMs, testing the HAC-mediated restoration of dystrophin expression.
DYS-HAC is the first vector carrying the whole dystrophin genomic locus, including all the associated regulatory elements.22 This potentially allows proper activation of the complex mechanism regulating dystrophin expression, for instance, the activities of seven different promoters driving transcription of tissue-specific isoforms and exon-skipping and exon-scrambling events, which are finely regulated in both development- and tissue-specific manner.1 The possibility to restore dystrophin expression in a tissue-specific manner, following native regulation mechanisms, makes DYS-HAC a promising tool for the treatment of DMD also at cardiac muscle level.
First, dystrophin expression was analyzed on healthy, DMD, and genetically corrected DYS-HAC hiPS cells during the cardiac differentiation procedure, by RT-PCR using specific primers designed to span exon–exon junctions localized inside the deleted genomic sequence of the DMD patient (from exon 4 to 43 of the muscle dystrophin isoform) (Figure 3a). mRNA from human heart and skeletal muscle was used as positive control.
Figure 3.

HAC-driven expression of dystrophin sequences originally deleted in DMD patients. (a) The genomic organization of the dystrophin gene: the gray vertical bars represent the exons; the green arrows indicate the promoters driving the expression of the different dystrophin isoforms within the gene1,2; primer pairs used to amplify exon–exon junctions inside the deleted region are indicated by red arrowheads, and those used to amplify specific isoforms are indicated by blue arrowheads. Exon number here reported refers to the muscle dystrophin isoform Dp427m. (b) RT-PCR of specific dystrophin sequences localized inside the patient with deletion of exons (Ex) 4–43. Primers were constructed to spam five different exon–exon junctions (Ex. J.). cDNA from human heart and skeletal muscle was used as positive control. Analyses were performed at three different stages of the differentiation process: undifferentiated colonies, differentiated EBs cultured in suspension (day 20), and adhered cells (day 24), for each hiPS cell lines (DMD, healthy, and DYS-HAC hiPS cells). cDNA from human tissues was used as positive control. cDNA, complementary DNA; DMD, Duchenne muscular dystrophy; DYS-HAC, human artificial chromosome carrying the whole dystrophin genomic sequence; EBs, embryoid bodies; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HAC, human artificial chromosome; hiPS cells, human induced pluripotent stem cells; RT-PCR, reverse transcriptase–polymerase chain reaction.
As expected, healthy hiPS cell–derived EBs displayed dystrophin expression both when cultured in suspension and after adhesion, whereas, as expected, in DMD hiPS cell–derived EBs, no dystrophin expression was observed in any condition (Figure 3b). In DYS-HAC hiPS cell–derived EBs, dystrophin expression was restored both when cultured in suspension and after adhesion. A positive result was obtained for each of the five exon junctions checked, distributed on the whole deleted genomic region.
HAC-driven expression of multiple dystrophin isoforms
In vivo, CMs are known to express the full-length muscle dystrophin isoform (Dp427m), together with other isoforms, such as the Dp260 (ref. 27) and Dp71, the smallest but multifunctional product of the DMD gene expressed in many tissues, including cardiac muscle. Dp71 has been shown to contribute to the proper clustering and anchoring of structural and signaling proteins to the plasma membrane and of nuclear envelope proteins to the inner nuclear membrane.28
The expression of different dystrophin isoforms such as Dp427m, Dp260, Dp140, and Dp71 was analyzed by RT-PCR during the cardiac differentiation procedure on healthy, DMD, and DYS-HAC hiPS cells (Figure 4a). mRNA from human tissues was used as positive control.
Figure 4.

HAC-driven expression of multiple dystrophin isoforms. (a) RT-PCR of dystrophin isoforms Dp427m, Dp260, Dp140, and Dp71 at three different stages of the differentiation process: undifferentiated colonies, differentiated EBs cultured in suspension (day 20), and adhered cells (day 24), for each hiPS cell lines (DMD, healthy, and DYS-HAC hiPS cells). cDNA from human tissues was used as positive control. (b) Real-time PCR for dystrophin isoforms present in cardiac tissue. Muscle-specific Dp427m is double checked with Dp427m-specific primer set spanning exons 1–3 and an all Dp427 isoform-specific primer set spanning exons 25–26. Data are presented as mean ± SD. cDNA, complementary DNA; DYS-HAC, human artificial chromosome carrying the whole dystrophin genomic sequence; EBs, embryoid bodies; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HAC, human artificial chromosome; hiPS cells, human induced pluripotent stem cells; RT-PCR, reverse transcriptase–polymerase chain reaction; DMD, Duchenne muscular dystrophy.
As expected, healthy hiPS cell–derived EBs (both in suspension and after adhesion) displayed the expression of all the four different dystrophin isoforms. The expression of isoform Dp140 indicates other cell types are present besides CM.29 In DMD hiPS cell–derived EBs, the expression of the Dp260 isoform, the promoter of which stands within the large genomic deletion (exons 4–43), was not observed, whereas isoforms Dp140 and Dp71 were still detectable as their promoters are downstream of the deleted area (intron 44 and intron 62, respectively). The amplicon relative to isoform Dp427m is still present, as the primers specific for this isoform are designed on the first transcribed exons (exons 1–3), upstream of the deletion, testifying a proper initiation of dystrophin transcription. On DYS-HAC hiPS cell–derived EBs (both in suspension and after adhesion), expression of all isoforms can be observed, notably with the restoration of Dp260 isoform transcript.
To assess the efficiency of HAC-driven recovery of dystrophin mRNA levels, we performed RT-PCR experiments targeted at the isoforms expressed in the cardiac tissue (Figure 4b). A set of primers targeting exons 25–26 identifying all Dp427 isoforms was used in addition to the Dp427m-specific one to assess the relative expression of Dp427 transcripts inside the deleted genomic region. Confirming the previous results of the RT-PCR, DMD hiPS cell–derived EBs did not display at all the expression of transcripts from inside the deleted genomic region, both in regard to Dp427 isoforms, the transcription of which is truncated after exon 4, and in regard to Dp260 isoform, the transcription of which cannot be initiated. The expression of the truncated Dp427m isoform and the short Dp71 isoform was variable among the different experiments, not reaching statistical significance (n = 3). Differentiated healthy and DYS-HAC hiPS cell–derived CMs displayed similar amounts of all transcripts tested, highlighting the proper function of the HAC in driving and regulating the transcription of different dystrophin isoforms.
Taken together, these analyses on mRNA transcripts show that, during the cardiac differentiation protocol, dystrophin expression is correctly restored by the DYS-HAC and multiple dystrophin isoforms are expressed.
HAC-mediated restoration of dystrophin protein expression and correct subcellular localization
In vivo, in physiological conditions, dystrophin is a key structural protein creating a bridge across the sarcolemma, which provides a flexible connection between the basal lamina of the extracellular matrix and the inner cytoskeleton. For this important role, dystrophin should be correctly folded and localized under the plasma membrane of skeletal and cardiac muscle cells.
For this reason, we verified the proper restoration of dystrophin expression at protein level and its subcellular localization by immunofluorescence and confocal microscopy. Analyses were performed 4 days after EB adhesion on substrate with a physiological stiffness of 15 kPa (Figure 5a).
Figure 5.
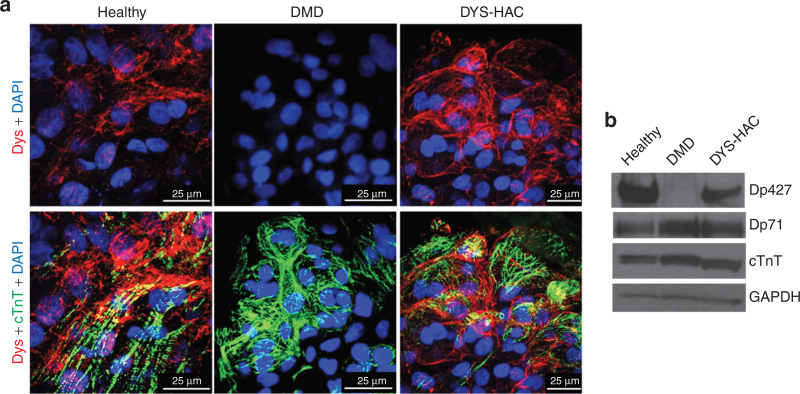
HAC-mediated restoration of dystrophin protein expression and correct subcellular localization. (a) Confocal microscopic analysis of dystrophin and cardiac troponin T expression on CMs obtained on day 24 of the differentiation procedure from DMD, healthy, and DYS-HAC hiPS cells. Nuclei are counterstained with DAPI. (b) Western blot analysis for dystrophin isoforms with a polyclonal antibody and for cardiac marker troponin T detected in two isoforms in the samples from human hiPS cell–derived cardiomyocytes. CMs, cardiomyocytes; DAPI, 4′,6-diamidino-2-phenylindole; DMD, Duchenne muscular dystrophy; DYS-HAC, human artificial chromosome carrying the whole dystrophin genomic sequence; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HAC, human artificial chromosome; hiPS cells, human induced pluripotent stem cells.
DYS-HAC hiPS cell–derived EBs displayed a clear dystrophin expression at membrane localization, drawing the boundaries of cTnT-positive CMs. A similar dystrophin staining was observed in healthy hiPS cell–derived EBs. On the other hand, as expected, in DMD hiPS cell–derived EBs, no dystrophin staining was observed. These results were confirmed by western blot analysis, in which full-length dystrophin was not observed in DMD hiPS cell–derived CMs, whereas its expression was perfectly restored in CMs corrected with the DYS-HAC (Figure 5b). Although much less abundant, other dystrophin isoforms were also detectable in the DYS-HAC–corrected cells (see Supplementary Figure S2).
These results demonstrate that the DYS-HAC restores the expression of dystrophin at protein level, which can correctly localize at membrane level in genetically corrected, hiPS cell–derived CMs.
Discussion
The derivation of genetically corrected, patient-specific hiPS cells has been recently shown to be a promising strategy for modeling genetic diseases. In particular, An et al.30 reported the derivation of hiPS cells from Huntington’s disease patients’ fibroblasts that were genetically corrected by an homologous recombination approach and differentiated into DARPP-32–positive neurons. Tedesco et al.25 reprogrammed fibroblasts and myoblasts from limb-girdle muscular dystrophy patients in hiPS cells and developed a protocol for the derivation of mesoangioblast-like cells that were genetically corrected in vitro with a lentiviral vector carrying the human α-sarcoglycan gene. The huge potential of hiPS cell technology has also been demonstrated in modeling human cardiovascular diseases. Some pioneering proof-of-concept studies on patients with inherited arrhythmogenic diseases, most notably different subtypes of long QT syndrome, demonstrated how hiPS cell–derived cardiac cells reproduce the clinical phenotype “in the petri dish.”14,31
In this work we derived, for the first time to our knowledge, human CMs (hCMs) from DMD patient–specific hiPS cells and genetically corrected hCMs from DYS-HAC–containing hiPS cells.
Different procedures for the cardiac differentiation of pluripotent stem cells have been reported in literature, mainly reproducing in vitro the processes of embryonic development through the stimulation with soluble cytokines, driving pluripotent stem cells toward early mesoderm specification until the derivation of cardiac progenitor cells.26,32,33 However, the major drawback of their application, both in vitro and in vivo, is their immature phenotype.
To study the restoration of dystrophin expression and its correct localization in cardiac cells, the derivation of hCMs characterized by a functionally and structurally mature phenotype is of paramount importance.
We developed an ad hoc optimized differentiation procedure integrating a cytokine-based EBs differentiation with a subsequent step of adhesion-dependent maturation. Increasing evidence demonstrate that adhesion and substrate sensing are key requirements for the process of muscle cell functional differentiation. In our previous work, we have shown that the development of sarcomeric structures of human striated muscles is influenced by the substrate on which the cells are cultured.34 Furthermore, in our group, it has been recently demonstrated that adhesion on substrate with a physiological stiffness promotes functional maturation of hCMs, allowing a shortening of calcium transients.36
Using the procedure developed, we derived hCMs presenting expression of cardiac-specific markers, a defined sarcomeric organization and calcium transients lasting less than 1 second. In these conditions, dystrophin has been observed to correctly localize at membrane level on both healthy and genetically corrected hCMs.
Certain variability has been observed in the expression of cardiac muscle markers along the differentiation process, for the different hiPS cell lines. This could be explained since, as widely reported in literature, hiPS cell lines derived from different cells and different clones can have a different ability to differentiate toward the cardiac lineage. In addition, it has been recently demonstrated that in vitro cultured DMD skeletal muscle cells display a delay in the appearance of typical myogenic markers.35 Similarly, we observed a delay in the expression of the late cardiac differentiation marker ventricular myosin light chain in DMD-derived cardiac cells, compared with the healthy and genetically corrected (DYS-HAC) ones. Further analyses could be performed to specifically address this issue in cardiac muscle.
hiPS cell–derived CMs represent a unique platform for testing in vitro the efficiency of the DYS-HAC in restoring a proper dystrophin expression at cardiac level, in a patient-specific manner.
DYS-HAC is a potential tool for use in DMD gene correction, and its ability to properly restore dystrophin expression on differentiated human cells has been recently reported.25 However, its efficiency in restoring different isoforms’ expression on differentiated hCMs has not been extensively investigated so far. For the first time, we demonstrated that DYS-HAC (i) does not hinder and is stably maintained during cardiac differentiation of hiPS cells; (ii) allows a proper dystrophin expression restoration during the cardiac differentiation procedure of hiPS cells (in particular, the expression of specific sequences deleted in the patient was observed); (iii) drives the transcription of multiple dystrophin isoforms in CMs at similar expression levels compared with healthy hiPS cell–derived CMs, in particular, the full-length muscle isoform Dp427m and the cardiac isoform Dp260, which are not expressed in the DMD patient with deletion of exons 4–43; and (iv) allows the recovery of full-length dystrophin protein that localizes properly at the cell membrane. These results highlight DYS-HAC as a potential tool for use in gene correction of DMD patient cells.
Finally, the coupling of DMD CMs with specific technologies for testing cardiac functionality, such as measurement of force generation or performance under stressed condition, will provide a unique platform for studying in vitro the pathogenesis of DMD-associated cardiac disease and its correction.
Materials and Methods
hiPS cell culturing and cardiac differentiation
hiPS cells, obtained as previously reported,23 were cultured on a feeder layer of mitomycin-C–inactivated murine embryonic fibroblasts. Composition of the culture medium was as follows: Dulbecco's modified Eagle medium/F12 (Life Technologies, Carlsbad, CA) containing 20% knockout serum (Life Technologies), 2 mmol/l of l-glutamine (Life Technologies), 0.1 mmol/l of nonessential amino acids (Life Technologies), 0.1 mmol/l of 2-mercaptoethanol (Life Technologies), 50 units and 50 mg/ml of penicillin and streptomycin (Life Technologies), and 4 ng/ml of basic fibroblast growth factor (Peprotech, Rocky Hill, NJ).
To maintain the pluripotent state for a high number of passages and avoid chromosomal aberrations, the colonies were passed as described below—usually once a week. hiPS cells were washed with phosphate-buffered saline (PBS) and treated with 1 ml of Collagenase, trypsin, knockout serum solution for 30 seconds. Collagenase, trypsin, knockout serum solution was prepared as follows: 5 ml of 2.5% trypsin (Life Technologies), 5 ml of 1 mg/ml collagenase IV (Sigma-Aldrich, St Louis, MO), 0.5 ml of 0.1 mol/l CaCl2 (Sigma-Aldrich), and 10 ml of knockout serum were all added to 30 ml of distilled water. The mechanical separation of colonies was performed using a cutting pipette through a stereomicroscope. The selected undifferentiated pieces were then replated for expansion onto dishes containing fresh murine embryonic fibroblast feeders or moved to ultralow adhesion plates to be cultured in suspension for EB generation.
The obtained EBs were differentiated in suspension using a protocol adapted from Kattman et al.26 Briefly, for EB formation, the detached and separated colonies were maintained for 24 hours in basal medium (StemPRO-34; Life Technologies), 2 mmol/l of l-glutamine (Life Technologies), 150 µg/ml of transferrin (Roche, Basel, Switzerland), 50 µg/ml of ascorbic acid (Sigma-Aldrich), 0.4 mmol/l of monothioglycerol (Sigma-Aldrich), 50 units and 50 mg/ml of penicillin and streptomycin (Life Technologies) supplemented with 10 ng/ml of human bone morphogenetic protein 4 (R&D Systems, Minneapolis, MN). From day 1 to day 4, EBs were cultured in basal medium with 10 ng/ml of human bone morphogenetic protein 4, 5 ng/ml of human basic fibroblast growth factor (R&D), and 6 ng/ml of hActivin A (R&D). From day 4 to day 8, the EB culture medium consisted of basal medium and 10 ng/ml of human vascular endothelial growth factor (R&D) and 150 ng/ml of human DICKKOPF-1 (R&D). Finally, from day 8 to day 14, the EB culture medium consisted of basal medium and 10 ng/ml of human vascular endothelial growth factor and 5 ng/ml of human basic fibroblast growth factor. Cultures were maintained in a 5% CO2, 5% O2, and 90% N2 environment for the first 16 days and then transferred to a 5% CO2 air environment.
The obtained EBs were maintained in suspension using the last medium described until day 20 and then seeded on hydrogel substrates with a physiological stiffness of 15 kPa (prepared as previously described)34,36,37, functionalized with 100 µg/ml of laminin (Becton Dickinson, Franklin Lakes, NJ), and cultured in these conditions for additional 4 days.
Immunofluorescence
A standard immunohistochemistry protocol was used. Briefly, cells were fixed with PBS containing 2% paraformaldehyde (Sigma-Aldrich) for 7 minutes, permeabilized with PBS containing 0.5% Triton X-100 (Sigma-Aldrich), and blocked in PBS containing 2% horse serum for 45 minutes, at room temperature. Primary antibodies were applied for 1 hour at 37 °C. Cells were washed in PBS (Life Technologies) and incubated with fluorescence-conjugated secondary antibodies against mouse, rabbit, or goat, depending on primary antibody used, for 45 minutes at 37 °C. Finally, nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich), and samples were mounted with Elvanol (Sigma-Aldrich) and viewed under Leica TCS SP5 fluorescence confocal microscope (Leica Microsystems, Wetzlar, Germany). Primary antibodies used were the following: mouse monoclonal anti-cTnT (Thermo Scientific, Waltham, MA; #MS-295-P; 1:100 dilution), mouse monoclonal anti-α-actinin (Sigma-Aldrich; #A7811; 1:100 dilution), rabbit polyclonal anti-dystrophin (Abcam, Cambridge, UK; #ab15277; 1:200 dilution), mouse monoclonal anti-Cx43 (Millipore; #MAB3067; 1:100 dilution), goat polyclonal anti-SERCA2a (Santa Cruz; #SC 8094; 1:200 dilution), and goat polyclonal anti-GATA4 (Santa Cruz; #SC 1237; 1:200 dilution). Secondary antibodies used were the following: goat anti-mouse (Life Technologies; #A11005 and #A11001; 1:200 dilution), goat anti-rabbit (Life Technologies; #A11012 and A1108; 1:200 dilution), and donkey anti-goat (Jackson ImmunoLab, West Grove, PA; #705-165-003; 1:300 dilution). All antibodies were diluted in 3% bovine serum albumin (Sigma-Aldrich).
Calcium measurements
Confocal calcium measurements were performed as previously reported in Martewicz et al.38 Briefly, CMs were loaded in serum-free Dulbecco’s modified Eagle medium containing 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 25 mmol/l (Life Technologies) and supplemented with 2,5 mmol/l of fluorescent calcium dye Fluo-4 AM (Life Technologies) for 20 minutes at 37 °C in the presence of 2 mmol/l of Pluronic F-127 (Life Technologies) and 20 mmol/l of sulfinpyrazone (Sigma-Aldrich), then incubated for additional 10 minutes at 37 °C without Fluo-4 AM, and added with 0.2 mmol/l of di-8-ANEPPS (Life Technologies). Cell dynamics were obtained in recording solution: NaCl, 125 mmol/l; KCl, 5 mmol/l; Na3PO4, 1 mmol/l; MgSO4, 1 mmol/l; 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 20 mmol/l; CaCl2, 2 mmol/l; and glucose, 5.5 mmol/l, to pH 7.4 with NaOH. Line scans were acquired with a Leica TCS SP5 fluorescence confocal microscope using a 63× oil immersion objective, with 488-nm Ar laser line as an excitation source and 400-Hz acquisition frequency. Line scans were then analyzed using ImageJ software (version number 1.46; National Institutes of Health, Bethesda, MD) to obtain calcium transient profile. For evaluating the calcium reuptake rate after contraction, the half-life of the calcium decay was considered. Half-life of the calcium decay was calculated by fitting a first-order exponential decay to the calcium reuptake phase of the calcium transient profile. For the calcium release phase, the time to peak value was calculated considering the time from baseline to a minimum of the second derivative of the calcium transient. All numerical data were manipulated with Origin 8.1 software (Origin Lab, Northampton, MA).
Reverse transcriptase–polymerase chain reaction
Total RNA from hiPS cell colonies and differentiated EBs was purified with RNeasy Mini Kit (Qiagen, Venlo, Netherlands), in accordance with the manufacturer’s instructions, or with TRIzol reagent (Life Technologies) and treated using a Turbo DNA-free kit (Life Technologies) to remove genomic DNA contamination. For the cardiac marker analyses, first-strand complementary DNA (cDNA) synthesis was performed using an oligo-(dT)20 primer and the cDNA Reverse Transcription Kit (Life Technologies). PCR was performed with cDNA using AmpliTaq Gold (Life Technologies). Amplifications were performed with an annealing temperature of 55 or 58 °C for 30–35 cycles. For the dystrophin isoform analyses, cDNA retrotranscription was carried out with High Capacity cDNA Reverse Transcription Kit (Life Technologies), followed by RT-PCR using Platinum Taq Polymerase (Life Technologies) with annealing temperature of 60 °C for 35 cycles. All the amplicons were resolved by electrophoresis on a 2% agarose gel, followed by staining with SYBR Safe Gel. Primer sequences are given in Table 2.
Table 2. Primer sequences.
| Forward primer | Reverse primer | |
|---|---|---|
| Dystrophin isoform Dp427m (exons 1–3) | TCGCTGCCTTGATATACACTTTTCA | GGTTCTCAATATGCTGCTTCCCA |
| Dystrophin isoform Dp260 | AGGAAGCTGCGAAATCTGTCTTAC | GGCAGACTGGATGCTCTGTTCA |
| Dystrophin isoform Dp140 | ACCGAAAGAGGTTTTTGCACACC | ACTGGCATCTGTTTTTGAGGATTGC |
| Dystrophin isoform Dp71 | CATGAGGGAACAGCTCAAAGGC | CAGTCTTCGGAGTTTCATGGCA |
| Dystrophin exon junctions 3–4 | CCTGACAGGGCAAAAACTGCCAA | TGTGTGGCTGACTGCTGGCAA |
| Dystrophin exon junctions 9–10 | CGGAGCCCATTTCCTTCACAGCATT | CCGGCCCTGATGGGCTGTCA |
| Dystrophin exon junction 22–23 | GACTCGGGGAATTGCAGGCTT | GGGCAGGCCATTCCTCCTTCA |
| Dystrophin exon junction 25–26 | GGCCTGCCCTTGGGGATTCA | TCTGGCATAGACCTGTTGGCACA |
| Dystrophin exon junction 34–35 | TGCCTGGGGAAAGGCTACTCA | GCAGTGGTCACCGCGGTTTG |
| NKX2.5 | GCGATTATGCAGCGTGCAATGAGT | AACATAAATACGGGTGGGTGCGTG |
| cTnT | TTCACCAAAGATCTGCTCCTCGCT | TTATTACTGGTGTGGAGTGGGTGTGG |
| MLC2v | ACATCATCACCCACGGAGAAGAGA | ATTGGAACATGGCCTCTGGATGGA |
| GAPDH RT-PCR | CCCCTTCATTGACCTCAACTACA | TTGCTGATGATCTTGAGGCTGT |
| GAPDH real-time PCR | GAAGGTGAAGGTCGGAGTCAAC | CAGAGTTAAAAGCAGCCCTGGT |
cTnT, cardiac troponin T; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MLC2v, ventricular myosin light chain; RT-PCR, reverse transcriptase–polymerase chain reaction.
Real-time PCR
Real-time PCR on cDNA retrotranscribed with High Capacity cDNA Reverse Transcription Kit was carried out with Power SYBR Green PCR Master Mix (Life Technologies) in a 7000 System thermal cycler platform (Life Technologies). Annealing temperature for all primer sets was 60 °C (sequences reported in Table 2). Relative amount of transcripts was calculated with Pfaffl method relative to healthy hiPS cell–derived EB expression levels. Amplification efficiency for all primer sets was >1.9.
Western blot analyses
Detection of dystrophin (antibody 1:500; overnight + 4 °C; Abcam; #ab15277), cTnT (antibody 1:500; 60 minutes at room temperature; Thermoscientific #MS-295-P), and glyceraldehyde 3-phosphate dehydrogenase (antibody 1:2,000; 60 minutes at room temperature; Abcam; #ab8245) was carried out after protein lysates were resolved in a NuPAGE 3–8% Tris-acetate polyacrylamide gel (Life Technologies) and transferred for 6 hours at 4°C on a polyvinylidene difluoride membrane (Life Technologies) with a BioRad cassette. Detection was performed with Novex ECL Kit (Life Technologies). Anti-rabbit (Life Technologies) and anti-mouse (BioRad) secondary horseradish peroxidase–conjugated antibodies were used.
Acknowledgments
This work was supported by: Progetti di eccellenza CARIPARO grants to NE, Città della Speranza to MS, DIRPRGR10 Progetti Giovani Studiosi 2010 University of Padova Department of Industrial Engineering to ES, Fondazione Ing. Aldo Gini to SZ. We thank Motonobu Katoh (Department of Biomedical Science, Institute of Regenerative Medicine and Biofunction, Tottori University, Yonago, Japan) for helping with experimental work.
The authors declare no conflict of interest.
References
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve. 2011;44:8–19. doi: 10.1002/mus.22097. [DOI] [PubMed] [Google Scholar]
- Fayssoil A, Nardi O, Orlikowski D, Annane D. Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail Rev. 2010;15:103–107. doi: 10.1007/s10741-009-9156-8. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang YS, Schaniel C, Lee DF. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32:952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Barad L, Zeevi-Levin N, Shick R, Shtrichman R, Lorber A. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to ß-adrenergic stimulation. J Cell Mol Med. 2012;16:468–482. doi: 10.1111/j.1582-4934.2011.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Wei H, Lu J, Ho S, Zhang G, Sun X. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:1122–1133. doi: 10.1093/eurheartj/ehs226. [DOI] [PubMed] [Google Scholar]
- Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi-Levin N, Itskovitz-Eldor J, Binah O. Cardiomyocytes derived from human pluripotent stem cells for drug screening. Pharmacol Ther. 2012;134:180–188. doi: 10.1016/j.pharmthera.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322–325. doi: 10.1038/nature11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Duan D. Progress in gene therapy of dystrophic heart disease. Gene Ther. 2012;19:678–685. doi: 10.1038/gt.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QL, Yokota T, Takeda S, Garcia L, Muntoni F, Partridge T. The status of exon skipping as a therapeutic approach to duchenne muscular dystrophy. Mol Ther. 2011;19:9–15. doi: 10.1038/mt.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick B, Yue Y, Long C, Duan D. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res. 2008;102:121–130. doi: 10.1161/CIRCRESAHA.107.162982. [DOI] [PubMed] [Google Scholar]
- Janssen PM, Hiranandani N, Mays TA, Rafael-Fortney JA. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2005;289:H2373–H2378. doi: 10.1152/ajpheart.00448.2005. [DOI] [PubMed] [Google Scholar]
- Shin JH, Bostick B, Yue Y, Hajjar R, Duan D. SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J Transl Med. 2011;9:132. doi: 10.1186/1479-5876-9-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, Hoshiya H, Takiguchi M, Abe S, Iida Y, Osaki M. Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Ther. 2011;18:384–393. doi: 10.1038/gt.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiya H, Kazuki Y, Abe S, Takiguchi M, Kajitani N, Watanabe Y. A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin gene. Mol Ther. 2009;17:309–317. doi: 10.1038/mt.2008.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol Ther. 2010;18:386–393. doi: 10.1038/mt.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedesco FS, Hoshiya H, D’Antona G, Gerli MF, Messina G, Antonini S. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci Transl Med. 2011;3:96ra78. doi: 10.1126/scitranslmed.3002342. [DOI] [PubMed] [Google Scholar]
- Tedesco FS, Gerli MF, Perani L, Benedetti S, Ungaro F, Cassano M. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med. 2012;4:140ra89. doi: 10.1126/scitranslmed.3003541. [DOI] [PubMed] [Google Scholar]
- Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- D’Souza VN, Nguyen TM, Morris GE, Karges W, Pillers DA, Ray PN. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum Mol Genet. 1995;4:837–842. doi: 10.1093/hmg/4.5.837. [DOI] [PubMed] [Google Scholar]
- Tadayoni R, Rendon A, Soria-Jasso LE, Cisneros B. Dystrophin Dp71: the smallest but multifunctional product of the Duchenne muscular dystrophy gene. Mol Neurobiol. 2012;45:43–60. doi: 10.1007/s12035-011-8218-9. [DOI] [PubMed] [Google Scholar]
- Shiba Y, Hauch KD, Laflamme MA. Cardiac applications for human pluripotent stem cells. Curr Pharm Des. 2009;15:2791–2806. doi: 10.2174/138161209788923804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11:253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Wei H, Ma D, Sun X, Liew R. Clinical applications of patient-specific induced pluripotent stem cells in cardiovascular medicine. Heart. 2012;98:443–449. doi: 10.1136/heartjnl-2011-301317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–2740. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Serena E, Zatti S, Reghelin E, Pasut A, Cimetta E, Elvassore N. Soft substrates drive optimal differentiation of human healthy and dystrophic myotubes. Integr Biol (Camb) 2010;2:193–201. doi: 10.1039/b921401a. [DOI] [PubMed] [Google Scholar]
- Martone J, De Angelis FG, Bozzoni I. U1 snRNA as an effective vector for stable expression of antisense molecules and for the inhibition of the splicing reaction. Methods Mol Biol. 2012;867:239–257. doi: 10.1007/978-1-61779-767-5_16. [DOI] [PubMed] [Google Scholar]
- Serena E, Cimetta E, Zatti S, Zaglia T, Zagallo M, Keller G. Micro-arrayed human embryonic stem cells-derived cardiomyocytes for in vitro functional assay. PLoS ONE. 2012;7:e48483. doi: 10.1371/journal.pone.0048483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatti S, Zoso A, Serena E, Luni C, Cimetta E, Elvassore N. Micropatterning topology on soft substrates affects myoblast proliferation and differentiation. Langmuir. 2012;28:2718–2726. doi: 10.1021/la204776e. [DOI] [PubMed] [Google Scholar]
- Martewicz S, Michielin F, Serena E, Zambon A, Mongillo M, Elvassore N. Reversible alteration of calcium dynamics in cardiomyocytes during acute hypoxia transient in a microfluidic platform. Integr Biol (Camb) 2012;4:153–164. doi: 10.1039/c1ib00087j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.