

Abstract
Gene transfer has therapeutic potential for treating HIV-1 infection by generating cells that are resistant to the virus. We have engineered a novel self-inactivating lentiviral vector, LVsh5/C46, using two viral-entry inhibitors to block early steps of HIV-1 cycle. The LVsh5/C46 vector encodes a short hairpin RNA (shRNA) for downregulation of CCR5, in combination with the HIV-1 fusion inhibitor, C46. We demonstrate here the effective delivery of LVsh5/C46 to human T cell lines, peripheral blood mononuclear cells, primary CD4+ T lymphocytes, and CD34+ hematopoietic stem/progenitor cells (HSPC). CCR5-targeted shRNA (sh5) and C46 peptide were stably expressed in the target cells and were able to effectively protect gene-modified cells against infection with CCR5- and CXCR4-tropic strains of HIV-1. LVsh5/C46 treatment was nontoxic as assessed by cell growth and viability, was noninflammatory, and had no adverse effect on HSPC differentiation. LVsh5/C46 could be produced at a scale sufficient for clinical development and resulted in active viral particles with very low mutagenic potential and the absence of replication-competent lentivirus. Based on these in vitro results, plus additional in vivo safety and efficacy data, LVsh5/C46 is now being tested in a phase 1/2 clinical trial for the treatment of HIV-1 disease.
Introduction
HIV-1 continues to be a major global public health issue, having claimed more than 25 million lives over the past three decades. It is estimated that 34 million individuals around the world are currently living with HIV-1. Standard treatment for HIV-1 infection is highly active antiretroviral therapy, which can reduce plasma viral loads to undetectable levels for years at a time.1–3 During this time, however, HIV-1 persists in various cellular reservoirs, and discontinuation of antiretroviral therapy can lead to rapid rebound of viral loads causing renewed disease progression toward AIDS.4–6 While antiretroviral therapy is effective at reducing viral load and maintaining CD4+ T-lymphocyte counts, strict adherence by the individual is required to maintain effectiveness; however, side effects of antiretroviral therapy can be severe, long-term complications can develop, and HIV-1 resistance to the antiretroviral regimen can also develop.7–10
A promising alternative approach is cell-delivered gene therapy, in which anti–HIV-1 agents are delivered into target cells with the intention to interfere with the HIV-1 life cycle. Infusion of the genetically engineered HIV-1–resistant cells to patients has the potential to control HIV-1 infection, slow disease progression, repair damage to the immune system, and reduce reservoirs of infected and latently infected cells.11–13 Other approaches that have been tested include vaccines, immunotherapy, adoptive immunotherapy, and vectored immunoprophylaxis. HIV-1 gene therapy has been applied targeting early life cycle steps before integration, such as HIV-1 binding, fusion/entry, and reverse transcription, or later steps, including integration, transcription, translation, maturation, or virion assembly.12 Some of these approaches were tested in clinical trials using gene agents such as silencing dominant negative rev, env antisense RNA, ribozymes, Rev response element (RRE) decoy, fusion inhibitors, short hairpin RNA, and zinc finger nucleases.12–14
One promising strategy of preventing HIV-1 entry is based on suppression of the HIV-1 coreceptor, C-C chemokine receptor type 5 (CCR5). Genetic and molecular studies on human populations have demonstrated that individuals homozygous for a defective CCR5 gene, CCR5∆32, are protected from HIV-1 infection,15–18 and heterozygous individuals with a 50% reduction in the expression level of CCR5 on the cell surface have a substantially reduced rate of disease progression.19,20 Homozygous CCR5∆32 is a stable genetic trait with a frequency of 1.4% in the Caucasian population.21 These individuals are healthy apart from the potential for increased pathogenicity of West Nile Virus infection.22
A functional cure for HIV-1 infection has been demonstrated recently in the “Berlin patient” case, where a HIV-1–positive individual, with concurrent acute myeloid leukemia, was treated by transplant of homozygous CCR5Δ32 allogeneic hematopoietic stem/progenitor cells (HSPC).23 Reconstitution of the immune system with cells protected from HIV-1 infection led to substantial attenuation of HIV-1 replication and an increase in CD4+ T-cell counts. The CCR5∆32 donor cells nearly completely replaced the recipient cells within 61 days, and the patient’s viral load has remained undetectable in the absence of antiretroviral therapy.24 However, due to the low prevalence of homozygous CCR5∆32 genotype and limited availability of donors, more practical approaches are currently being sought. Blocking virus–CCR5 interaction by inhibiting or eliminating CCR5 expression is being investigated by a number of groups that include the use of ribozymes directed to CCR525–28, single-chain intrabodies,27,29 RNA interference,30–37 and zinc finger nuclease.38–40
A specific short hairpin RNA to CCR5 was previously demonstrated to effectively inhibit CCR5 expression and thereby protect primary human CD4+ T lymphocytes from CCR5-tropic HIV-1 infection in culture.31,41 Expression of this potent anti-CCR5 shRNA (CCR5 shRNA1005, or here termed sh5) was subsequently optimized using the human H1 promoter in a lentiviral vector to stably inhibit HIV-1 replication.42 The H1-CCR5 shRNA 1005 vector was shown to be noncytotoxic and effective in stable downregulation of CCR5 in human primary peripheral blood mononuclear cells (PMBCs) in vitro,42 and in vivo using the humanized bone marrow–liver–thymus (BLT) mouse model36 as well as in nonhuman primates introduced through hematopoietic stem cell transplant.41
C46 is an HIV-1 entry inhibitor derived from the C-terminal heptad repeat of HIV-1 gp41 modified to be expressed on the cell surface. C46, like other gp41-derived C peptides, blocks HIV-1 fusion to the cellular membrane by interacting with the N-terminal coiled-coil domain of the HIV-1 gp41 intermediate structure and preventing the six-helix bundle formation. In vitro studies have shown that membrane-anchored C46 protein effectively protects cells against a broad range of HIV-1 isolates by blocking entry of the virus to the target cells.43–46 The safety of C46 has been tested previously in a phase 1 clinical trial in which autologous T cells, transduced with a retroviral vector expressing C46, were infused into HIV-1–positive patients without adverse effects.47
It is well established that HIV-1 can rapidly develop resistance to monotherapy by emergence of point mutations in virus variants, whereas combined therapy shows better clinical outcome.10,48–50 In patients treated with CCR5 inhibitors, R5 variants of HIV-1 can evolve to use the blocked receptor. In addition, there are X4-tropic strains of HIV-1 that use the alternate coreceptor, CXCR451–53. The role of these X4 viruses in HIV-1 pathogenesis is as yet unclear54–57; however, effective genetic therapeutic applications for HIV-1 infection will likely require combinations of multiple reagents directed against HIV-1.
In the present study, we have developed a self-inactivating (SIN) lentiviral vector construct for gene transfer, LVsh5/C46, to express a combination of two anti–HIV-1 genes: sh5, an shRNA to the HIV-1 coreceptor CCR5, and C46, the antiviral fusion inhibitor peptide. We assessed expression of the anti–HIV-1 transgenes in cell culture and any impact on viability or phenotype and efficacy of inhibiting HIV-1 infection. We show here the ability of LVsh5/C46-modified cells to stably downregulate CCR5 and express C46. LVsh5/C46 was well tolerated when introduced into hematopoietic cells based on their viability, ability to proliferate, differentiate, and retain their normal phenotype. Moreover, we show that treatment of hematopoietic cells with LVsh5/C46 provides protection from the establishment of HIV-1 infection and replication from multiple strains, clades, and tropisms of HIV-1.
Results
Engineering and production of LVsh5/C46 vector
To construct the LVsh5/C46 plasmid, we utilized FG12, a SIN lentiviral plasmid that is based on the HIV-1 backbone with modified 5′ and 3′ HIV-1 long terminal repeats.31 First, the HIV-1 fusion inhibitor C4643 was introduced into the FG12 plasmid downstream of the human Ubiquitin C (UbC) promoter, replacing the enhanced green fluorescent protein sequence. Next, a previously characterized shRNA against CCR5 (1005), driven by the human H1 RNA polymerase III promoter,41 was inserted upstream of the UbC promoter (Figure 1a). The two viral-entry inhibitors, incorporated into a single construct, were then tested in vitro for their ability to be expressed in target cells, their effect on the treated cells, and their potency in suppressing HIV-1 replication.
Figure 1.

Expression of the dual-therapeutic anti–HIV-1 genes in T cell lines and in human primary cultures. (a) Schematic representation of LVsh5/C46 lentiviral vector. The CCR5 shRNA (1005)/FG12 vector (previously described)41 was digested by NdeI/XhoI to excise a fragment containing CCR5 shRNA (sh5) under the human H1 RNA polymerase III promoter. The insert was cloned into the same sites in an FG11F vector encoding membrane-anchored C4643, under the Ubiquitin C promoter (UbC). Other components of the vector include 5′ and 3′ modified HIV-1 long terminal repeats (LTRs), a central polypurine tract (cPPT), and a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE). (b) PBMCs and Molt4/CCR5 cells were transduced with LVsh5/C46 at MOI of 1 and 2, respectively, in triplicate, and expression of sh5 and C46 was assessed by flow cytometry. CCR5 shRNA (sh5) expression was demonstrated by downregulation of CCR5 expression evidenced by CD195 staining. Cell-surface expression of C46 was detected by staining with 2F5 antibody. (c) The T cell line CEM.NKR.CCR5 was transduced with LVsh5/C46, and the expression of CCR5 and C46 was similarly assessed over 8 weeks in culture by flow cytometry. (d, e) Quantification of integrated LVsh5/C46 DNA and LVsh5/C46-mediated C46 mRNA synthesis in transduced PBMCs. Cells were treated with LVsh5/C46 at MOIs of 1, 5, and 10 (in duplicate), resulting in transduction efficiencies of 35, 62.5, and 74% (respectively) at 4 days postinfection as assessed by flow cytometry (data not shown). Genomic DNA and RNA were isolated at 8 days posttransduction. C46 DNA copy number per cell was determined by quantitative PCR and was normalized to β-globin (d). C46 RNA transcript levels were measured by RT-qPCR using C46 primers and were normalized to β2-microglobulin mRNA as a measure of relative C46 expression (e). PBMC, peripheral blood mononuclear cell; shRNA, short hairpin RNA.
Small-scale production of LVsh5/C46 viral vector was used during the initial characterization studies followed by preclinical testing of LVsh5/C46 preparations generated using methodology and procedures consistent with good laboratory practice (GLP) and good manufacturing practice (GMP). Comprehensive testing for purity, safety, and functionality was conducted for a 251 ml pilot lot of GLP vector and two large-scale batches (25 l each) of GMP-grade vector. Small-scale virus production routinely yielded a titer of 3–5 × 106 infectious viral particles per milliliter (ivp/ml), whereas GLP grade and GMP-grade productions of LVsh5/C46 vector yielded 8 × 107 ivp/ml and 1–2 × 108 ivp/ml, respectively. LVsh5/C46 virus particles were introduced into various cell lines, PBMCs, CD4+ T lymphocytes, and hematopoietic stem cells for detailed preclinical characterization.
Expression of LVsh5/C46 in hematopoietic cells
Gene delivery of the dual anti–HIV-1 agents was first assessed in target cells transduced with small-scale vector preparations of LVsh5/C46. PBMCs and the CCR5-expressing T cell line Molt4/CCR5 were transduced with LVsh5/C46, and cell-surface expression of C46 and CCR5 was assessed by fluorescence-activated cell sorting (FACS) after 4 days. At a multiplicity of infection (MOI) of 1, 39.35% of PBMCs (33.9 + 5.45%) were transduced as determined by expression of the C46 peptide (Figure 1b). At this MOI, CCR5 expression was reduced from 58.3% in control cells (56.3 + 2.03%) to 19.65% in LVsh5/C46 transduced cells (14.2 + 5.45%) (Figure 1b). When Molt4/CCR5 cells were transduced at an MOI of 2, 92.9% of the cells expressed C46 peptide, and CCR5 expression was decreased from 86.98 to 17.94% posttransduction (Figure 1b). These results demonstrated successful delivery of LVsh5/C46 vector into cells, ability of the modified cells to express sh5 short hairpin RNA and C46 peptide, and successful downregulation of CCR5.
To assess longer-term stability of the integrated LVsh5/C46 in the modified cells, the T cell line CEM.NKR.CCR5 was transduced with LVsh5/C46, and a time course of CCR5 and C46 expression was analyzed by flow cytometry. As shown in Figure 1c, after 2 months in culture, 68.5% of cells maintained C46 peptide expression (48.8 + 19.7%), and CCR5 expression was decreased from 98.5% on day 0 to 34.1% at week 8, indicating that LVsh5/C46 vector persisted in the cells over time and effectively expressed the therapeutic anti–HIV-1 genes.
To quantify LVsh5/C46 gene transfer, PBMCs were transduced with increasing doses of LVsh5/C46 followed by evaluation of vector copy number and RNA transcript synthesis in the transduced cells. Transduction at MOI of 1 resulted in one copy of LVsh5/C46 per cell, while higher doses of LVsh5/C46 vector (MOIs of 5 and 10) led to an average of 1.5–2 copies of LVsh5/C46 viral DNA per cell (Figure 1d). Furthermore, we observed a dose-dependent increase in C46 RNA synthesis in response to elevated LVsh5/C46 dose as quantified by reverse transcription–quantitative PCR (RT-qPCR) (Figure 1e). This data demonstrated the ability to define the preferred dose of LVsh5/C46 vector delivered into human primary cells by correlating MOI with the integrated vector copy number and the RNA transcripts of C46 transgene.
To further evaluate the therapeutic potential of LVsh5/C46, we purified CD4+ T lymphocytes and CD34+ HSPC and demonstrated that the intended target cells were genetically modified in vitro to produce the anti–HIV-1 gene agents. Purification of CD4+ T lymphocytes was achieved by isolation of PBMCs, followed by a CD8 depletion step and CD3/CD28 stimulation and activation. The purification process yielded purity of 88.6% CD4+ cells (Figure 2a, lower right panel). Human CD34+ HSPC were isolated from granulocyte colony-stimulating factor mobilized peripheral blood using CliniMACS with CD34-microbeads as positive selection, which yielded purity of over 99% (Figure 2b). Purified CD4+ T lymphocytes and CD34+ HSPC cells were then treated with LVsh5/C46 vector at increasing MOIs (0.5–10), and the cell-surface C46 expression was determined by FACS analysis. A dose-dependent increase in the number of cells expressing C46 was observed in response to increasing doses of LVsh5/C46 (Figure 2c,d). Transduction efficiency in CD4+ cells ranged between 10 and 50% and between 2 and 15% in CD34+ cells. The doses of LVsh5/C46 chosen for the subsequent studies were MOI of 1 for CD4+ transduction and MOI of 5 for CD34+ HSPC. In large-scale experiments using GMP-grade LVsh5/C46 vector, improved transduction efficiencies were observed. Over 40% transduction efficiency was obtained in CD4+ T lymphocytes transduced at MOI of 1 and in CD34+ HSPC transduced at MOI of 5 (Figure 2e).
Figure 2.

Introducing LVsh5/C46 vector into target cells. (a) Human CD4+ T lymphocytes were isolated from PBMCs by CD8 depletion followed by selection and expansion using CD3/CD28 beads. Cells were stained with CD4, CD3, and CD8 antibodies and analyzed by flow cytometry. Purified CD4+ T-lymphocyte cells are shown as CD4+/CD3+/CD8− fraction in the lower right panel. (b) CD34+ HSPC were isolated from G-CSF mobilized peripheral blood, using CD34+ microbeads. Positive fraction was stained with CD34 antibody and analyzed by flow cytometry. (c, d) Purified cells were treated with LVsh5/C46 at the increasing doses (MOIs as indicated), in triplicate, and the percentage of (c) C46-expressing CD4+ cells and (d) CD34+ cells was determined by flow cytometry. (e) Purified CD4+ T lymphocytes and CD34+ HSPC cells were treated with GMP-grade LVsh5/C46 at MOI of 1 and 5, respectively, and LVsh5/C46 transduction was analyzed by flow cytometry, measuring the percentage of cells expressing cell-surface C46. G-CSF, granulocyte colony-stimulating factor; GMP, good manufacturing practice; HSPC, hematopoietic stem/progenitor cell; MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cell.
To further evaluate the degree of gene transfer in the transduced cells, CD4+ and CD34+ cells were treated with GMP-grade LVsh5/C46 at MOIs of 1 and 5, respectively. Genomic DNA was extracted, and quantitative PCR was performed to determine C46 copy number per cell. As shown in Figure 3a, an average of 2–2.5 copies of integrated LVsh5/C46 vector DNA was observed in CD4+ and CD34+ transduced cells. Expression of CCR5 shRNA (sh5) was also detected in the transduced CD4+ and CD34+ using RT-qPCR (Figure 3b). Taken together, these results validate the ability of LVsh5/C46 to genetically modify hematopoietic target cells, by stable integration and sustained expression of C46 and sh5.
Figure 3.
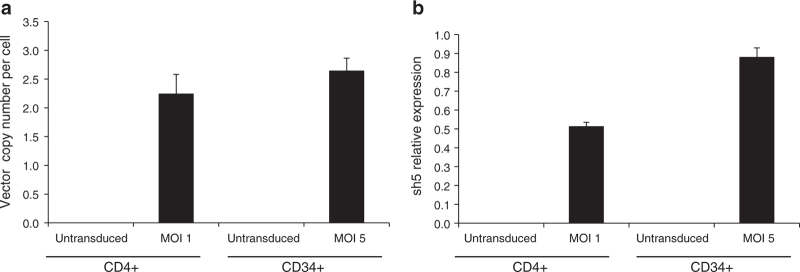
LVsh5/C46 genomic DNA integration and LVsh5/C46-mediated shRNA expression in hematopoietic cells. CD4+ T lymphocytes and CD34+ HSPC were transduced with LVsh5/C46 at MOIs of 1 and 5, respectively. (a) Genomic DNA was isolated from the transduced cells, and C46 DNA copy number normalized to β-globin was determined by quantitative PCR. (b) RNA was extracted from the transduced cells, and CCR5 shRNA transcript level was determined by RT-qPCR using sh5 primers and was normalized to the expression of RNU38B microRNA. HSPC, hematopoietic stem/progenitor cell; MOI, multiplicity of infection; RT-qPCR, real-time quantitative PCR; shRNA, short hairpin RNA.
LVsh5/C46-modified HSPC retain their full differential capacity
To determine whether CD34+ HSPC transduced with LVsh5/C46 could maintain their differentiation capacity, colony-forming assays were performed. CD34+ cells were transduced with a high dose of LVsh5/C46 (MOI of 10), and after 2 weeks in methylcellulose culture, the colony-forming units (CFU) were enumerated and characterized. Quantification of colonies of erythroid (CFU-E), myeloid (CFU-GM), and multiple lineage origin (CFU-GEMM) generated from LVsh5/C46-transduced cells showed no significant difference compared to untransduced cells (Figure 4a). Similar results were obtained when HSPC were transduced with either singular anti–HIV-1 genes (C46 or sh5) or the dual agents (LVsh5/C46). Transduced cells generated comparable numbers of differentiated colonies, with no obvious effects of either C46 or sh5 expression (Figure 4b–d). These findings demonstrate that LVsh5/C46-modified HSPC retain their capacity for multilineage hematopoietic differentiation in vitro, without lineage skewing, even at a relatively high dose of LVsh5/C46.
Figure 4.

LVsh5/C46-modified CD34+ HSPC maintain their multilineage hematopoietic differentiation potential. CD34+ HSPC from three healthy donors were transduced with the indicated lentiviral vectors, and 2 weeks posttransduction, methylcellulose colony-forming unit (CFU) assays were performed. Colonies were enumerated for CFU erythrocyte (CFU-E); CFU granulocyte/monocyte (CFU-GM), and CFU granulocyte/erythrocyte/monocyte/megakaryocyte (CFU-GEMM) and compared to untransduced HSPC. (a) CD34+ cells were transduced with LVsh5/C46 vector at MOI of 10, and the relative colony counts were plotted against untransduced cells. (b–d) CD34+ cells were transduced with lentiviral vectors expressing sh5 alone, sh5 fused to enhanced green fluorescent protein reporter gene, C46 alone, or LVsh5/C46 (sh5/C46), and CFU assays were performed. Colonies from the lineages (b) CFU-GM, (c) CFU-E, and (e) CFU-GEMM were scored, and the relative colony counts were plotted against untransduced cells. HSPC, hematopoietic stem/progenitor cell; MOI, multiplicity of infection.
LVsh5/C46-modified cells retain their normal phenotype
To investigate the effect of LVsh5/C46 on the phenotype of the treated cells, PBMCs were treated with LVsh5/C46 at MOIs of 1 and 10, and analyzed after 7–8 days in culture. We first determined whether apoptosis was induced in LVsh5/C46-modified cells. Similar activity of Caspase 3/7 was found in transduced PBMCs compared to untransduced cells, at low and high MOIs of LVsh5/C46, indicating that neither LVsh5/C46 transduction nor expression of sh5 or C46 induced programmed cell death (Figure 5a). We then assessed the influence of LVsh5/C46 on cell growth as measured by the number of metabolically active cells and observed comparable activities in untreated and LVsh5/C46-treated cells (Figure 5b). In addition, PBMCs were treated with LVsh5/C46 or with lentiviral vectors expressing the single genes (sh5 or C46), and cell viability was assessed and enumerated for a period of 12 days (see Supplementary Figure S1a,b). Proliferation was found to be similar between transduced and untransduced cells with no impact on viability.
Figure 5.

LVsh5/C46 vector does not induce deleterious effect on cells. PBMCs from three to five healthy donors were transduced with LVsh5/C46 (sh5/C46) at MOIs of 1 or 10 or and were compared to untransduced cells and to cells transduced with a control lentivirus vector encoding eGFP (enhanced green fluorescent protein) at MOI of 1. In different experiments, transduction efficiency was determined by flow cytometry to be 15–44% at an MOI of 1 and 36–64% at an MOI of 10. Cells were assessed for apoptosis, proliferation, and inflammation; results were normalized to untransduced cells; and Student’s t-test was performed. (a) Apoptosis was not induced in LVsh5/C46-transduced PBMCs. Caspase 3/7 activities were assessed 7 days posttransduction by luminescence assay (n = 3). Staurosporine was used as a positive control. (b) LVsh5/C46 did not alter viability or proliferative capacity of modified PBMCs. Cells were cultured for 7 days, and proliferation rate was assessed (n = 5). IL-2 was used as a positive control. (c–e) Transduced PBMCs were cultured for 8 days, and the level of the cytokines (c) interferon-γ (n = 3), (d) interleukin-6 (n = 4), and (e) TNF-α (n = 4) was determined. Cells treated with PHA were used as positive controls. *P < 0.05 compared to untransduced cells. **P < 0.01 compared to untransduced cells. MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cell; TNF, tumor necrosis factor.
To further examine the cellular response to LVsh5/C46, the levels of the cytokines interferon γ, interleukin-6, and tumor necrosis factor α were measured in transduced PBMCs and control cells. As shown in Figure 5c–e, there were no significant differences in the intracellular production of these proinflammatory cytokines between LVsh5/C46-treated PBMCs and untreated cells, indicating that inflammatory signaling was not activated by either viral transduction or LVsh5/C46-mediated expression of sh5 or C46.
Stability of LVsh5/C46 vector in transduced cells
To evaluate the stability of the viral vector in modified cells, VERO cells (monkey kidney epithelial) were transduced with LVsh5/C46, and the structure and integrity of the integrated vector was evaluated. Genomic DNA extracted from the transduced cells and subjected to southern blot analysis revealed two major forms of the integrated LVsh5/C46 DNA: a nonspliced 3.8 kb viral vector and the anticipated spliced form of 3 kb, due to splicing of the UbC intron, as described previously58,59 (see Supplementary Figure S2a,b). Sequencing analysis of the 3 kb band confirmed splicing event within the UbC region (data not shown). Apart from the expected splicing, the overall structural organization of the integrated LVsh5/C46 vector was kept intact and was genetically stable, with no rearrangements, duplications, insertions, or unexpected deletions.
Low risk of mutagenic potential in LVsh5/C46-modified hematopoietic cells
To assess the mutagenic potential of LVsh5/C46, in vitro immortalization assays were performed. Primary murine hematopoietic cells (Lin−) were cultured in the presence of LVsh5/C46 at MOIs of 20, 40, and 80, and the incidence of cell transformation was calculated as replating frequency per vector copy number. LVsh5/C46 vector was compared to two positive control vectors shown previously to induce in vitro immortalization.60 lv-SF, a lentiviral vector in SIN configuration under the control of a strong internal retroviral promoter of the spleen focus forming virus, and RSF91, a gamma-retroviral vector with internal or long terminal repeat– contained spleen focus forming virus promoter sequences. A mock infected sample cultured without a viral vector served as a measure of spontaneous immortalization and routinely scored negative. In three independent experiments, transformation frequency per vector copy number in cells treated with LVsh5/C46 vector was strongly reduced compared to positive controls (see Supplementary Figure S3). These results demonstrate a significantly reduced risk of insertional mutagenesis and genotoxicity.
No evidence of replication-competent lentivirus in LVsh5/C46-transduced cells
To further evaluate the safety of LVsh5/C46 vector as a potential therapeutic agent, the risk of replication-competent lentivirus (RCL) development was analyzed. LVsh5/C46 is a SIN lentiviral vector, in which modifications to the HIV-1 long terminal repeats significantly reduce the likelihood that RCL will develop. RCL testing has been performed on samples of LVsh5/C46 viral production batches (large-scale GLP and GMP). Five percent of the total batch volumes were used to inoculate C8166-45 cells, a highly infectable cell line.61 After a culturing period of a minimum of 21 days to allow potential amplification of any RCL, the supernatants were collected and were incubated with fresh permissive cells. Supernatants were collected for p24 ELISA to detect HIV-1 capsid protein, and genomic DNA was used for psi-gag PCR to detect recombination. Results from all batches tested showed no detectable quantities of p24 antigen or HIV-1 DNA, hence, confirming the absence of RCL in the LVsh5/C46-modified cells (see Supplementary Table S1). Moreover, in a separate RCL assay, in which postproduction HEK293T cells were cocultured with the C8166-45 cell line, no RCL was detected in any of the LVsh5/C46 test samples, as indicated by the absence of detectable quantities of p24 protein and HIV-1 psi-gag recombination (see Supplementary Table S1). These results demonstrate that the likelihood of RCL development using clinical grade of LVsh5/C46 lentiviral vector is extremely low.
Modification of T cell lines with LVsh5/C46 effectively inhibits HIV-1 replication
The ability of the LVsh5/C46 vector to confer cellular resistance from HIV-1 infection was evaluated by conducting HIV-1 challenge assays in vitro. First, the T cell line Molt4/CCR5 was transduced with LVsh5/C46 at MOI of 5 followed by infection with CCR5 (R5)-tropic HIV-1 strain BaL or CXCR4 (X4)-tropic strain NL4-3. Two weeks postinfection, the appearance of viral p24 antigen in the medium was measured, and the infection level of LVsh5/C46-modified cells was compared to untransduced control cells. As shown in Figure 6a, while untransduced Molt4/CCR5 were susceptible to BaL (R5) and NL4-3 (X4) strains, viral infection was strongly suppressed in LVsh5/C46-modified cells, indicating that cells expressing LVsh5/C46 were significantly more resistant to HIV-1 infection compared to control cells. Similarly, when the LVsh5/C46-modified Molt4/CCR5 cells were infected with a dual R5/X4 tropic HIV-1 strain, SF2, viral titer was reduced dramatically relative to untransduced cells, exhibiting ~100-fold inhibition of viral replication (Figure 6b). This observation was consistent when transduced cells were exposed to low or high dose of SF2 strain. These results demonstrate the ability of LVsh5/C46 to provide strong resistance against R5-, X4-, and R5/X4-tropic HIV-1 strains.
Figure 6.

LVsh5/C46 inhibited R5-, X4-, and dual-tropic HIV-1 infection in Molt4/CCR5 cells. (a) Cells were transduced with LVsh5/C46 at MOI of 5, with transduction efficiency of ~80–100%, in three individual experiments. Cells were then challenged with the HIV-1 viral strains BaL (R5-tropic) and NL4-3 (X4-tropic), and viral replication was measured by p24 antigen. (b) LVsh5/C46-transduced Molt4/CCR5 cells were infected with increasing doses of SF2 virus (R5/X4) as indicated, and viral replication was determined by p24 protein levels. Results are from one representative experiment. (c) Molt4/CCR5 were transduced with the indicated lentiviral vectors (sh5, C46, or sh5/C46), followed by infection with BaL strain (R5-tropic) at MOI of 0.2, and p24 protein levels were assessed 7 and 10 days later. Results are from one representative experiment. (d) Cells were transduced with sh5, C46, or LVsh5/C46 (sh5/C46) lentiviral vectors at MOIs of 1 and 5 and were challenged with NL4-3 (X4-tropic) or BaL (R5-tropic) HIV-1 viruses. HIV-1 infection was measured by p24 protein levels on day 13 postinfection and was compared to untransduced cells. *P = 0.006 compared to untransduced cells. **P = 0.011 compared to untransduced cells. MOI, multiplicity of infection.
We next tested the potency of the dual-therapeutic vector to single vectors containing either sh5 or C46 alone. Molt4/CCR5 cells were transduced with either LVsh5/C46 or lentiviral vectors containing sh5 or C46 genes, followed by infection with the R5-tropic strain BaL, and infection level was evaluated 7 and 10 days postinfection by p24 assay. In cells expressing either sh5 or C46 alone, BaL infection was inhibited; nonetheless, LVsh5/C46 was more effective than either vector expressing single agents (Figure 6c).
The Molt4/CCR5 cells were then transduced with low or high MOIs of sh5, C46, or LVsh5/C46 lentiviral vectors constructs, followed by exposure to either X4-tropic NL4-3 or R5-tropic BaL, and infection level was analyzed. As expected, sh5 vector was able to inhibit replication of R5-tropic but not X4-tropic strain, whereas C46 provided a stronger resistance against R5-tropic and X4 HIV-1–tropic strains. The combined LVsh5/C46 exhibited the strongest viral inhibition, reducing p24 concentration from 6,400 to 2 ng/ml in BaL infection and from 2,048 to 4 ng/ml in NL4-3. The response to higher dose of LVsh5/C46 resulted in the same effect (Figure 6d). Taken together, these results demonstrate that LVsh5/C46, as a dual anti–HIV-1 agents in a single lentiviral construct, has additive effect compared to the single agents, delivering robust inhibition of a broad range of HIV-1 strains.
LVsh5/C46-treated PBMCs inhibit replication of R5- and X4-tropic HIV-1 strains
We next assessed the ability of LVsh5/C46 to provide resistance to primary human PBMC cultures from HIV-1. PBMCs were treated with either LVsh5/C46 or vectors containing the single anti–HIV-1 agents: sh5 and C46, and the expression of C46 and downregulation of CCR5 by sh5 was confirmed 4 days posttransduction by flow cytometry (Figure 7a). Based on this analysis, transduction efficiency was estimated to be 43.7% for LVsh5/C46, 49.4% for C46 alone, 67.5% for sh5/eGFP, and 51.7% for eGFP control vector. Transduced PBMCs infected with R5-tropic HIV-1 strain NFNSX inhibited viral infection, whereas when infected with the X4-tropic strain NL4-3, only cells expressing C46 or the dual-therapeutic LVsh5/C46 vector were capable of inhibiting HIV-1 infection (Figure 7b). To further test the ability of PBMCs to resist HIV-1 infection, cells were transduced with LVsh5/C46 at MOI of 5 and reached transduction efficiency of 20–50%. After 48 hours, the transduced cells were infected with HIV-1 virus strains, and the level of p24 antigen was evaluated relative to control untransduced cells. Results from four independent experiments of cells challenged with the laboratory strains BaL (R5-tropic), SF162 (R5-tropic), and Bru (X4-tropic) showed effective inhibition of viral replication in all three strains of HIV-1 (Figure 7c) demonstrating again the potency of LVsh5/C46 to inhibit HIV-1 infection. Clinical isolates of HIV-1 from Clade B and Clade D subgroups were tested to further characterize LVsh5/C46-medited viral inhibition. Clade B is the most predominant subtype of HIV-1 that is found in the developed Western World, whereas Clade D is generally limited to East and Central Africa. PBMCs were infected with clinical isolate 92US723 (R5/X4 dual tropic, Clade B), and 92UG021 (X4-tropic, Clade D), in three independent assays. Infection of LVsh5/C46 modified PBMCs with Clade B clinical HIV-1 isolate led to an average of 83% inhibition of viral replication, whereas 67% inhibition was observed with Clade D isolate (Figure 7d). Altogether, these results demonstrated that delivery of the dual anti–HIV-1-agents sh5 and C46 to hematopoietic cells mediated by the LVsh5/C46 lentiviral vector inhibit a broad range of HIV-1 isolates, clades, and tropism.
Figure 7.
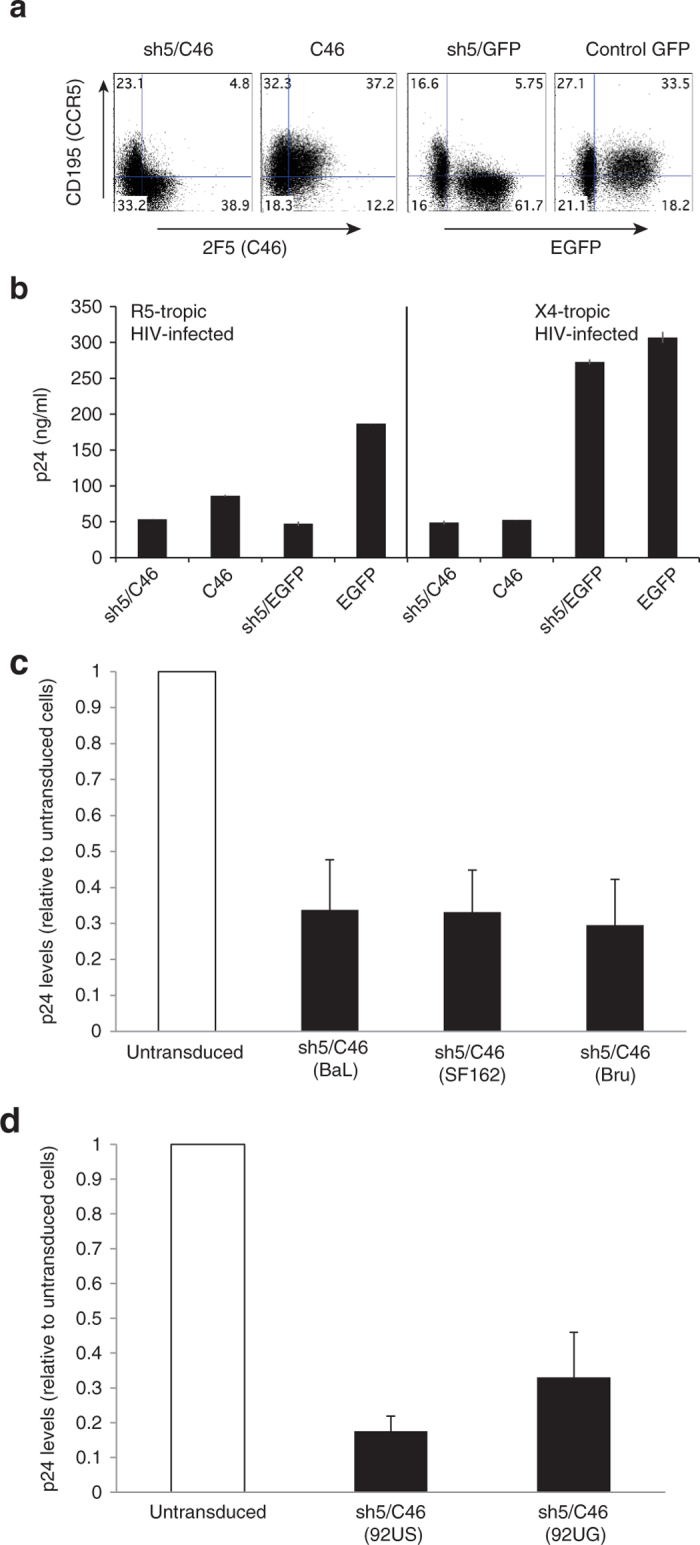
LVsh5/C46-modified PBMCs inhibit infection from laboratory and clinical strains of HIV-1. (a, b) PBMCs were transduced with the LVsh5/C46 (sh5/C46), C46 alone, sh5/GFP, or GFP vectors, at MOIs of 0.5, and 4 days later, transduction efficiency was determined by flow cytometry, (a) measuring CCR5 and C46 expression, or CCR5 and enhanced green fluorescent protein. (b) Cells were then infected with R5-tropic strain NFNSX or X4-tropic NL4-3, at MOIs of 0.02, and p24 protein levels was measured to assess HIV-1 replication. (c) PBMCs were transduced with LVsh5/C46 (sh5/C46) at MOI of five in four independent experiments, and 48 hours later were challenged with the HIV-1 laboratory strains BaL (R5-), SF162 (R5-), or Bru (X4-tropic) at MOIs of 0.14, 0.16, and 0.17, respectively. Four days postinfection, p24 levels were assessed and plotted relative to untransduced cells (this was done due to high variability between experiments and donor variability). Average p24 readings were: untransduced-BaL: 134,935 ng/ml (SE: 80,245)/transduced-BaL: 24,159 ng/ml (SE: 5,665). Untransduced-SF162: 72,153 ng/ml (SE: 30,041)/transduced-SF162: 20,121 ng/ml (SE: 6,181). Untransduced-Bru: 106,066 ng/ml (SE: 39,585)/transduced-Bru: 19,371 ng/ml (SE: 5,033). (d) PBMCs were transduced with LVsh5/C46 (sh5/C46) at MOI of 5, and 48 hours later were challenged with the HIV-1 clinical isolates 92US723 (R5/X4-tropic, Clade B) or 92UG021(X4-tropic, Clade D) in three independent experiments. Viral infection levels were determined relative to untransduced cells. Average p24 levels were: untransduced-92US723: 94,986 ng/ml (SE: 60,533)/transduced-92US723: 20,148 ng/ml (SE: 15,764). Untransduced-92UG021: 45,804 ng/ml (SE: 16,988)/transduced-92UG021: 15,536 ng/ml (SE: 9,401). GFP, green fluorescent protein; MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cell.
Discussion
In this preclinical study, we have assessed the therapeutic potential of a lentiviral vector (LVsh5/C46) containing two anti–HIV-1 agents that target viral entry. We have demonstrated the ability to stably introduce LVsh5/C46 into various hematopoietic cells, including CD4+ T lymphocytes and CD34+ HSPC, to allow for expression of a CCR5-targeted shRNA (sh5) and C46. Treatment of target cells with the LVsh5/C46 vector did not show any indication of toxicity. In transduced PBMC cultures, cell viability and proliferation were normal, apoptosis was not induced, and no mark of inflammation was observed. LVsh5/C46-treated CD34+ cells maintained their ability to differentiate into various hematopoietic lineages with no sign of lineage skewing. Moreover, LVsh5/C46-modified cells showed profound resistance to R5-, X4-, and dual-tropic strains of HIV-1.
Gene therapy for HIV-1 relies on genetic modification of hematopoietic cells in order to generate long-lasting HIV-1–resistant cells that would replenish and stabilize the patient’s immune system. Transplantation of manipulated CD4+ T cells would provide a pool of T lymphocytes with resistance to HIV-1 infection. A recent study by Scholler et al.62 has shown detection of engineered T lymphocytes 11 years after infusion that suggests modified cells may persist for decades, with continued expression and function that may contribute to prolonged survival. Genetic modification of CD34+ cells would generate HIV-1–resistant progenitor cells with the capacity to self-renew and differentiate into all hematopoietic lineages, including CD4+ T cells, macrophages, and dendritic cells, which are the natural targets of HIV-1. The general concept is that in HIV-1–infected individuals, the modified cells, even at low-to-moderate levels of gene modification, would have a selective survival advantage over unmodified cells, allowing them to proliferate and expand over time, and as a consequence will minimize viral loads and reduce viral reservoirs.
In this work, we transduced T cell lines, PBMCs, and CD34+ HSPC to assess toxicity, phenotype, and in the former two cell types protection from HIV-1. We did not test protection of CD34+ HSPC in the present experiments but as a continuation in a humanized mouse model (manuscript in preparation).
Recent clinical applications using lentiviral vectors for treatment of genetic diseases, such as β-thalassemia and adrenoleukodystrophy, have also shown these vectors to be safe and efficient.63–65 A theoretical concern with the safety of lentiviral vector usage is the development of RCL. To date, RCL has not been observed in any gene therapy clinical trial. LVsh5/C46, as a SIN vector, includes modifications designed to contribute to the safety of the vector. These features minimize the homology between the vector and the HIV-1 sequences, decrease the capacity of RCL formation, and reduce the likelihood of mobilization of the vector. Moreover, by separating the packaging genes into three plasmids, the number of recombination events required to produce a competent replicative virus increases. LVsh5/C46 pseudovirus batches did not show any evidence for the emergence of replicative particles in transduced cells (see Supplementary Table S1). Likewise, integration analysis of LVsh5/C46 provirus demonstrated absence of predominant integration sites (data not shown). Taken together, these data support the safety profile of LVsh5/C46 for clinical use.
The stability of the delivered LVsh5/C46 vector into target cells was confirmed by southern blot and sequencing analyses, with no unforeseen alterations to the genetic elements of LVsh5/C46. Splicing of the 812 bp intron within the 5′ UTR of the human UbC promoter was well documented previously.58,59 In the present study, the spliced UbC promoter was found to be sufficient to drive expression of C46 peptide to a detectable level and was effective in inhibiting HIV-1 infection.
We have demonstrated efficient delivery of LVsh5/C46 to various hematopoietic cells. PBMCs transduced at MOI of 1 resulted in transduction efficiency of ~34% with 1 copy of LVsh5/C46 vector per cell, and this level of transduction was sufficient to inhibit HIV-1 in challenge assays (Figures 1b,d and 7). Based on mathematical modeling, we have previously shown that a level of gene marking of ~10–20% in hematopoietic stem cells would be sufficient to have a “curative” effect, i.e., impact on viral load and CD4+ lymphocyte counts.66 For large-scale studies, an MOI of 1 for CD4+ T lymphocytes and MOI of 5 for CD34+ HSPC were chosen as the optimal doses, with the ideal of a therapeutic target of <5 copies of LVsh5/C46 per cell (Figure 3a).
Expression of the integrated LVsh5/C46 transgenes C46 and sh5 in cells over time was examined in the T cell line CEM.NKR.CCR5 (Figure 1c), due to the difficulties of maintaining primary cultures for extended period of time in culture. These results suggest that coexpression of the two anti–HIV-1 genes has no influence on their stability over time. An et al.41 have previously demonstrated stable expression of CCR5 shRNA in nonhuman primates for over 14 months posttransplantation of CD34+ HSPC, and Younan et al. have recently shown C46-mediated protection from simian-human immunodeficiency virus in a nonhuman primate study transplanting autologous ex vivo modified CD34+ HSPC.67
Various gene therapies have been developed to inhibit HIV-1 infection by blocking postintegration steps or early steps in the HIV-1 life cycle that occur prior to genome integration. Preintegration strategies are thought to have an advantage since production of HIV-1–resistant cells may lead to selective expansion of these modified cells over infected cells, which could prevent establishment of chronic HIV-1 infection and limit the viral reservoir. Entry inhibitors also have the potential to prevent the generation of escape mutants in the treated cells, a process that occurs during reverse transcription.68 Previous computational modeling studies have demonstrated that inhibition of preintegration steps is more likely to provide more effective outcomes.69–72
CCR5 is the predominant coreceptor used by HIV-1 during both initial infection and subsequent infection.73,74 The “Berlin patient” case has provided evidence that knocking down CCR5 expression has the potential to cure HIV-1. Currently, a gene therapy trial using CCR5 zinc finger nuclease is being conducted, and preliminary results indicate that treatment is generally well tolerated.75,76 Despite the promising results obtained with CCR5 knockdown, these strategies do not protect against X4-tropic HIV-1 or dual R5/X4 tropisms, which emerge with disease progression.21,77 For that reason, a dual-therapy design has been chosen in which CCR5 shRNA sh5 and the membrane-anchored C46 peptide have been engineered into a single lentiviral vector. C46, shown previously to effectively inhibit a broad range of viral isolates, was the first HIV-1 entry inhibitor to be tested in a clinical trial and was found to be safe with no major toxicities.47 Recently, Kimpel et al.78 have evaluated three leading genetic strategies for their potency to inhibit HIV-1 replication and demonstrated C46 to be the most robust in HIV-1 inhibition in T cells compared to tat/rev shRNA and RNA antisense against HIV-1 envelope. The combinatorial therapy of CCR5 shRNA and C46 peptide was found in this study to have a potentially synergistic effect on HIV-1 inhibition compared to the function of the singular genes (Figure 6c,d), which is supported by previous studies using a similar combinatorial approaches.79 Although lentiviral vectors expressing sh5 alone or C46 alone were able to protect PBMCs against R5- or X4-tropic HIV-1 (respectively), this effect was observed only at a low level of infection, as indicated by the level of p24 protein in control cells (Figure 7b). When infection levels were high, the dual-therapeutic vector displayed the most prominent inhibition of HIV-1 replication (Figure 6c,d).
LVsh5/C46 has also been characterized for safety and efficacy in vivo using a variety of humanized mouse models. During a GLP pharmacology/toxicology study, LVsh5/C46-modified human CD34+ HSPC transplanted into NSG mice displayed normal hematopoietic engraftment and differentiation that included a safe vector integration site profile; also, a humanized bone marrow–liver–thymus (BLT) mouse model study supported the ability of LVsh5/C46-modified hematopoietic cells to protect CD4+ T cells from HIV-1 pathogenesis in vivo and reduce viral load within peripheral blood and tissues (manuscript in preparation).
Taken together, the preclinical safety and efficacy studies presented herein, LVsh5/C46 vector is now being evaluated in a phase 1/2 clinical trial in which autologous hematopoietic cells are transduced ex vivo, followed by infusion back into HIV+ subjects (Clinical Trials NCT01734850). LVsh5/C46 anti–HIV-1 genes are designed to protect not only the mature T lymphocyte and macrophage populations but also the HSPC reservoirs that give rise to those progeny cells. This is the first clinical trial using RNA interference to downregulate CCR5 expression in combination with the fusion inhibitor C46 peptide in T cells and HSPC.
Materials and Methods
Cell isolation and culture
Human primary PBMCs
Buffy coats were obtained from healthy donors from the Australian Red Cross Blood Service. PBMCs were isolated from buffy coats by Ficoll Paque Plus (GE Healthcare, Uppsala, Sweden, #17-1440-02) density-gradient centrifugation, and the isolated cells were cultured in RPMI (Life Technologies, Carlsbad, CA, #72400047) supplemented with 20% fetal bovine serum and PHA-P for 48 hours. On the day of transduction, 10 U/ml rhIL-2 was added.
CD4+ cells
CD8+ cells were depleted from isolated PBMCs via CD8 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany, #130-045-201) and a VarioMACS separator (Miltenyi Biotec). CD3+ T lymphocytes were positively selected and isolated by CD3/CD28-CTS beads (Life Technologies, #402.03D). The CD4+/CD3+ lymphocytes were then cultured for 48 hours with the CD3/CD28 beads in X-vivo 15 serum-free media (Lonza, 04-744Q) supplemented with 20 mmol/l HEPES (Life Technologies, #15630106), 5% fetal bovine serum (Life Technologies, 10099141), and 100 U/ml rh IL-2 (Roche, Mannheim, Germany #1147528001). Cells were transduced overnight on RetroNectin (TaKaRa, Japan, #T100B) coated plates at various MOI, as indicated in the experimental methodology.
CD34+ cells
Primary human CD34+ HSPC were purchased commercially from AllCells; cells were directly isolated from granulocyte colony-stimulating factor (Neupogen) mobilized peripheral blood apheresis products with a CliniMACS instrument and CD34+ microbeads (Miltenyi Biotec) according to manufacturer’s specifications. Purified CD34+ HSPC were prestimulated overnight in 50 ng/ml each of SCF, TPO, and Flt3L (R&D Systems, Minneapolis, MN #255-SC, #288-TP, and #308-FK) and transduced on 2.5 mg/cm2 RetroNectin-coated plates.
Molt4/CCR5 and CEM.NKR.CCR5 cell lines
Cells were cultured in RPMI supplemented with 10% fetal bovine serum. Molt4/CCR5 cultures were also supplemented with 1 mg/ml geneticin (Life Technologies, #10131-035).
HEK293T cell line
These cell lines were used for viral production and were cultured in Dulbecco’s Modification of Eagles Medium and 10% fetal calf serum.
LVsh5/C46 plasmid construction
To clone LVsh5/C46 (Cal-1) lentiviral plasmid, the FG12 plasmid31 was first modified by adding multiple cloning sites to generate the FG11 plasmid.36 The membrane-anchored C46 sequence (containing a signal peptide, the C46 peptide, a hinge, and a membrane-spanning domain)43 was then cloned into BamHI/EcoRI sites within the FG11 plasmid downstream of the UbC promoter to produce FG/C46. The FG/C46 plasmid was digested with NdeI/XhoI, leaving a fragment of 7.5 kb, containing the UbC promoter and the C46 peptide. Next, the FG12 H1shRNACCR5 lentiviral vector41 was digested with NdeI/XhoI, and the 2.4 kb fragment, containing the human H1 RNA polymerase III promoter and CCR5 shRNA (1005) sequence was ligated into the same sites of FG/C46.
Lentiviral vector production and transduction
To produce pseudotype virus, the lentiviral SIN vectors were cotransfected with three helper plasmids: gag/pol helper plasmid, the HIV-1 Rev plasmid, and the VSV-G envelope plasmid. The four-plasmid system was transfected into HEK293T cells by calcium phosphate method (Clontech, Mountain View, CA), as described previously.80 For small-scale production, virus-containing media (VCM) was collected 24 and 48 hours posttransfection and pooled. VCM was concentrated by ultracentrifugation (25,000 rpm) over a 20% sucrose solution. Large-scale virus was produced in a manufacturing facility under either GLP or GMP conditions, based on a method published previously.81 VCM was purified by Mustang Q followed by concentration using tangential flow filtration filters.
Lentiviral vectors were diluted (1/8–1/512 for unconcentrated samples or 1/10–1/30,000 for concentrated samples), and 1 ml used to transduce 1 × 105 293T cells in a 12-well plate. Seventy-two hours posttransduction, cells were analyzed for enhanced green fluorescent protein expression or stained with 2F5 for cell-surface expression of C46. Samples were transduced in duplicate.
Using the percentage of positive cells, titer was calculated according to the following formula:
| Titer(ivp/ml) = [cell number × % positive cells(GFP+ or 2F5+/100)× VCM dilution factor]/VCM volume (ml) |
MOI was calculated by dividing the number of virus particles by the number of cells.
RCL detection
RCL rapid analysis was performed on the large-scale GLP and GMP viral production batches made by Indiana University Vector Production Facility. The assay was modeled on guidelines recommended by the US Food and Drug Administration for detecting replication-competent retrovirus. In the amplification phase, a test sample of concentrated LVsh5/C46 lentiviral vector, representing 5% of the total volume, was used to inoculate a cell line C8166-45 (derived from human umbilical cord blood lymphocytes). The inoculated cells were cultured for a minimum of 21 days to allow potential amplification of any RCL, and the supernatant was collected. In the indicator phase, fresh C8166-45 cells were infected with the supernatant from the amplification step and passaged for 7 days. At the end of this phase, supernatants and genomic DNA were collected for ELISA and PCR, respectively. Presence of RCL in the test sample was indicated by detection of p24 antigen in the indicator cell supernatants and psi-gag sequences in indicator cell DNA61.
In the coculture, RCL assay postproduction cells were used as a test sample. 1 × 108 of HEK293T cells that were used to produce the two batches of the GMP LVsh5/C46 vector were cocultivated with C8166-45 cells, followed by the amplification and induction phases as described above to detect RCL.
Viral isolates
NL4-3 and NFNXS were prepared by calcium phosphate transfection of 293T cells with either NL4-3 or NFNXS plasmid DNA. VCM was collected 72 hours posttransfection, and aliquots stored at −80 °C.
BaL, SF162, Bru, 92UG021, and 92US723 were prepared by infecting cell-free supernatant from PBMCs onto fresh uninfected PBMCs in the presence of 8 µg/ml Polybrene. Infections were performed over 2–2.5 hours with gentle rocking, followed by one wash of the cells and culture in RPMI containing 20% fetal bovine serum and 100 U/ml IL-2. Fresh culture media and PBMCs were added from days 7 through 21. Supernatant was collected every 3 days from day 14, and aliquots stored at −80 °C.
BaL and SF2 used in Molt4/CCR5 experiments were prepared by infecting PM-1 cells with cell culture supernatant containing either BaL or SF2 in the presence of 8 µg/ml Polybrene. Infections were performed over 2–2.5 hours with gentle rocking. At completion of infection, cells were washed once and put into culture. Fresh media and PM-1 cells were added from days 7 through 21. Supernatant was collected every 3 days from day 14, and aliquots stored at −80 °C. Titers for all virus batches were determined using Perkin Elmer HIV-1 P24 ELISA.
Flow cytometry (FACS)
Up to 1 × 106 cells were transferred to FACS tubes, washed in 1 ml of phosphate-buffered saline, and stained with antibodies according to the manufacturer’s recommendations. Cells were incubated for 20–30 minutes (room temperature or 4 °C), washed in 1 ml of phosphate-buffered saline, and fixed in 2% paraformaldehyde for 30 minutes at 4 °C prior to data acquisition on an LSRII (Becton Dickinson, San Jose, CA) using FACS Diva software. Data were analyzed using FlowJo Software (TreeStar, Ashland, OR). As controls, unstained samples and/or relevant isotype control antibodies were used.
For C46 peptide expression, cells were stained for 30 minutes with 1 µg 2F5 (Polymun Scientific, Klosterneuburg, Austria, #AB001), a monoclonal antibody directed against HIV-1 gp41, conjugated to PE using a Zenon PE Human IgG1 labeling kit (Molecular Probes, Eugene, OR, Z25455), followed by two rounds of signal amplification using a PE-FASER kit (Miltenyi Biotec, #130091764). As a control for background fluorescence, untransduced cells were stained with 2F5.
For endogenous CCR5 expression, CD195 (APC or PE-Cy7 fluorophores; Becton Dickinson) was used for staining. The fluorescently stained cells were detected by flow cytometry. Transduction efficiency was determined by calculating the percentage of C46-positive cells.
Evaluation of cell purity
CD4+ cells were stained with CD4, CD3, and CD8 antibodies (CD4 FITC or PE, CD3 PerCP-Cy5.5 and CD8 PE, Becton Dickinson) and analyzed by flow cytometry to calculate the percentage of positive cells. Purity of CD34+ was determined by using anti-CD34 antibodies (Becton Dickinson).
Genomic DNA isolation and real-time quantitative PCR analysis
Genomic DNA from transduced cells was isolated using the Invitrogen Purelink Genomic DNA Mini Kit according to the manufacturer’s instructions. Genomic DNA from each sample (50 ng of genomic DNA per reaction) was analyzed using validated qPCR assays to detect C46 transgene and β-globin (control gene). Primer and probe sequences for this assay are as follows: C46 sense: 5′-CACAGCCTGATCGAGGAGAG-3′; C46 antisense: 5′ GTCCTGCCACTGGTGGTG-3′; C46 probe: 5′-CACTCCACGCAGCACTTCCGCTCG-3′ (5′ 6-FAM, Int ZEN 3′ Iowa Black FQ). β-Globin sense: 5′-CAACCTCAAACAGACACCATGG-3′; β-globin antisense: 5′-TCCACGTTCACCTTGCCC-3′; β-globin probe: 5′-CTCCTGAGGAGAAGTCTGCCGTTACTGCC-3′ (5′ TEXAS RED 3′ BHQ-2). Singleplex PCR reactions were set up in duplicate in 25 µl reaction volumes using Agilent Brilliant II master mix in a 96-well plate on the Stratagene Mx3000P real-time PCR platform.
Thermocycling conditions were 50 °C 2 minutes, 95 °C 10 minutes, 40 × (95 °C 15 seconds; 60 °C 1 minute).
Data analysis was performed with Stratagene qPCR MxPro software. Serial dilutions of LVsh5/C46 plasmid, with known concentration in a background of 10 ng/µl genomic DNA, were used to generate standard curves. Known amounts of genomic DNA (equated to cell number) was serially diluted in H2O to generate a β-globin standard curve; copy number of C46 transgene was determined from the plasmid standard curve and normalized to cell numbers determined using the genomic DNA β-globin standard curve.
RNA isolation and RT-qPCRanalysis
C46 RT-qPCR
Total RNA was isolated from cells using Invitrogen Purelink RNA Mini Kit according to the manufacturer’s instructions. RNA (20 ng per reaction) was first converted to cDNA using Invitrogen Superscript VILO cDNA synthesis kit. Thermocycling conditions were 25 °C 10 minutes, 45 °C 60 minutes, 85 °C 5 minutes, 4 °C hold. cDNA was diluted 1:20 and 5 µl added to Singleplex validated PCR reactions set up in duplicate in 25 µl reaction volumes using Agilent Brilliant II master mix in a 96-well plate on the Stratagene Mx3000P real-time PCR platform.
Taqman primer and probe sequences for this assay are as follows: C46: 5′ CACAGCCTGATCGAGGAGAG; C46: 3′ GTCCTGCCACTGGTGGTG; C46 probe: CACTCCACGCAGCACTTCCGCTCG (5′ 6-FAM, Int ZEN 3′ Iowa Black FQ).
β2M sense: 5′-GCTGGCGCTACTCTCTCTTTCT-3′; β2M antisense: 5′-GGATGGCGTGAGTAAACCTGAA-3′; β2M probe: 5′-CCTGGAGGCTATCCAGCGTACTCCAAAG-3′ (5′ HEX 3′ BHQ-1).
Thermocycling conditions were 50 °C 2 minutes, 95 °C 10 minutes, 40 × (95 °C 15 seconds; 60 °C 1 minute).
Data analysis was performed with Stratagene qPCR MxPro software. LVsh5/C46 plasmid with known concentration was serially diluted in a background of 10 ng/µl total PBMC RNA to generate standard curves. Known amounts of total PBMC RNA were serially diluted in H2O, converted to cDNA, and diluted in 1:20 in H2O to generate a β2-microglobulin standard curve. Copy number of C46 transgene was determined from the plasmid standard curve and normalized to β2M levels as determined from starting input amount RNA to determine relative C46 expression.
sh5 RT-qPCR
Total RNA was extracted using either Qiagen miRNeasy Kit or Ambion mirVana miRNA Isolation kit according to the manufacturer’s instructions. Extracted RNA (10 ng per reaction) was run in duplicate in Taqman custom designed small RNA Assay and microRNA assays to determine the relative expression of sh5. cDNA was generated using a Taqman MicroRNA reverse transcription kit. Thermocycling was as follows: 16 °C 30 minutes; 42 °C 30 minutes, 85 °C 5 minutes; hold 4 °C. cDNA was diluted 1 in 5 in nuclease-free water, and 5 µl added to each custom Taqman PCR assay set up with Taqman Universal Master Mix No UNG in 20 µl reactions in 96-well plates on the Stratagene Mx3000P. Thermocycling conditions were 50 °C for 2 minutes, 95 °C for 10 minutes, 40 × (95 °C for 15 seconds; 60 °C for 1 minute).
Standard curves were generated using synthetic RNA Oligos for both sh5 and RNU38B (control gene used to normalize sh5). RNA Oligos of known copy number (107–101) were diluted 10-fold in a background of 10 ng/μl tRNA. Data analysis was performed with Stratagene qPCR MxPro software. Expression of sh5 was determined from the sh5 standard curve and normalized to RNU38B.
Apoptosis assay
Apoptosis assays were performed using the Caspase-Glo 3/7 Assay (Promega, #G8091), measuring caspase-3 and -7 activities, which play key effector roles in apoptosis. Cells were transduced overnight on 2.5 µg/cm2 RetroNectin-coated plates and the following day transferred to 96-well plates at 5 × 104 cells per well in 100 µl culture volume with three replicates per condition. Seven days posttransduction, 100 µl of Caspase-Glo 3/7 Reagent was added to each well, and the plates incubated for a further 2–3 hours at room temperature. Luminescence was measured on a Fluostar Optima (BMG Labtech, Ortenberg, Germany). As a positive control for apoptosis, cells were incubated with 40 µmol/l staurosporine. Fold change in luminescence was determined relative to the untransduced control.
Proliferation assay
Proliferation assays were performed using the Premix WST-1 Cell Proliferation Assay System (Takara, #MK400), which measures cell proliferation based on the enzymatic cleavage of tetrazolium salt (WST-1) to a water-soluble dye that can be detected by absorbance at 450 nm. Cells were transduced overnight on 2.5 µg/cm2 RetroNectin-coated plates, and the following day transferred to 96-well plates at 5 × 104 cells per well in 100 µl culture volume with 6 replicates per condition. Seven days posttransduction, 10 µl of Premix WST-1 was added to each well, and the plates were incubated for a further 2.5–4 h at 37 °C. Absorbance was measured at 450 nm on a Fluostar Optima (BMG Labtech). As a positive control for proliferation, cells were incubated with 100 ng/ml rhIL-2. Negative controls were culture media alone (blank) and untransduced cells. Fold change in absorption was determined relative to the untransduced control.
Inflammation response assay
Inflammation response assays were performed on culture supernatants using Quantikine Human IFN-γ ELISA kit (R&D Systems, #DIF50), Quantikine Human IL-6 ELISA kit (R&D Systems, #D6050), and VeriKine Human INF-α ELISA kit (PBL Interferon Source, Piscataway, NJ #41100). Cells were transduced overnight on 2.5 µg/cm2 RetroNectin-coated plates, and the following day transferred to 96-well plates at 5 × 104 cellsper well in 100 µl culture volume with 3 replicates per condition. At 7 days posttransduction, supernatant was removed from the culture and stored at −80 °C for batching of analysis. ELISA assays were performed according to manufacturer’s instructions. Standard curves for each cytokine to be measured were prepared from the standards supplied with the kits. Absorbance at 450 nm was measured on a Fluostar Optima (BMG LabTech). Cytokine levels in the sample supernatants were read off the standard curve, and fold changes were determined for the transduced samples relative to untransduced cells.
HIV-1 challenge assays
HIV-1 challenges on PBMCs were performed by pelleting 5 × 105 cells in HIV-1 viral supernatant containing 8 µg/ml polybrene. Cells were then incubated at 37 °C for 2–2.5 hours with gentle tapping every 15 minutes. Postincubation, cells were washed once and put into cultures of 1.5 ml in a six-well plate. The cultures were then sampled for p24 at either day 4 or 6.
HIV-1 challenges on Molt4/CCR5 cells were performed by pelleting 1 × 106 cells in HIV-1 viral supernatant containing 8 µg/ml Polybrene. Cells were then incubated at 37 °C for 2–2.5 hours with gentle rocking. Postincubation, cells were washed once and put into cultures of 3 ml per T25. Cultures were sampled on days 7 or 8, and 13 or 14, and fresh media added after sampling.
Evaluation of p24 by ELISA assay
p24 was determined by PerkinElmer HIV-1 p24 ELISA following the manufacturer’s instructions.
Analysis of integrated vector genomes
VERO cells were transduced with GMP-grade LVsh5/C46vector, genomic DNA was then isolated, and digested with NotI/Bsu36I and subjected to southern blot analysis using an 890 bp probe positioning at the 5′ end of the LVsh5/C46 sequence (NotI/PstI). The LVsh5/C46 plasmid, digested with NotI/Bsu36I, was used as a positive control.
CFU assay
Cells are plated at 500 cells per dish in methylcellulose media (Methylcult Optimum, Stem Cell Technologies) to enable colonies to form from individual cells. Cultures are incubated in a humidified dish for 14–16 days and then scored.
Acknowledgments
Funding for this work was provided in large part by Calimmune. O.W., M.B., M.M., H.I., F.D., J.Z., N.K., J.C.C., L.B., J.B., B.B., and G.P.S. are all employed by Calimmune. A.H., T.N., and R.K. were employed by Calimmune. Funding for Figure 7a,b were provided by NIH grants (NHLBI 1R01HL086409, 3R01HL086409-03S1, and NIAID 1R01AI100652-01A1 (D.S.A.) and AI55281 (I.S.Y.C.)).
O.W., M.B., M.M., H.I., A.H., F.D., B.B., D.S.A., J.B., L.B., and G.P.S. were all involved in the concept of the study. M.B., M.M., H.I., T.N., B.B., D.S.A., L.B., and G.P.S. designed the experiments. M.B. and M.M. performed cell culture and primary cell isolation, viral production, and viral isolates. M.M. performed flow cytometry analyses and apoptosis, proliferation, and inflammation assays. H.I. and A.H. performed DNA and RNA extractions, RT-qPCR, and real-time quantitative PCR analyses. M.B. performed challenge assays. D.S.A., I.S.Y.C., and J.B. performed and were responsible for flow cytometry and the NFNSX and NL4-3 challenge assays (Figure 7a,b). K.C. generated GMP-grade vector and conducted southern blot analysis and RCL assay. Troy Hawkins at Indiana University also assisted with southern blot analysis and sequence confirmation. M.R. and C. B. conducted the in vitro immortalization assay. M.B., M.M., H.I., B.B., J.Z., N.K., and J.C.C. performed at-scale CD4+ cell and CD34+ cell isolations and GMP-grade transduction assays. O.W. wrote the manuscript, with assistance from G.P.S., B.B., M.B., and R.K..
The authors O.W., M.B., M.M., H.I., A.H., F.D., T.N., R.K., J.Z., N.K., J.C.C., L.B., J.B., B.B., and G.P.S. declare employment by Calimmune. K.C. is a founder of Rimedion but is not employed by the company and has no intellectual property interest related to this study. I.S.Y.C. declares a financial interest in Calimmune.
References
- Günthard HF, Frost SD, Leigh-Brown AJ, Ignacio CC, Kee K, Perelson AS. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. J Virol. 1999;73:9404–9412. doi: 10.1128/jvi.73.11.9404-9412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud D, Siberry GK, Ahonkhai A, Kajdas J, Monie D, Hutton N. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J Virol. 2004;78:968–979. doi: 10.1128/JVI.78.2.968-979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff CT, Ray SC, Kwon P, Zinn R, Pendleton A, Hutton N. Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J Virol. 2002;76:9481–9492. doi: 10.1128/JVI.76.18.9481-9492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Davey RT, Jr, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- Davey RT, Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. 1999;96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan PR, Whaley M, Montaner JS. Rate of HIV-1 RNA rebound upon stopping antiretroviral therapy. AIDS. 1999;13:F59–F62. doi: 10.1097/00002030-199905280-00001. [DOI] [PubMed] [Google Scholar]
- Nolan D, Reiss P, Mallal S. Adverse effects of antiretroviral therapy for HIV infection: a review of selected topics. Expert Opin Drug Saf. 2005;4:201–218. doi: 10.1517/14740338.4.2.201. [DOI] [PubMed] [Google Scholar]
- Shafer RW, Schapiro JM. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 2008;10:67–84. [PMC free article] [PubMed] [Google Scholar]
- Reust CE. Common adverse effects of antiretroviral therapy for HIV disease. Am Fam Physician. 2011;83:1443–1451. [PubMed] [Google Scholar]
- Thompson MA, Aberg JA, Hoy JF, Telenti A, Benson C, Cahn P. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA panel. JAMA. 2012;308:387–402. doi: 10.1001/jama.2012.7961. [DOI] [PubMed] [Google Scholar]
- Mitsuyasu RT, Zack JA, Macpherson JL, Symonds GP. Phase I/II clinical trials using gene-modified adult hematopoietic stem cells for HIV: lessons learnt. Stem Cells Int. 2011;2011:393698. doi: 10.4061/2011/393698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi JJ, June CH, Kohn DB. Genetic therapies against HIV. Nat Biotechnol. 2007;25:1444–1454. doi: 10.1038/nbt1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds GP, Johnstone HA, Millington ML, Boyd MP, Burke BP, Breton LR. The use of cell-delivered gene therapy for the treatment of HIV/AIDS. Immunol Res. 2010;48:84–98. doi: 10.1007/s12026-010-8169-7. [DOI] [PubMed] [Google Scholar]
- Chung J, Rossi JJ, Jung U. Current progress and challenges in HIV gene therapy. Future Virol. 2011;6:1319–1328. doi: 10.2217/fvl.11.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Rosenberg PS, Goedert JJ, Ashton LJ, Benfield TL, Buchbinder SP, International Meta-Analysis of HIV Host Genetics Effects of CCR5-Delta32, CCR2-64I, and SDF-1 3’A alleles on HIV-1 disease progression: an international meta-analysis of individual-patient data. Ann Intern Med. 2001;135:782–795. doi: 10.7326/0003-4819-135-9-200111060-00008. [DOI] [PubMed] [Google Scholar]
- O’Brien SJ, Nelson GW. Human genes that limit AIDS. Nat Genet. 2004;36:565–574. doi: 10.1038/ng1369. [DOI] [PubMed] [Google Scholar]
- Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2:1240–1243. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E. Evidence for the cure of HIV infection by CCR5?32/?32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- Feng Y, Leavitt M, Tritz R, Duarte E, Kang D, Mamounas M. Inhibition of CCR5-dependent HIV-1 infection by hairpin ribozyme gene therapy against CC-chemokine receptor 5. Virology. 2000;276:271–278. doi: 10.1006/viro.2000.0536. [DOI] [PubMed] [Google Scholar]
- DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med. 2010;2:36ra43. doi: 10.1126/scitranslmed.3000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordelier P, Kulkowsky JW, Ko C, Matskevitch AA, McKee HJ, Rossi JJ. Protecting from R5-tropic HIV: individual and combined effectiveness of a hammerhead ribozyme and a single-chain Fv antibody that targets CCR5. Gene Ther. 2004;11:1627–1637. doi: 10.1038/sj.gt.3302329. [DOI] [PubMed] [Google Scholar]
- Bai J, Gorantla S, Banda N, Cagnon L, Rossi J, Akkina R. Characterization of anti-CCR5 ribozyme-transduced CD34+ hematopoietic progenitor cells in vitro and in a SCID-hu mouse model in vivo. Mol Ther. 2000;1:244–254. doi: 10.1006/mthe.2000.0038. [DOI] [PubMed] [Google Scholar]
- Steinberger P, Andris-Widhopf J, Bühler B, Torbett BE, Barbas CF., 3rd Functional deletion of the CCR5 receptor by intracellular immunization produces cells that are refractory to CCR5-dependent HIV-1 infection and cell fusion. Proc Natl Acad Sci USA. 2000;97:805–810. doi: 10.1073/pnas.97.2.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Banerjea A, Akkina R. Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides. 2003;13:303–312. doi: 10.1089/154545703322616989. [DOI] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci USA. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Akkina R. HIV-1 resistance conferred by siRNA cosuppression of CXCR4 and CCR5 coreceptors by a bispecific lentiviral vector. AIDS Res Ther. 2005;2:1. doi: 10.1186/1742-6405-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JS, Javien J, Nolta JA, Bauer G. Preintegration HIV-1 inhibition by a combination lentiviral vector containing a chimeric TRIM5 alpha protein, a CCR5 shRNA, and a TAR decoy. Mol Ther. 2009;17:2103–2114. doi: 10.1038/mt.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butticaz C, Ciuffi A, Muñoz M, Thomas J, Bridge A, Pebernard S. Protection from HIV-1 infection of primary CD4 T cells by CCR5 silencing is effective for the full spectrum of CCR5 expression. Antivir Ther (Lond) 2003;8:373–377. [PubMed] [Google Scholar]
- Kim SS, Peer D, Kumar P, Subramanya S, Wu H, Asthana D. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol Ther. 2010;18:370–376. doi: 10.1038/mt.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood. 2010;115:1534–1544. doi: 10.1182/blood-2009-04-215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M, Kamata M, Chen KN, Pariente N, An DS, Chen IS. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J Gene Med. 2010;12:255–265. doi: 10.1002/jgm.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briefs B. Trial watch: novel HIV gene therapy enters Phase I trial. Nat Rev Drug Disc. 2009;267;8 doi: 10.1038/nrd2862. [DOI] [PubMed] [Google Scholar]
- An DS, Donahue RE, Kamata M, Poon B, Metzger M, Mao SH. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci USA. 2007;104:13110–13115. doi: 10.1073/pnas.0705474104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Kamata M, Kittipongdaja P, Chen KN, Kim S, Pang S. Characterization of a potent non-cytotoxic shRNA directed to the HIV-1 co-receptor CCR5. Genet Vaccines Ther. 2009;7:8. doi: 10.1186/1479-0556-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer M, Brandenburg G, Martinius H, Schult-Dietrich P, Melikyan G, Kunert R. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J Virol. 2004;78:568–575. doi: 10.1128/JVI.78.2.568-575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildinger M, Dittmar MT, Schult-Dietrich P, Fehse B, Schnierle BS, Thaler S. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J Virol. 2001;75:3038–3042. doi: 10.1128/JVI.75.6.3038-3042.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrengel S, Hermann F, Hagmann I, Oberwinkler H, Scrivano L, Hoffmann C. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J Virol. 2005;79:10237–10246. doi: 10.1128/JVI.79.16.10237-10246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn RC, Hermann FG, Kim EY, Rett MD, Wolinsky SM, Johnson RP. Efficient entry inhibition of human and nonhuman primate immunodeficiency virus by cell surface-expressed gp41-derived peptides. Gene Ther. 2008;15:1210–1222. doi: 10.1038/gt.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lunzen J, Glaunsinger T, Stahmer I, von Baehr V, Baum C, Schilz A. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol Ther. 2007;15:1024–1033. doi: 10.1038/mt.sj.6300124. [DOI] [PubMed] [Google Scholar]
- Chen TK, Aldrovandi GM. Review of HIV antiretroviral drug resistance. Pediatr Infect Dis J. 2008;27:749–752. doi: 10.1097/INF.0b013e3181846e2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane PA. New developments in HIV drug resistance. J Antimicrob Chemother. 2009;64 suppl. 1:i37–i40. doi: 10.1093/jac/dkp258. [DOI] [PubMed] [Google Scholar]
- Buchacz K, Baker R, Ward DJ, Palella FJ, Chmiel JS, Young B. Trends in decline of antiretroviral resistance among ARV-experienced patients in the HIV outpatient study: 1999-2008. AIDS Res Treat. 2012;2012:230290. doi: 10.1155/2012/230290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- Endres MJ, Clapham PR, Marsh M, Ahuja M, Turner JD, McKnight A. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell. 1996;87:745–756. doi: 10.1016/s0092-8674(00)81393-8. [DOI] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Langford SE, Ananworanich J, Cooper DA. Predictors of disease progression in HIV infection: a review. AIDS Res Ther. 2007;4:11. doi: 10.1186/1742-6405-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray N, Doms RW. HIV-1 coreceptors and their inhibitors. Curr Top Microbiol Immunol. 2006;303:97–120. doi: 10.1007/978-3-540-33397-5_5. [DOI] [PubMed] [Google Scholar]
- Regoes RR, Bonhoeffer S. The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 2005;13:269–277. doi: 10.1016/j.tim.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Tsibris AM, Kuritzkes DR. Chemokine antagonists as therapeutics: focus on HIV-1. Annu Rev Med. 2007;58:445–459. doi: 10.1146/annurev.med.58.080105.102908. [DOI] [PubMed] [Google Scholar]
- Nenoi M, Mita K, Ichimura S, Cartwright IL, Takahashi E, Yamauchi M. Heterogeneous structure of the polyubiquitin gene UbC of HeLa S3 cells. Gene. 1996;175:179–185. doi: 10.1016/0378-1119(96)00145-x. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Crinelli R, Giacomini E, Carloni E, Magnani M. A potent enhancer element in the 5’-UTR intron is crucial for transcriptional regulation of the human ubiquitin C gene. Gene. 2009;448:88–101. doi: 10.1016/j.gene.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17:1919–1928. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornetta K, Yao J, Jasti A, Koop S, Douglas M, Hsu D. Replication-competent lentivirus analysis of clinical grade vector products. Mol Ther. 2011;19:557–566. doi: 10.1038/mt.2010.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F. Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Seymour LW, Thrasher AJ. Gene therapy matures in the clinic. Nat Biotechnol. 2012;30:588–593. doi: 10.1038/nbt.2290. [DOI] [PubMed] [Google Scholar]
- Murray JM, Fanning GC, Macpherson JL, Evans LA, Pond SM, Symonds GP. Mathematical modelling of the impact of haematopoietic stem cell-delivered gene therapy for HIV. J Gene Med. 2009;11:1077–1086. doi: 10.1002/jgm.1401. [DOI] [PubMed] [Google Scholar]
- Younan PM, Polacino P, Kowalski JP, Peterson CW, Maurice NJ, Williams NP. Positive selection of mC46-expressing CD4+ T cells and maintenance of virus specific immunity in a primate AIDS model. Blood. 2013;122:179–187. doi: 10.1182/blood-2013-01-482224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen SG, Shimizu S, An DS. Stem cell-based anti-HIV gene therapy. Virology. 2011;411:260–272. doi: 10.1016/j.virol.2010.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Laer D, Hasselmann S, Hasselmann K. Gene therapy for HIV infection: what does it need to make it work? J Gene Med. 2006;8:658–667. doi: 10.1002/jgm.908. [DOI] [PubMed] [Google Scholar]
- Applegate TL, Birkett DJ, Mcintyre GJ, Jaramillo AB, Symonds G, Murray JM. In silico modeling indicates the development of HIV-1 resistance to multiple shRNA gene therapy differs to standard antiretroviral therapy. Retrovirology. 2010;7:83. doi: 10.1186/1742-4690-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviran S, Shah PS, Schaffer DV, Arkin AP. Computational models of HIV-1 resistance to gene therapy elucidate therapy design principles. PLoS Comput Biol. 2010;6:e1000883. doi: 10.1371/journal.pcbi.1000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund O, Lund OS, Gram G, Nielsen SD, Schønning K, Nielsen JO. Gene therapy of T helper cells in HIV infection: mathematical model of the criteria for clinical effect. Bull Math Biol. 1997;59:725–745. doi: 10.1007/BF02458427. [DOI] [PubMed] [Google Scholar]
- Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol. 1992;66:1354–1360. doi: 10.1128/jvi.66.3.1354-1360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon P, June C. Chemokine receptor 5 knockout strategies. Curr Opin HIV AIDS. 2011;6:74–79. doi: 10.1097/COH.0b013e32834122d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton KM, Burch BD, Soriano-Sarabia N, Margolis DM. Prospects for treatment of latent HIV. Clin Pharmacol Ther. 2013;93:46–56. doi: 10.1038/clpt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1–infected individuals. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpel J, Braun SE, Qiu G, Wong FE, Conolle M, Schmitz JE. Survival of the fittest: positive selection of CD4+ T cells expressing a membrane-bound fusion inhibitor following HIV-1 infection. PLoS ONE. 2010;5:e12357. doi: 10.1371/journal.pone.0012357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CW, Younan P, Jerome KR, Kiem HP. Combinatorial anti-HIV gene therapy: using a multipronged approach to reach beyond HAART. Gene Ther. 2013;20:695–702. doi: 10.1038/gt.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeot PE, Duperrier K, Nègre D, Boson B, Rigal D, Cosset FL. High levels of transduction of human dendritic cells with optimized SIV vectors. Mol Ther. 2002;5:283–290. doi: 10.1006/mthe.2002.0541. [DOI] [PubMed] [Google Scholar]
- Leath A, Cornetta K. Developing novel lentiviral vectors into clinical products. Meth Enzymol. 2012;507:89–108. doi: 10.1016/B978-0-12-386509-0.00005-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.