

Preface
Focal adhesion kinase (FAK) is a cytoplasmic protein tyrosine kinase that is over-expressed and activated in several advanced-stage solid cancers. FAK promotes tumor progression and metastasis through effects on cancer cells, as well as stromal cells of the tumor microenvironment. FAK’s kinase-dependent and –independent functions control cell movement, invasion, survival, gene expression, and cancer stem cell self-renewal. Small molecule FAK inhibitors decrease tumor growth and metastasis in several preclinical models and possess initial clinical activity in patients with limited adverse events. We discuss FAK signaling effects on both tumor and stromal cell biology that provide rationale and support for future therapeutic opportunities.
Keywords: focal adhesion kinase, tumor growth, metastasis, tumor microenvironment, FAK inhibitors
Introduction
Focal Adhesion Kinase (FAK) is a multi-functional regulator of cell signaling within the tumor microenvironment1–3. During development and in various tumors, FAK promotes cell motility, survival and proliferation through kinase-dependent and -independent mechanisms. In the past few years, phase I and II clinical trials have been initiated with FAK inhibitors; yet, some of FAK’s functions in tumorigenesis remain under investigation.
Chromosomal region 8q24.3, which encompasses PTK2 (encoding FAK), is linked to ovarian cancer susceptibility4. Large databases such as The Cancer Genome Atlas show that FAK mRNA levels are elevated in serous ovarian tumors (~37%)5 and invasive breast cancers (~26%)6 with correlations to poor overall patient survival7, 8. Increased FAK mRNA levels are also found in several other human malignancies (Figure 1A)3. Studies with tumor tissue arrays find that FAK activation, as determined by phosphospecific antibody recognition of the FAK tyrosine (Y) 397 auto-phosphorylation site, increases with tumor progression3. However, unlike classical oncogenes such as Ras or PI3-kinase (PI3K), only a few missense mutations within PTK2 are found in tumors5. Instead, elevated FAK activity is associated with PTK2 amplification, consistent with a model whereby increased FAK dimerization induced by higher FAK levels contributes to catalytic activation9.
Figure 1. FAK expression in cancer and FAK domain structure.

(A) Percent of tumor samples with elevated focal adhesion kinase (FAK) mRNA. The Cancer Genome Atlas was quiered using the cBioPortal (www.cbioportal.org). Search criteria included mRNA expression data (Z-scores for all genes) and tumor datasets with mRNA data. Numbers of tumors analyzed (n) is shown on the X axis. (B) FAK consists of a central kinase domain flanked by a protein4.1-ezrin-radixin-moesin (FERM) homology domain on the N-terminal side and a C-terminal focal adhesion targeting (FAT) domain. Both terminal domains are separated from the kinase domain by a linker region containing proline-rich regions (PRR). Important tyrosine (Y) phosphorylation (P) sites are indicated; Y397, K454 and H58 play crucial roles in FAK activation. FAK binding partners are shown at their interaction sites within FAK. Binding of these proteins affects outcomes like cell motility (orange), cell survival (yellow) or both functions (orange/yellow). Roles involving FAK activation are shown in grey, important contributions to the tumor environment in green.
Here, we discuss advances in understanding FAK signaling connections in tumor and stromal cells. We cover the intricate roles of FAK in tumor invasion, growth, and metastasis. We highlight genetic mouse models used to elucidate new roles for FAK in endothelial cells (ECs) and discuss how stromal FAK signaling contributes to tumor progression. Finally, we summarize new translational developments using small molecule FAK inhibitors.
FAK regulation
Control of FAK expression
Nuclear factor κB (NFκB) and p53 are well-characterized transcription factors that activate and repress the PTK2 promoter, respectively10, 11. Other transcription factors such as Nanog12, Argonaute2 (Ago2)13, and PEA314 also increase PTK2 promoter activity. Nanog promotes FAK expression in colon carcinoma cells and as part of a signaling loop, Nanog activity is increased by FAK phosphorylation12. Ago2, a part of the cellular RNA interference machinery, is amplified in hepatocellular carcinoma and induces FAK transcription13. Ago2-silencing reduces FAK levels and concomitantly blocks tumorigenesis and metastasis in mice. Elevated PEA3 and FAK levels correlate with metastatic stages in human oral squamous cell carcinoma14. PEA3 induces FAK expression in vitro and silencing of either PEA3 or FAK reduces metastasis of human melanoma xenografts. Given the complexity and size of the PTK2 promoter region, it is likely that transcription factor combinatorial effects regulate PTK2 transcription.
FAK is also subject to alternative splicing as PTK2 with deletion of exon 33 (FAK amino acids 956–982), identified in a subset of breast and thyroid patient samples, results in enhanced cell motility and invasion15. However, this deletion likely disrupts FAK linkage to integrins and it is unclear how truncated FAK may function. PTK2 with deletion of exon 26, also occurring in breast cancer, removes a FAK C-terminal domain caspase cleavage site and results in increased FAK protein stability and anti-apoptotic signaling16. Interestingly, alternative splicing or increased FAK mRNA expression does not always translate into elevated FAK protein levels17. FAK mRNA turnover mediated by microRNA-7 blocks orthotopic breast carcinoma growth and lung metastasis in mice, and microRNA-7 expression in breast cancer patient samples inversely correlates to cancer stage18. At the protein level, FAK is subject to proteasomal or calpain-mediated degradation19. Poly-ubiquitination by the E3 ligase mitsugumin 53 (also known as TRIM72) promotes FAK proteasomal degradation during myogenesis, but this has not been tested in tumor cells20. However, in general, FAK protein levels are elevated in advanced stage solid tumors. Together, these results support the notion that elevated FAK expression is connected to several tumor-associated phenotypes.
Regulation of FAK activity
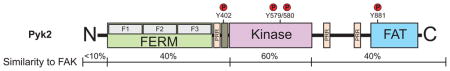
FAK is a cytoplasmic tyrosine kinase that associates with receptors at the plasma membrane and with distinct protein complexes in the nucleus21. Elucidating the regulatory mechanism(s) of how FAK associates with these distinct signaling complexes is key to understanding FAK biological function. FAK domain structure (Figure 1B) consists of an N-terminal FERM (band 4.1-ezrin-radixin-moesin) domain, a central kinase region, proline-rich domains, and a C-terminal focal adhesion targeting domain2, 19. Proline-rich tyrosine kinase 2 (Pyk2), a FAK ortholog with ~45% amino acid sequence identity, can compensate for some FAK functions after FAK loss in knockout (KO) mouse models (Box 1)22–24.
Box 1. Commonalities and differences between Pyk2 and FAK.
Proline-rich tyrosine kinase-2 (Pyk2) shares a similar domain organization with focal adhesion kinase (FAK) (see figure), with 60% sequence identity in the central kinase domain, and the conserved arrangement of proline-rich regions (PRRs) and tyrosine phosphorylation sites. Phosphorylation of Pyk2 Y402 and Y881 create Src-homology-2 (SH2) binding sites for Src and growth-factor-receptor-bound-2 (GRB2), respectively. Pyk2 and FAK contain C-terminal focal adhesion (FA) targeting (FAT) domains that bind to paxillin19, 136. However, Pyk2 shows peri-nuclear distribution and, unlike FAK, is not strongly localized to FAs. Substitution of the Pyk2 C-terminal domain with that of FAK facilitates co-localization of a Pyk2-FAK chimera to β1 integrin-containing FA, indicating that there are significant binding differences between FAK and Pyk219. For instance, the FAK FAT domain uniquely binds the integrin-associated protein talin3. Although Pyk2 can be activated by integrins, this is highly dependent on Src activation. Upon FAK KO in fibroblasts, increased Pyk2 levels induce an intrinsic mechanism promoting cell survival23. Consistent with findings in ovarian carcinoma cells, this is in part mediated through nuclear translocation and selective regulation of the p53 tumor suppressor by Pyk2. Phosphorylation of Pyk2 Y881 has been proposed as a prognostic marker for non-small-cell lung cancer progression137. In this cancer type, Pyk2 is positively correlated to the expression of cancer stem cell markers, indicating a possible mode of action. In glioblastoma cells, Pyk2 is regulated by a specific microRNA, miR-23b, that does not target FAK138 and supports the idea of differential Pyk2 and FAK functions. Pyk2 signaling upon FAK knockout has also been linked to increased Rho GTPase activation22, facilitation of angiogenesis24, regulation of macrophage motility101, and control of tumorigenic outgrowth mediated by mammary cancer stem cells139. Due to these functions, Pyk2 activity could compromise the outcome of FAK-targeted therapy; indeed FAK-selective inhibitors have been shown to enhance Pyk2 tyrosine phosphorylation in endothelial cells24.
The best-characterized mechanism promoting FAK activation involves integrin receptor clustering upon cell binding to extracellular matrix (ECM) proteins which may involve FAK dimerization9. This leads to FAK auto-phosphorylation at Y397, binding of Src-family kinases to the phosphorylated site, Src-mediated phosphorylation of the FAK kinase domain activation loop (Y576/577), and formation of an activated FAK-Src complex1–3. Indirect interactions between the FAK C-terminal domain and integrins at focal adhesions (FAs) mediate the integrin/FAK linkage, as over-expression of a FAK C-terminal fragment blocks integrin-mediated FAK activation19. Alternatively, the activation loop within the FAK kinase domain is also directly phosphorylated by the RET receptor, thereby enhancing FAK kinase activity25. FAK phosphorylation at Y397 can also occur in trans9.
Recent studies show that FAK FERM domain plays a prominent role in the intra-molecular regulation of FAK activity by binding to the kinase domain and blocking FAK Y397 site accessibility and auto-phosphorylation21, 26, 27. Studies with a fluorescent biosensor revealed FAK FERM conformational changes upon phosphoinositide lipid interaction and upon cell binding to ECM27, 28. FAK FERM domain interaction with membrane-associated proteins such as tetraspan TM4SF5 or growth factor receptors can also induce FAK activation29, 30. FAK activity is also increased via FAK FERM domain alterations induced by changes in pH and increased cell-ECM tension31–34. Local pH increases promote FERM conformational changes via His58 deprotonation, mediated in part by plasma membrane sodium/hydrogen transporters like NHE132. Increased intracellular pH commonly occurs in cancer and increased matrix stiffness or forces associated with collagen fiber cross-linking trigger increase FAK Y397 phosphorylation and tumor progression in mouse breast cancer models32, 33. It is likely that context-dependent stimuli trigger FAK activation through steps involving the FERM domain, FAK dimerization, or other yet to be determined mechanisms.
FAK in tumor cells
FAK is at the intersection of various signaling pathways promoting cancer growth and metastasis (Figure 2). This includes kinase-dependent control of cell motility19, invasion35, cell survival11, 36, and transcriptional events promoting epithelial-mesenchymal transition (EMT)37–39. Additionally, kinase-independent scaffolding functions of FAK can influence cell survival or breast cancer stem cell proliferation11, 40, 41. Understanding the balance between kinase-dependent and -independent functions is key to the interpretation of FAK-related phenotypes. Conditional tissue-specific FAK floxed mouse models and chemical FAK inhibitors have allowed the delineation of several FAK-associated signaling pathways (Table 1). For example, several groups have used polyoma virus middle T antigen (PyMT)-driven breast tumor models combined with tissue-specific FAK knockout through the mouse mammary tumor virus (MMTV) promoter (MMTV-PyMT model)42–45 to assess FAK function in tumor progression.
Figure 2. FAK connections to tumor growth and metastasis.

FAK drives cancer growth and metastasis through kinase-dependent (green) or -independent (orange) functions. FAK is activated by receptor tyrosine kinases (RTK), intracellular pH changes (H+), integrins, G-protein coupled receptors (GPCR) and cytokine receptors. The exact mechanisms are not always clear (indicated by ‘?’). Oxidative stress and FAK catalytic inhibition increase FAK nuclear localization. (A) Active FAK increases cell motility through effects on Arp2/3, Rho guanine nucleotide exchange factors (RhoGEF), talin or cortactin, and Src- or PI3-kinase (PI3K) mediated signaling. This drives cytoskeletal remodeling, focal adhesion (FA) formation and turnover, and expression and cell-surface presentation of matrix metalloproteinases (MMPs), enhancing cell invasion and tumor metastasis. (B) Kinase-independent scaffolding of endophilin A2 induces the expression of endothelial-mesenchymal transition (EMT) markers. FAK affects survival and proliferation through kinase-dependent and –independent roles to promote tumor growth. (C) FAK induces cell cycle progression through cyclin D1 involving Krüppel-like factor 8 (KLF8), Src–ERK, or JNK signaling. Signaling through PI3K/Akt mediates inhibition of apoptosis through transcriptional effects by NFkB or YB-1. (D) Nuclear FAK acts as a scaffold for p53 and Mdm2 in a kinase-independent manner, increasing p53 poly-ubiquitination (Ub) and degradation, thereby promoting cell survival.
Table 1. Mouse models available to characterize FAK function in cancer.
Shown are characteristics of common mouse models for the dissection of FAK function.
| Model | Type | Affected cells | Promoter | Inducer | Phenotype | Reference |
|---|---|---|---|---|---|---|
| FAK flox/flox knock-out | Cell lineage-specific k.o. | Endothelial cells | PDGFb | Cre-ERT | Impact neovascularization, tumor growth and angiogenesis | (92) |
| Endothelial cells | SCL | Cre-ERT | (24)(82) | |||
| Endothelial cells | Tie2 | Cre-ERT | (81)(83) | |||
| Hematopoietic cells | MX1 | Cre-ERT | Increased HSC pool, impaired cytokine-induced growth survival, and anti-apoptotic signaling in myeloid and erythroid lineages | (104)(105) | ||
| Keratinocytes | K14 | Cre-ERT | Reduced papilloma formation/progression | (114)(117) | ||
| Mammary epithelial cells | MMTV | Cre-ERT | Reduced breast tumor formation and progression, suppression of mammary cancer stem cells | (45)(73)(74) | ||
| Megakaryocytes, Platelets | Pf4 | Cre-ERT | Increased bleeding time, megakaryopoiesis and decreased platelet spreading | (106) | ||
| Myeloid cells | Lysozyme M | Cre-ERT | Modified neutrophil and macrophage functions | (100)(101) | ||
| Prostate cells | Probasin | Cre-ERT | Reduced androgen-independent formation of neuroendocrine carcinoma. No change in progression to adenocarcinoma. | (115) | ||
| FAK hetero-zygote | Global deletion of one allele | All | None | none | Increased tumor angiogenesis | (93) |
| FAK-KD knock-in | Cell lineage-specific k.i. | Endothelial cells | SCL, Tie2 | Cre-ERT | Disruption of AJ formation during development. Reduced VP and VEGF-induced tumor cell extravasation/ metastasis | (69)(76)(88) |
k.o. knock-out, k.i. knock-in, VP: vascular permeability, HSC: hematopoietic stem cell.
FAK promotes invasive cell phenotypes
Tumor cell invasion into the surrounding microenvironment is a key step in cancer progression, allowing cancer cells to form metastases at secondary locations. This requires transition to a motile phenotype through changes in FA and cytoskeletal dynamics, and alterations in matrix metalloproteinase (MMP) expression or activation to facilitate ECM invasion. EMT, which is driven by a transcriptional program, supports the progression to these invasive properties.
Canonical FAK signaling is linked to FA formation and turnover2, 19, 46. FAK recruitment and activation at nascent FAs involves binding to the Rho guanine nucleotide exchange factor (GEF) ARHGEF28 (p190RhoGEF or Rgnef), and a FAK-ARHGEF28 signaling complex promotes local invasion of orthotopic colon carcinomas in mice47, 48. FAK-ARHGEF28 dimerization and the associated increase in FAK activation is thereby linked to elevated tyrosine phosphorylation of paxillin, an adaptor protein involved in FA maturation49. FAK also recruits the integrin activating adapter protein talin to nascent FAs, occurring independently of direct talin binding to β integrins50.
FAK not only promotes FA formation-maturation, but also drives FA turnover through control of targeted FA protein proteolysis. Mutations that disrupt FAK-talin binding inhibit proteolytic talin cleavage, preventing efficient FA turnover50. Protein cleavage is mediated by proteases like calpain-2 or caspase-8 containing multi-protein complexes51. Additionally, FAK proline-rich sites facilitate interaction with the actin-binding protein cortactin, whose phosphorylation by FAK contributes to FA turnover52. In head and neck cancer, blocking of an integrin/FAK/cortactin/JNK1 signaling cascade through specific antibodies against β1 integrins renders cells sensitive to radiotherapy and delays xenograft growth53. Interestingly, FAK depletion decreases the abundance of tyrosine-phosphorylated proteins at FAs, while simultaneously increasing their levels at invadopodia in a Src-dependent manner54. However, increased invadopodia formation is not sufficient to promote tumor cell invasion in the absence of FAK, indicating that increased FAK-mediated cell motility underlies an invasive cell phenotype.
Dynamic rearrangement of the actin cytoskeleton is another integral component of cell movement and cell protrusion. FAK-associated proteins such as talin and cortactin bind actin and link FAs to changes in actin dynamics19. Additionally, catalytically inactive FAK associates with Arp2/3 through its FERM domain and enhances f-actin polymerization in cooperation with N-WASP55, 56. These events are proposed to occur prior to integrin-mediated FAK activation. In this model, the FAK FERM domain may function as scaffold to direct Arp2/3 activity to cell protrusions preceding mature FA formation.
MMP expression and activation at cell protrusions facilitates matrix invasion of motile cells. FAK activity increases MMP-9 expression and spontaneous breast carcinoma metastasis in a syngeneic and orthotopic mouse model57. Other studies show that MMP regulation and surface presentation in cancer cells involves multiple downstream pathways such as p130Cas58 and the PI3K/Akt/mTOR cascade59. Although mTOR effects on MMPs are not clearly understood, the FAK-p130Cas complex targets MMP-14 to FAs and promotes MMP-14 membrane surface presentation58. Knockdown of FAK or p130Cas does not alter the generation of pancreatic carcinoma protrusions, but prevents ECM degradation. During in vivo metastasis of human breast cancer cell xenografts in mice, MMP-14 function also has been proposed to require FAK signaling through an alternative pathway involving activation of Krüppel-like factor 860.
EMT-like transcriptional programs have been shown to drive tumor cell motility and invasion, and FAK signaling contributes to this EMT profile change61. FAK re-expression in FAK-null cells drives Snail1-induced EMT37. FAK scaffolding increases endophilin A2 phosphorylation leading to alterations in EMT markers, including MMPs, and affecting PyMT-induced breast tumor progression38. Indirect factors such as micro-RNA-7-mediated reduction of FAK expression results in loss of mesenchymal markers along with increased E-cadherin expression in breast tumor models18. Additionally, FAK may also act in a proximal manner to affect the dynamics of E-cadherin internalization in tumor cells39. Over-expression of a FAK mutant unable to be phosphorylated on multiple sites in colon carcinoma cells blocks Src-induced E-cadherin internalization62 and pharmacological inhibition of FAK activity increases cell-cell adhesion strength in part by stabilizing E-cadherin surface expression63. Together, these results implicate FAK in both the cell surface and transcriptional regulation of E-cadherin levels in tumor cells.
FAK drives tumor survival and growth
FAK promotes cell survival through kinase-dependent and -independent linkages. Kinase-dependent pathways include signaling through the PI3K/Akt cascade (Figure 2)3. Integrins and other extracellular stimuli induce FAK-survival signals to prevent anoikis and other types of cell death3. In ovarian cancer, tumor ascites prevents death-inducing signals via activation of an integrin αvβ5/FAK/Akt signaling pathway64. Moreover, FAK activity was shown to promote anchorage-independent survival of murine ovarian carcinoma cells, independent of effects on Src kinase activity8.
FAK signaling is also associated with ovarian cancer resistance to paclitaxel-induced cell death65. Pharmacological FAK inhibition enhances chemosensitivity in taxane-resistant cells by decreasing YB-1 transcription factor phosphorylation in an Akt-dependent manner65. Additionally, phosphorylation of FAK Y861 by protein tyrosine kinase 6 has been postulated to initiate an Akt-dependent anti-anoikis cascade66. Simplistically, increased FAK activity-mediated survival signals in vitro correspond to increased tumor growth. However, it remains undetermined whether mutational activation of the PI3K or Akt signaling pathways, commonly occurring in tumors, may bypass effects of FAK inhibition. Additionally, FAK functions downstream of G-protein-linked receptors for stress hormones like norepinephrine, and prevents ovarian cancer cell anoikis7.
FAK activity also enhances cell cycle progression (Figure 2)46 and recent studies have linked this to a matrix stiffness-sensitive signaling linkage between FAK, p130Cas, Rac GTPase, and the actin binding protein lamellipodin67. However, studies with transgenic mice show that kinase-dead (KD) FAK-expressing fibroblasts and ECs grow normally68, 69. Thus, FAK kinase activity is not essential for proliferation of all cell types. Yet, tumor cells require FAK activity in the processes of extravasation and proliferation of micro-metastases in foreign tissue environments35, 70. Although conditional FAK knockout (KO) in the intestinal epithelium of transgenic mice shows that FAK is dispensable for normal intestinal homeostasis, these mice require FAK for intestinal regeneration following DNA damage71. Reduced cyclin D1 levels associated with decreased epithelial proliferation also occur in the colonic epithelium of FAK knockout mice during mucosal wound healing72. Additionally, double deletion of FAK and the tumor suppressor adenomatous polyposis coli (APC) suppresses tumor formation induced by APC loss71. Although it has been proposed that FAK inhibition (as opposed to FAK KO) may suppress colorectal cancer tumor formation, the mechanisms connecting FAK activity to cell proliferative responses remain unresolved.
A reduction in cyclin D1 levels upon FAK loss is associated with decreased ERK activity in mammary epithelial cells73. Similarly, FAK deletion in the mouse PyMT breast tumor model decreases Src-mediated p130Cas activation and signaling to ERK43. Ex vivo knockout and subsequent transient re-expression of FAK in cells isolated from PyMT-induced breast tumors showed that FAK Y397 auto-phosphorylation, catalytic activity, and the integrity of FAK proline-rich region 2 (Figure 1B) are needed for cell proliferation as well as anoikis resistance45. In human breast carcinoma cell lines, FAK knockdown prevented tumor growth driven by oncogenic mutations in the Ras and PI3K signaling pathways45. These studies support the notion that FAK serves as a regulator for cell intrinsic signals promoting proliferation.
The location of FAK signaling complexes controlling cell survival and growth is also varied. Studies with FAK-KD mutants and pharmacological inhibitors support a kinase-independent scaffolding role in the nucleus38, 40, 68. In vitro, FAK nuclear translocation occurs after oxidative stress74 and upon pharmacological FAK inhibitor treatment75 (Figure 2). Nuclear FAK restricts p53 tumor-suppressive functions by promoting Mdm2 E3 ligase-dependent ubiquitination and degradation of p5311, 40. This prevents p53 transcriptional activity, reducing the levels of targets like the p21CIP1 cell cycle inhibitor76. FAK/p53 regulation may also release p53-induced inhibition of the PTK2 promoter, increasing FAK mRNA transcription. Interestingly, in a squamous cell carcinoma mouse model, FAK loss increases cell resistance to DNA damage after ionizing radiation, associated with p53-mediated induction of DNA repair77. Although this study raises the issue whether FAK inhibition in combination with radiation may be clinically disadvantageous, alternative conclusions find that endothelial FAK knockout acts to sensitize tumors to DNA-damaging therapy78.
FAK control of cancer stem cells
Cancer stem cells or tissue-specific progenitor cells can facilitate tumor growth, and in certain cancers, FAK signaling has been linked to the maintenance of these cell types. In the MMTV-PyMT mouse model, conditional embryonic FAK deletion suppresses mammary cancer stem cell (MaCSC) generation44. FAK loss reduces number and size of mammospheres and MaCSC surface markers. In the PyMT breast tumor model, FAK effects on MaCSC-associated markers are linked to FAK scaffolding effects on endophilin A238. Subsequent studies showed that FAK regulates MaCSC and normal progenitor cell activities via both kinase-dependent and -independent mechanisms41. In a conditional FAK-KD knockin mouse, loss of FAK kinase activity impairs luminal progenitor proliferation and reduces the MaCSC number, but does not affect FAK scaffolding functions required for basal mammary stem cell self-renewal41. This suggests that pharmacological FAK inhibition may be effective in only the subset of human breast cancer subtypes arising from luminal progenitor cells. However, this needs to be tested further as orthotopic tumor growth and spontaneous metastasis of basal-like murine 4T1 and human MDA-MB-231 breast carcinoma grafts is prevented by FAK inhibition in mice79. Inhibiting FAK also induces apoptosis in precursor B cells with a deletion in the Ikaros transcription factor80. Ikaros loss prevents B cell differentiation and locks precursor cells in a state of high adhesion-dependent proliferation, a process associated with B cell acute lymphoblastic leukemia80. Precursor B cells rely on integrin/FAK signaling as major driver of cell proliferation, survival, and self-renewal. Thus, FAK may be a key signaling protein downstream of integrins in the control of stem cell proliferation.
FAK in the stromal microenvironment
Signals between tumors and cells in the surrounding microenvironment can drive tumor progression. At sites of micro-metastases, tumor cells need to adapt to a new microenvironment and/or modify it. As mentioned above, non-cellular microenvironment cues, such as matrix composition or stiffness, cytokines, growth factors, integrins and pH changes trigger FAK activity that influences various aspects of tumor growth and metastasis. In this section, we will focus on the cellular microenvironment component, as FAK signaling plays important roles within vascular and non-vascular stromal cells in the tumor microenvironment.
Endothelial FAK in the control of progression
Several transgenic mouse models support the importance of FAK expression and activity in ECs during vascular development and tumor angiogenesis (Table 1). EC proliferation and survival are fundamental events promoting angiogenesis. Global, as well as EC-specific FAK-KO81–83, FAK-KD68, 76, or deletion of residues surrounding FAK Y39784 result in early embryonic lethality associated with multiple vascular defects, such as hemorrhage and edema. In vitro, primary ECs from these mice exhibit defects in survival, proliferation, sprouting, migration, and tubulogenesis.
EC FAK is considered a therapeutic target in vascular disease85, 86. In tumor-associated ECs, FAK mRNA and protein are elevated87 and FAK Y397 phosphorylation is increased88. Stimulated changes in EC migration are a fundamental component of angiogenesis and FAK activation downstream of growth factor, integrin, and cytokine receptors contributes to EC motility86. Pharmacological FAK inhibition prevents EC motility and tubulogenesis in vitro, aortic sprouting ex vivo, and growth factor-stimulated angiogenesis in mice24, 89. In a proteomic screen analyzing invading versus non-invading ECs in 3D-collagen matrices, pro-angiogenic factors promote the association of RACK1 and vimentin with FAK during endothelial invasion90. This linkage is hypothesized to mediate changes in EC shape and FA formation, initial steps in tumor neovascularization.
Orthotopic glioma implantation in adult mice with EC-specific FAK deletion results in tumor vascular normalization associated with reduced vascular permeability (VP), partial restoration of EC-EC and astrocyte-EC interactions91. Similarly, EC FAK deletion in melanoma and lung carcinoma-bearing mice results in tumor growth inhibition by impairing VEGF-induced angiogenesis92. Surprisingly, increased melanoma and lung carcinoma growth occurs in FAK heterozygous compared to wildtype mice93. This FAK heterozygous phenotype was associated with increased angiogenesis, but it is unclear whether this represents compensatory mechanisms or reduced FAK activity. In contrast, pharmacological FAK inhibition prevents EC sprouting in a dose-dependent manner24 and FAK inhibitors are potent anti-angiogenic agents89. Furthermore, FAK inhibition reduces tumor angiogenesis in animal models of human colon94, ovarian8, 88, 95 and hepatocellular carcinoma96, supporting a stimulating role of FAK activity in angiogenesis.
During development, EC specific FAK-KO results in decreased EC proliferation and survival, and increased apoptosis81, 83. FAK-KO results in reduced VEGF-stimulated Akt phosphorylation, associated with reduced EC proliferation and increased cell death92. It remains unclear, whether FAK plays differential roles in maintaining basal EC proliferation and survival, as opposed to stimulated events occurring during angiogenesis. In adult mice with a developed vasculature, EC FAK-KO does not trigger apoptosis, in part because Pyk2 is expressed and compensates for FAK loss in promoting cell survival23, 24 (Box 1). However, as mice with double FAK- and Pyk2-KO within ECs have not been described, it remains undetermined whether FAK-KD expression in a Pyk2-null background may function to promote EC survival and, if so, whether this would be dependent on a FAK-KD scaffolding or nuclear function. These experiments would provide important fundamental insights, as increased FAK nuclear translocation occurs upon treatment of ECs with FAK inhibitors75. Future studies need to determine the specific molecular roles of kinase-inhibited FAK in potentially promoting EC survival, but preventing angiogenesis.
The vasculature of tumors is often disorganized, tortuous, and leaky86. These changes are associated with alterations in EC adherens junctions (AJs) that maintain vascular barrier function. During development, global or EC-specific FAK-KD expression results in disorganized EC patterning and defective blood vessel morphogenesis68, 76 (Table 1). In human ECs, knockdown of FAK enhances AJ stability, associated with enhanced cell membrane localization of vascular endothelial cadherin (VE-cadherin)97. Although it was reported that conditional deletion of FAK in mouse endothelium disrupts lung barrier function in part through RhoA activity deregulation82, this phenotype has not been observed in two other EC FAK-KO mouse models24, 91. However, in other cell types, elevated RhoA activity occurs upon FAK-KO22, 98 and has been linked to alterations in Pyk2 and ARHGEF28 signaling22. Additionally, loss of FAK activity disrupted AJ formation during development76, but it remains unclear whether this is distinct from the observed embryonic lethal phenotype. In adult mice, pharmacological or genetic inhibition of FAK activity does not alter basal vascular barrier formation, but instead prevents paracellular permeability increases by VEGF69. These results support both kinase-dependent and -independent connections of FAK to AJ regulation.
VEGF promotes VP via tension-independent FAK activation, rapid FAK localization to EC cell-cell junctions, binding of the FAK FERM domain to VE-cadherin, and direct FAK phosphorylation of β-catenin, facilitating VE-cadherin-β-catenin dissociation and EC AJ breakdown69. In glioma studies, FAK expression is essential for tumor-induced VP in the brain of mice91. The signaling pathway promoting FAK activation downstream of VEGFR is different from that triggered by integrins, as VEGF-stimulated FAK activation and binding to VE-cadherin is regulated by a conformational change within the FAK FERM domain69. However, the molecular mechanisms underlying this regulation remain undetermined. Subsequent studies revealed that FAK kinase activity is required for Src translocation to AJs, and that FAK controls VE-cadherin Y658 phosphorylation, required to promote VEGF-stimulated VP and tumor cell extravasation88. Notably, FAK-KD expression and VE-cadherin Y658F mutation in ECs prevents tumor cell transmigration across EC barriers. Importantly, EC-specific FAK-KD expression in mice decreases VEGF-enhanced tumor cell extravasation in vivo, and EC FAK-KD expression prevents spontaneous orthotopic melanoma metastasis without affecting primary tumor growth88. While the mechanism for this anti-metastatic effect is not known, tumor- and VEGF-associated VE-cadherin internalization within ECs depends on FAK activity. Mechanistically, these results are consistent with roles of EC FAK in modulating expression of VCAM-1, a protein mediating the adhesion of immune cells to ECs75, and E-selectin99. The latter is proposed as a mechanism to prepare vascular microenvironment sites for the seeding of metastatic disease (Figure 3).
Figure 3. Regulation of vascular permeability and extravasation processes by endothelial cell FAK.

Tumor cells, via the secretion of growth factors and the activation EC-specific receptors (1) induce conformational FAK activation through multiple mechanisms. (2) FAK signaling promotes ERK/MAPK signaling cascade activation (3) leading to GATA-4-dependent transcriptional expression of VCAM-1 (4)75, a surface protein that can facilitate immune cell adhesion to ECs (5). EC FAK also promotes E-selectin expression (6) favoring tumor cell adhesion to ECs (7) and the lodging of metastatic cancer cells within sites of vascular hyper-permeability99. FAK activation can occur downstream of integrin receptor binding to matrix proteins consisting of a FAK-Src multi-protein complex (8). In response to VEGF signals, FAK promotes the localization of Src to adherens junctions; key sites that maintain vascular barrier integrity (9). FAK binding via FERM domain to the VE-cadherin (VEC) cytoplasmic tail, and FAK-dependent pY658 VEC and pY142 β-catenin phosphorylation (10) promotes VEC/β-catenin dissociation and VEC internalization/degradation. Loss of VEC from the cell surface leads to increased vascular permeability (11), allows for tumor cell transmigration across EC barriers, and leads to increased tumor cell metastasis (12)69, 88.
Overall, these studies reveal new roles for EC-specific FAK activity in the control of metastasis. Further studies are needed to determine whether anti-tumor growth effects of FAK inhibitors are primarily mediated through signaling inhibition within tumor cells or through effects on EC function. FAK inhibition may possess beneficial effects for patients in the control of tumor and other vascular pathologies by preventing VP and angiogenesis without negative effects on EC survival.
FAK promotion of tumor growth via effects in non-vascular stromal cells
In addition to FAK signaling within tumor and ECs, non-vascular stromal FAK functions also contribute to multiple aspects of tumor progression (Figure 4). Neutrophils and macrophages are major effectors of immune responses in conditions that induce inflammation, including cancer. Myeloid-specific (lysozyme M Cre) FAK-KO in mice decreases the capability of neutrophils to kill pathogens and triggers accelerated spontaneous death100. Other studies question whether FAK is expressed in neutrophils, and have used a similar myeloid-specific FAK-KO model to study macrophage function101. Primary FAK-KO mouse bone marrow macrophages have impaired directional chemotaxis in vitro and exhibit decreased monocyte recruitment to inflammatory sites in vivo. Pyk2 loss or combined FAK/Pyk2 knockdown have similar effects to FAK-KO, supporting overlapping signaling roles in macrophages101. Tumor-associated macrophages (TAMs) are key contributors to tumor progression and cancer-related inflammation. In a mouse model of pancreatic ductal adenocarcinoma, FAK inhibitor administration does not alter tumor angiogenesis, necrosis or apoptosis, but results in fewer TAMs within tumors and decreased primary tumor size102. In breast cancer mouse models, pharmacologic FAK inhibition decreases tumor growth, associated with diminished leukocyte and macrophage tumor infiltration79, 103. Other studies have used MX1-Cre to delete FAK in hematopoietic cells104, 105 and PF4-Cre to delete FAK in megakaryocyte lineages106. Resulting mouse and cell phenotypes are variable and have not yet been integrated into tumor studies (Table 1). Since potential “off-target” effects have been proposed to account for the inhibitory effects of FAK inhibitors on platelet spreading107, and most hematopoietic cells express Pyk2, combined Pyk2-FAK-KO or FAK-KD transgenic models are needed to further our understanding of tumor-immunological roles for FAK signaling.
Figure 4. Tumor microenvironmental impact of FAK signals.

FAK is an important regulator of EC, neutrophil, platelet, macrophage and fibroblast signaling in the tumor microenvironment leading to the increase (green boxes) or decrease (red boxes) of stromal cell functions. In ECs, FAK inhibits apoptosis and increases proliferation. EC FAK also contributes to the formation of abnormal vasculature via the increase of cell migration, survival, and vascular permeability (VP). Moreover, as described in detail in Figure 3, EC FAK is a key regulator of vascular permeability and tumor intra/extravasation leading to metastasis. FAK stimulates macrophage and fibroblast migration. FAK promotes the differentiation (dotted arrow) of macrophages. This occurs in stimulus-specific fashion where FAK activation either promotes or reverses (red/green box) fibroblast differentiation into CAF. For both macrophages and fibroblasts, FAK activity positively impacts cell recruitment to the tumor site. FAK promotes spreading, adhesion, and survival of stromal cells; with concomitant regulation ECM synthesis/remodeling to promote tumor progression. References are available in the corresponding text. VP: vascular permeability, TC: tumor cell, EC: endothelial cell, TAM: tumor-associated macrophage and CAF: cancer-associated fibroblast.
Cancer-associated fibroblasts (CAFs) influence tumor progression through mechanisms that are not fully understood. In a breast carcinoma model, tumor-secreted lysyl-oxidase-like-2 (LoxL2) activates fibroblasts and promotes α-smooth muscle actin expression in a FAK-dependent manner via Akt activation108. This signaling was blocked in vitro by pharmacologic inhibition of FAK but not of Src108. Since LoxL2 catalyzes matrix cross-linking, the effects on FAK may also be mediated through increased tissue tension33. Accordingly, FAK expression and activity are elevated in fibroblasts from lung fibrosis patients. Interestingly, FAK inhibition in a bleomycin-induced fibrosis mouse model shows marked abrogation of lung fibrosis109. Although early studies suggested that FAK expression might inhibit fibroblast differentiation and α-SMA expression, the potential role of compensatory Pyk2 levels in FAK-null fibroblasts was not addressed110. Finally, similar to TAMs, FAK inhibition in a pancreatic ductal adenocarcinoma model decreased CAF recruitment and tumor size102. Together, these studies support the notion that FAK promotes pro-tumor CAF functions.
FAK also influences the tumor microenvironment indirectly. FAK signaling within breast carcinoma cells regulates VEGF expression that, as described above, promotes VP and angiogenesis111. In acute myeloid leukemia, FAK expression and activity are important in the production of interleukin-6, -8, SDF-1 and angiopoietin-1, factors crucial for mesenchymal stromal stem cell maintenance112. FAK inhibition decreases TNFα-induced breast cancer cell interleukin-6 production and is correlated with reduced tumor-associated splenomegaly and tumor-associated CD45+ cells in a syngeneic model79. Overall, there is emerging evidence for the importance of FAK signaling as regulator of EC, hematopoietic cell, platelet, macrophage, and fibroblast signaling in the tumor microenvironment. Importantly, phenotypes associated with FAK inhibition show that there are multiple points of regulation for FAK function not only in tumor cells but also in the tumor microenvironment.
FAK in clinical applications
FAK functions drive various tumor-promoting signaling pathways (Figure 2)3, 94, 113, 114 and small molecule FAK inhibitors are emerging as promising chemotherapeutics, as FAK inhibition in mouse models prevents tumor growth, metastasis, vascular permeability, and angiogenesis8, 69, 79, 88, 89, 95, 102, 103, 115. Despite similarities between FAK- and Src-associated signaling pathways116, 117, unique FAK substrates have been identified and combined treatment with FAK and Src inhibitors shows enhanced anti-tumor activity in non-small cell lung cancer models118. FAK inhibitors have also exhibited enhanced activity in combination with cytotoxic drugs65, 95 or agents targeting angiogenesis, like the receptor tyrosine kinase inhibitor sunitinib96. This supports the notion that FAK inhibition will yield distinct responses and may act as a chemotherapy sensitizer.
However, the ability of Pyk2 to take over certain FAK functions after FAK deletion (Box 1) has to be factored into the design of FAK inhibitor therapies and dual FAK/Pyk2 inhibitors may yield different phenotypes. Similarly, FAK’s kinase-independent functions (Figure 2) have to be taken into consideration when designing or testing FAK kinase inhibitor therapy approaches. FAK’s scaffolding functions are not blocked, but possibly enhanced by FAK inhibition40, leading to potentially unpredictable therapy outcomes. Compounds in development against FAK can be sub-divided into ATP-competitive kinase inhibitors (KI), molecules blocking FAK catalytic activity by alternative means (aKI), and compounds targeting FAK scaffolding (scaffold inhibitors, SI) (Table 2).
Table 2. Anti-cancer compounds targeting FAK.
Shown are compounds currently in pre-clinical (PC) and clinical trials (I, Ib or II).
| Name | Alt. Name | Type | Specificity | Phase | Trial# | Reference |
|---|---|---|---|---|---|---|
| GSK2256098 | N.A. | N.A. | I | NCT01938443, NCT01138033, NCT00996671 | N.A. | |
| NVP-TAC544 | KI | FAK | PC | none | (24) | |
| PF 573,228 | PF-228 | KI | FAK | PC | none | (120) |
| TAE226 | NVP-226 | KI | FAK/Pyk2 | PC | none | (119) |
| VS-4718 | PND-1186 | KI | N.A. | I | NCT01849744 | (122) |
| VS-6062 | PF 562,271 PF-271 |
KI | FAK/Pyk2 | I | NCT00666926 | (121)(132) |
| VS-6063 | PF-04554878 defactinib | KI | N.A. | I/Ib, II | NCT01951690, NCT00787033, NCT01943292, NCT02004028, NCT01778803 | (65)(132) |
| 1H-Pyrrolo(2,3-b)pyridine | aKI | N.A. | PC | none | (128) | |
| Compound 1 and 2 | aKI | N.A. | PC | none | (127) | |
| Y15 | Compound 14 | aKI | FAK | PC | none | (130) |
| C4 | chloropyramine hydrochloride | SI | N.A. | PC | none | (130) |
| R2 | Roslins | SI | N.A. | PC | none | (131) |
| Y11 | SI | FAK | PC | none | (129) |
KI: ATP-competitive kinase inhibitors, aKI: molecules blocking enzymatic activity by alternative means, SI: protein scaffold inhibitors. N.A. Data not available.
Small molecule ATP-competitive kinase inhibitors
Small molecules inhibitors that bind within the active site of kinases compete with relatively high levels of ATP present in cells. Inhibitors are designed to make binding interactions with residues surrounding the ATP binding pocket of kinases. The best characterized cellular-active and selective nanomolar affinity FAK inhibitors are comprised of pyrimidine (TAE-226, PF 573,228, PF 562,271) or pyridine (VS-4718, previously known as PND-1186) ATP site hinge binders119–122. Despite highly conserved elements within the tyrosine kinase ATP binding pocket, FAK inhibitor selectivity is achieved through stabilization of the FAK kinase activation loop “DFG” motif into a helical conformation121, 123. The unusual kinase domain conformation and presence of Gly-563 preceding the DFG motif may confer loop flexibility as well as FAK selectivity124. Other pre-clinical FAK inhibitors, as discussed later, do not possess this type of target selectivity and no peer-reviewed information is available for the clinical-stage FAK inhibitor GSK-2256098.
In cell culture and animal models, these FAK inhibitors effectively decrease FAK Y397 auto-phosphorylation, prevent cell movement, but do not necessarily induce cell apoptosis in adherent culture conditions120, 122. Certain breast and ovarian tumor cells are resistant to growth inhibition at micromolar FAK inhibitor levels in 2D tissue culture conditions, but become sensitive to nanomolar FAK inhibitor concentrations when grown in a 3D anchorage-independent cell spheroid environment8, 122. This has been linked to cell type-specific dependence on integrin-ECM signals within spheroids. Particularly in mesothelioma cells with inactivating mutations in the NF2 gene, which encodes the Merlin tumor suppressor protein, survival and proliferation signals are mediated through cell-ECM rather than cadherin cell-cell contact signals125. Low Merlin protein levels are therefore predicted to serve as a biomarker for FAK inhibitor sensitivity in mesothelioma (Trial ID: NCT01870609) and possibly also in ovarian cancer126.
Alternative approaches to inhibit FAK function
New allosteric FAK inhibitors that bind to distinct kinase domain sites and do not directly compete with ATP binding are being developed127, 128. These compounds have the potential for high FAK specificity, but have not been rigorously tested in pre-clinical models. Several studies have identified small molecules via molecular docking analyses that may disrupt different FAK scaffolding protein-protein interactions. These include compounds of limited complexity (molecular weight < 300) termed C4, Y11, Y15, and R2 (Table 2). Proposed mechanisms are that C4 blocks FAK C-terminal domain interactions, Y11 and Y15 block access to the FAK Y397 site, and R2 blocks FAK interaction with p53129–131. These compounds act at micromolar concentrations in cells and show anti-tumor activity in xenograft mouse models. Although they have been shown to enhance the anti-tumor activity of other chemotherapeutics, questions remain about target selectivity.
FAK inhibitors in clinical trials
Pfizer (PF 562,271, Trial ID: NCT00666926) and GSK (GSK-2256098, Trial ID: NCT00996671) initiated phase I clinical trials with FAK inhibitors in 2008 and 2009, respectively. Both trials found that the compounds are tolerated with low adverse events. Notably, in the Pfizer trial, some patients exhibited stable disease while being treated with FAK inhibitor132. However, PF 562,271 (now termed VS-6062) displayed non-linear pharmacokinetics and was discontinued. PF-04554878, a later generation ATP site hinge binder, showed more favorable pharmacokinetics, and a phase I trial identified some ovarian, colorectal, and bile duct tumor patients exhibiting stable disease (Trial ID: NCT00787033)
Following the acquisition of the FAK inhibitor rights, Verastem initiated new trials (phase I and II) with PF-04554878 (now named VS-6063 or defactinib) and one phase I clinical study with PND-1186 (acquired from Poniard, now termed VS-4718) (Table 2). A phase II trial in patients with KRAS mutant non-small cell lung cancer (Trial ID: NCT01951690) is testing responses to VS-6063 treatment based on the status of tumor-associated INK4a/Arf and p53 mutations. This study is based upon the finding that FAK inhibitor sensitivity may be associated with the inactivation of INK4a/Arf as part of a RhoA GTPase feedback pathway that leads to FAK activation133. Additionally, as VS-6063 was found to enhance taxane-sensitivity of ovarian carcinoma cells65, a phase I/Ib study (Trial ID: NCT01778803) is evaluating the safety and effectiveness of VS-6063 as combinatorial treatment with paclitaxel. Finally, the inverse relationship between Merlin expression and FAK inhibitor sensitivity125 provides rationale for trials with VS-6063 (Trial ID: NCT01870609) and GSK2256098 (Trial ID: NCT01938443) in mesothelioma, where a large number of patients possess mutations in the Merlin gene. Results from these trials will show if Merlin status can provide a useful biomarker to predict patient response to FAK inhibitor therapy.
FAK’s future as a therapeutic target
One of the strongest rationales for FAK inhibition is that effects are mediated through alterations in both tumor and stromal cell biology. Known pathways are the prevention of cell motility, invasion, survival, and proliferation that are being driven by oncogenes and a variety of cell surface receptors. In addition to cancer, FAK inhibition may yield clinical benefits for vascular pathologies such as edema and in limiting inflammation. FAK inhibition in the prevention of vascular permeability may prevent tumor metastasis, could enhance chemotherapy drug delivery, and may help overcome chemo-resistance in patients.
Interestingly, a recent report suggests that EC FAK inactivation may enhance the effects of DNA damaging cancer treatments like doxorubicin or radiation therapy78. In this study, EC FAK loss prevented doxorubicin-stimulated NF-kB activation and the production of various cytokines, which act to protect tumor cells from DNA damage-driven apoptosis. Further studies are needed to determine if this connection is generalizable or dependent upon the loss of FAK activity. Nevertheless, FAK inhibitors may be promising drugs for combinatorial therapies including DNA damaging agents, an approach that may increase efficacy of these agents and overcome chemoresistance.
Lastly, FAK inhibitors may possess single agent activity in cancers where FAK expression and activity are amplified or where tumor cells become dependent on FAK-associated signals126, 134, 135. Examples include Ewing sarcoma134 or ovarian serous carcinoma135, where treatment with the FAK inhibitor PF-562,271 blocked in vitro and in vivo tumor growth in pre-clinical studies. Future research into the FAK-associated pathways will elucidate new chemotherapy combinations and biomarkers for patient stratification.
Key Points.
Focal Adhesion Kinase (FAK) is a non-receptor protein tyrosine kinase that drives tumor growth and metastasis through kinase-dependent and –independent pathways.
FAK promotes metastasis by regulating processes involved in tumor cell motility and invasion, including control of focal adhesion and cytoskeletal dynamics, as well as the regulation of matrix metalloproteinase (MMP) surface expression.
Tumor growth is enhanced through pro-proliferative and anti-apoptotic functions of FAK.
FAK is connected to cancer stem cell and progenitor cell maintenance through kinase-dependent and -independent functions. FAK signals contribute to the malignant outgrowth of these cells.
FAK favors tumor progression via the regulation of signaling pathways within cells of the tumor microenvironment, such as endothelial cells, hematopoietic cells, platelets, macrophages, and fibroblasts.
FAK activity promotes endothelial cell migration, proliferation, survival and stimulates tumor angiogenesis. FAK-mediated regulation of endothelial cell permeability can influence tumor metastasis.
FAK expression and activity in tumor and endothelial cells is frequently upregulated and correlated with a poor patient prognosis.
Several molecules targeting FAK kinase activity or its kinase-independent scaffolding function are under investigation in pre-clinical trials. Promising drug candidates in phase I or II clinical trials are small molecule ATP-competitive inhibitors.
Acknowledgments
We apologize to those authors whose work on FAK signaling has advanced the field, but we were unable to cite due to journal limitations. Studies in the Schlaepfer laboratory are funded by National Institutes of Health (NIH) grants CA102310 and CA180769. F. Sulzmaier is supported by NIH training grant (T32-CA121938). C. Jean is supported by an American Heart Association fellowship (12POST11760014). We thank Jonathan Pachter, Head of Research at Verastem Inc., for insightful discussions of ongoing FAK inhibitor clinical trials. We also thank Christine Lawson, Nichol L. Miller and Isabelle Tancioni for discussions and help in revising the manuscript.
Abbreviations
- AJ
adherens junction
- EC
endothelial cell
- ECM
Extracellular matrix
- EMT
epithelial-mesenchymal transition
- FA
focal adhesion
- FERM
band 4.1-ezrin-radixin-moesin
- GEF
guanine-nucleotide exchange factor
- MMP
matrix metalloproteinase
- KD
kinase-dead
- KO
knock out
- VP
vascular permeability
Glossary of terms
- Integrin receptor clustering (p.6)
The formation of multimeric membrane integrin clusters upon binding to extracellular matrix ligands, inducing the formation of multi-protein complexes at cytoplasmic integrin tails to drive focal adhesion formation and cytoskeletal rearrangement
- Focal Adhesion (p.6)
A multi-protein complex regulating cellular attachment by linking the actin cytoskeleton to components of the extracellular matrix via transmembrane receptors termed integrins
- Epithelial-mesenchymal transition (EMT) (p.7)
A cellular mechanism that allows polarized epithelial cells to acquire a mesenchymal phenotype characterized by increased cell migration and invasion and the ability to survive in adhesion independent conditions
- Floxed mouse models (p.7)
Transgenic insertion of loxP sites flanking a gene of interest. Induced expression of Cre recombinase catalyzes recombination between the loxP repeats and mediates the deletion of the gene of interest
- MMTV-PyMT model (p.7)
A mouse model with conditional expression of the polyoma virus middle T-antigen under the control of the mouse mammary tumor virus promoter, inducing the formation of mammary tumors
- Guanine nucleotide exchange factor (GEF) (p.7)
A protein that promotes the exchange of GDP for GTP on a GTPase, thereby facilitating its activation
- Invadopodia (p.8)
Specialized membrane protrusions (also known as an invasive pseudopodia) where active extracellular matrix degradation takes place
- Arp2/3 (p.8)
A seven-subunit protein complex involved in regulation of the actin cytoskeleton; mediates the nucleation of branched actin filaments
- N-WASP (Neural Wiskott-Aldrich syndrome protein) (p.8)
Promotes actin polymerization by stimulating the activity of the Arp2/3 complex
- Anoikis (p.10)
Cell death (apoptosis) induced by the loss of cell-matrix adhesion and a physiological mechanism to prevent cell displacement
- Mammospheres (p.11)
A collection of cells arising from a single cell of mammary origin through clonal growth in culture
- Vascular normalization (p.13)
The process of restoring normal vasculature form the classical cancer-associated tortuous and leaky vessels. This phenomenon involves increased vascular pericyte coverage and decreased VP and hypoxia, and results in decreased metastasis and increased blood perfusion, rendering vessels more efficient for oxygen and drug delivery
- Tumor cell extravasation (p.15)
The crucial step in tumor metastasis where tumor cells exit the vasculature to penetrate target organs. This requires tumor cell adhesion to the endothelium, spreading out across endothelial cells, and penetration of the basement membrane
- ATP site hinge (p.18)
A segment that connects the two lobes of a kinase domain. Hinge and kinase lobes form an interface creating the ATP binding pocket
Biographies
David D. Schlaepfer, Ph.D. is a Professor in The Moores UCSD Cancer Center in La Jolla, CA and has studied FAK for over 20 years. His group uses mouse models to study tumor-stromal interactions, mechanisms of metastasis, pre-clinical analyses of small molecule FAK inhibitors, and signaling linkages that promote ovarian tumor recurrence and progression.
Florian J. Sulzmaier, Ph.D. is a postdoctoral fellow at The Moores UCSD Cancer Center in La Jolla, CA. He completed his Ph.D. at the University of Hawaii at Manoa, studying kinase signaling pathways that control cell metastasis in cancers of the central nervous system. Florian is currently investigating the role of FAK in tumor epithelial-mesenchymal transition.
Christine Jean, Ph.D. is a postdoctoral fellow at The Moores UCSD Cancer Center in La Jolla, CA. She completed her Ph.D. in Toulouse France her interests revolve around the role of the microenvironment in modulating tumor growth and metastasis. She is currently investigating mechanisms of endothelial FAK signaling on tumor progression.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 2.Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123:1007–13. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 3.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 4.Goode EL, et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42:874–9. doi: 10.1038/ng.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sood AK, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest. 2010;120:1515–23. doi: 10.1172/JCI40802. Demonstrates that hormonal stress (increased levels of norepinephrine) protects ovarian cancer cells from anoikis in vitro and in vivo through FAK signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward KK, et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin Exp Metastasis. 2013;30:579–94. doi: 10.1007/s10585-012-9562-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brami-Cherrier K, et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014 doi: 10.1002/embj.201386399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corsi JM, Rouer E, Girault JA, Enslen H. Organization and post-transcriptional processing of focal adhesion kinase gene. BMC Genomics. 2006;7:198. doi: 10.1186/1471-2164-7-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cance WG, Golubovskaya VM. Focal adhesion kinase versus p53: apoptosis or survival? Sci Signal. 2008;1:pe22. doi: 10.1126/stke.120pe22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho B, et al. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J Biol Chem. 2012;287:18656–73. doi: 10.1074/jbc.M111.322883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng N, Li Y, Han ZG. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology. 2013;57:1906–18. doi: 10.1002/hep.26202. [DOI] [PubMed] [Google Scholar]
- 14.Li S, et al. Requirement of PEA3 for transcriptional activation of FAK gene in tumor metastasis. PLoS One. 2013;8:e79336. doi: 10.1371/journal.pone.0079336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang XQ, et al. Somatic mutational analysis of FAK in breast cancer: A novel gain-of-function mutation due to deletion of exon 33. Biochem Biophys Res Commun. 2014;443:363–9. doi: 10.1016/j.bbrc.2013.11.134. [DOI] [PubMed] [Google Scholar]
- 16.Yao L, et al. An aberrant spliced transcript of focal adhesion kinase is exclusively expressed in human breast cancer. J Transl Med. 2014;12:136. doi: 10.1186/1479-5876-12-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canel M, et al. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin Cancer Res. 2006;12:3272–9. doi: 10.1158/1078-0432.CCR-05-1583. [DOI] [PubMed] [Google Scholar]
- 18.Kong X, et al. MicroRNA-7 inhibits epithelial-to-mesenchymal transition and metastasis of breast cancer cells via targeting FAK expression. PLoS One. 2012;7:e41523. doi: 10.1371/journal.pone.0041523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen N, Yi JS, Park H, Lee JS, Ko YG. MG53 ubiquitinates focal adhesion kinase during skeletal myogenesis. J Biol Chem. 2013 doi: 10.1074/jbc.M113.525154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol. 2010;11:802–14. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- 22.Lim Y, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim ST, et al. Pyk2 inhibition of p53 as an adaptive and intrinsic mechanism facilitating cell proliferation and survival. J Biol Chem. 2010;285:1743–53. doi: 10.1074/jbc.M109.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weis SM, et al. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol. 2008;181:43–50. doi: 10.1083/jcb.200710038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plaza-Menacho I, et al. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J Biol Chem. 2011;286:17292–302. doi: 10.1074/jbc.M110.168500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper LA, Shen TL, Guan JL. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol Cell Biol. 2003;23:8030–41. doi: 10.1128/MCB.23.22.8030-8041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lietha D, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–87. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai X, et al. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28:201–14. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung O, et al. Tetraspan TM4SF5-dependent direct activation of FAK and metastatic potential of hepatocarcinoma cells. J Cell Sci. 2012;125:5960–73. doi: 10.1242/jcs.100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen TH, Chan PC, Chen CL, Chen HC. Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene. 2011;30:153–66. doi: 10.1038/onc.2010.398. [DOI] [PubMed] [Google Scholar]
- 31.Ritt M, Guan JL, Sivaramakrishnan S. Visualizing and manipulating focal adhesion kinase regulation in live cells. J Biol Chem. 2013;288:8875–86. doi: 10.1074/jbc.M112.421164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi CH, Webb BA, Chimenti MS, Jacobson MP, Barber DL. pH sensing by FAK-His58 regulates focal adhesion remodeling. J Cell Biol. 2013;202:849–59. doi: 10.1083/jcb.201302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seong J, et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc Natl Acad Sci U S A. 2013;110:19372–7. doi: 10.1073/pnas.1307405110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012;2:706–21. doi: 10.1158/2159-8290.CD-11-0239. Integrin-dependent FAK activation and signaling through ERK promote the proliferation of cancer cells after extravasation into the lung. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci. 2013;126:21–9. doi: 10.1242/jcs.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li XY, et al. Snail1 controls epithelial-mesenchymal lineage commitment in focal adhesion kinase-null embryonic cells. J Cell Biol. 2011;195:729–38. doi: 10.1083/jcb.201105103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan H, Zhao X, Sun S, Luo M, Guan JL. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J Biol Chem. 2013;288:3322–33. doi: 10.1074/jbc.M112.420497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393–401. doi: 10.1242/jcs.100115. [DOI] [PubMed] [Google Scholar]
- 40.Lim ST, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo M, et al. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res. 2013;73:5591–602. doi: 10.1158/0008-5472.CAN-13-1351. Demonstrates that FAK regulates mammary cancer stem cells (MaCSC) and luminal progenitors via kinase-dependent and -independent mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lahlou H, et al. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci U S A. 2007;104:20302–7. doi: 10.1073/pnas.0710091104. Mammary epithelial-specific deletion of FAK in PyMT mice prevents the progression of hyperplastic growth to malignant breast carcinomas. Demonstrates FAK’s role in the development of in vivo breast tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Provenzano PP, Inman DR, Eliceiri KW, Beggs HE, Keely PJ. Mammary epithelial-specific disruption of focal adhesion kinase retards tumor formation and metastasis in a transgenic mouse model of human breast cancer. Am J Pathol. 2008;173:1551–65. doi: 10.2353/ajpath.2008.080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo M, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009;69:466–74. doi: 10.1158/0008-5472.CAN-08-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pylayeva Y, et al. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252–66. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLean GW, et al. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–15. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 47.Miller NL, Lawson C, Chen XL, Lim ST, Schlaepfer DD. Rgnef (p190RhoGEF) knockout inhibits RhoA activity, focal adhesion establishment, and cell motility downstream of integrins. PLoS One. 2012;7:e37830. doi: 10.1371/journal.pone.0037830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller NL, et al. A non-canonical role for Rgnef in promoting integrin-stimulated focal adhesion kinase activation. J Cell Sci. 2013;126:5074–85. doi: 10.1242/jcs.135509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu HG, et al. p190RhoGEF (Rgnef) promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res. 2011;71:360–70. doi: 10.1158/0008-5472.CAN-10-2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawson C, et al. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J Cell Biol. 2012;196:223–32. doi: 10.1083/jcb.201108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barbero S, et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009;69:3755–63. doi: 10.1158/0008-5472.CAN-08-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomar A, Lawson C, Ghassemian M, Schlaepfer DD. Cortactin as a target for FAK in the regulation of focal adhesion dynamics. PLoS One. 2012;7:e44041. doi: 10.1371/journal.pone.0044041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eke I, et al. beta(1)Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J Clin Invest. 2012;122:1529–40. doi: 10.1172/JCI61350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan KT, Cortesio CL, Huttenlocher A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J Cell Biol. 2009;185:357–70. doi: 10.1083/jcb.200809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serrels B, et al. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol. 2007;9:1046–56. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- 56.Tang H, et al. Loss of Scar/WAVE complex promotes N-WASP- and FAK-dependent invasion. Curr Biol. 2013;23:107–17. doi: 10.1016/j.cub.2012.11.059. [DOI] [PubMed] [Google Scholar]
- 57.Mitra SK, Lim ST, Chi A, Schlaepfer DD. Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene. 2006;25:4429–40. doi: 10.1038/sj.onc.1209482. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J Cell Biol. 2012;196:375–85. doi: 10.1083/jcb.201105153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen JS, et al. Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis. 2013;34:10–9. doi: 10.1093/carcin/bgs274. [DOI] [PubMed] [Google Scholar]
- 60.Lu H, et al. KLF8 and FAK cooperatively enrich the active MMP14 on the cell surface required for the metastatic progression of breast cancer. Oncogene. 2013 doi: 10.1038/onc.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cicchini C, et al. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314:143–52. doi: 10.1016/j.yexcr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 62.Avizienyte E, et al. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat Cell Biol. 2002;4:632–8. doi: 10.1038/ncb829. [DOI] [PubMed] [Google Scholar]
- 63.Canel M, et al. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: effects on E-cadherin dynamics. Cancer Res. 2010;70:9413–22. doi: 10.1158/0008-5472.CAN-10-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lane D, Goncharenko-Khaider N, Rancourt C, Piche A. Ovarian cancer ascites protects from TRAIL-induced cell death through alphavbeta5 integrin-mediated focal adhesion kinase and Akt activation. Oncogene. 2010;29:3519–31. doi: 10.1038/onc.2010.107. [DOI] [PubMed] [Google Scholar]
- 65.Kang Y, et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J Natl Cancer Inst. 2013;105:1485–95. doi: 10.1093/jnci/djt210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng Y, et al. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene. 2013;32:4304–12. doi: 10.1038/onc.2012.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bae YH, et al. A FAK-Cas-Rac-Lamellipodin Signaling Module Transduces Extracellular Matrix Stiffness into Mechanosensitive Cell Cycling. Sci Signal. 2014;7:ra57. doi: 10.1126/scisignal.2004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lim ST, et al. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J Biol Chem. 2010;285:21526–36. doi: 10.1074/jbc.M110.129999. Studies (together with Reference #83) create a FAK kinase-dead (KD) knockin mouse models to test the importance of FAK enzymatic activity in development. FAK-KD is embryonically lethal due to defective blood vessel formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen XL, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell. 2012;22:146–57. doi: 10.1016/j.devcel.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A. 2009;106:10290–5. doi: 10.1073/pnas.0904227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ashton GH, et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev Cell. 2010;19:259–69. doi: 10.1016/j.devcel.2010.07.015. Shows that FAK is essential for Wnt/c-myc mediated regeneration of the intestinal epithelium and provides a novel pathway by which FAK promotes transformation of APC deficient epithelial cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Owen KA, Abshire MY, Tilghman RW, Casanova JE, Bouton AH. FAK regulates intestinal epithelial cell survival and proliferation during mucosal wound healing. PLoS One. 2011;6:e23123. doi: 10.1371/journal.pone.0023123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nagy T, et al. Mammary epithelial-specific deletion of the focal adhesion kinase gene leads to severe lobulo-alveolar hypoplasia and secretory immaturity of the murine mammary gland. J Biol Chem. 2007;282:31766–76. doi: 10.1074/jbc.M705403200. [DOI] [PubMed] [Google Scholar]
- 74.Luo SW, et al. Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. EMBO J. 2009;28:2568–82. doi: 10.1038/emboj.2009.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lim ST, et al. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J Cell Biol. 2012;197:907–19. doi: 10.1083/jcb.201109067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao X, Peng X, Sun S, Park AY, Guan JL. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J Cell Biol. 2010;189:955–65. doi: 10.1083/jcb.200912094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Graham K, Moran-Jones K, Sansom OJ, Brunton VG, Frame MC. FAK deletion promotes p53-mediated induction of p21, DNA-damage responses and radio-resistance in advanced squamous cancer cells. PLoS One. 2011;6:e27806. doi: 10.1371/journal.pone.0027806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tavora B, et al. Endothelial-FAK targeting sensitises tumours to DNA-damaging therapy. Nature. 2014 doi: 10.1038/nature13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Walsh C, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010;9:778–90. doi: 10.4161/cbt.9.10.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joshi I, et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. 2014;15:294–304. doi: 10.1038/ni.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Braren R, et al. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006;172:151–62. doi: 10.1083/jcb.200506184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schmidt TT, et al. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am J Physiol Lung Cell Mol Physiol. 2013;305:L291–300. doi: 10.1152/ajplung.00094.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shen TL, et al. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005;169:941–52. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Corsi JM, et al. Autophosphorylation-independent and -dependent functions of focal adhesion kinase during development. J Biol Chem. 2009;284:34769–76. doi: 10.1074/jbc.M109.067280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Infusino GA, Jacobson JR. Endothelial FAK as a therapeutic target in disease. Microvasc Res. 2012;83:89–96. doi: 10.1016/j.mvr.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Angelucci A, Bologna M. Targeting vascular cell migration as a strategy for blocking angiogenesis: the central role of focal adhesion protein tyrosine kinase family. Curr Pharm Des. 2007;13:2129–45. doi: 10.2174/138161207781039643. [DOI] [PubMed] [Google Scholar]
- 87.Lu C, et al. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007;67:1757–68. doi: 10.1158/0008-5472.CAN-06-3700. [DOI] [PubMed] [Google Scholar]
- 88.Jean C, et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J Cell Biol. 2014;204:247–63. doi: 10.1083/jcb.201307067. Pharmacological inhibition of FAK or endothelial-specific expression of FAK-KD prevents extravasation and spontaneous tumor spread by enhancing endothelial vessel barrier function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cabrita MA, et al. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol Oncol. 2011;5:517–26. doi: 10.1016/j.molonc.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dave JM, Kang H, Abbey CA, Maxwell SA, Bayless KJ. Proteomic profiling of endothelial invasion revealed receptor for activated C kinase 1 (RACK1) complexed with vimentin to regulate focal adhesion kinase (FAK) J Biol Chem. 2013;288:30720–33. doi: 10.1074/jbc.M113.512467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee J, Borboa AK, Chun HB, Baird A, Eliceiri BP. Conditional deletion of the focal adhesion kinase FAK alters remodeling of the blood-brain barrier in glioma. Cancer Res. 2010;70:10131–40. doi: 10.1158/0008-5472.CAN-10-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tavora B, et al. Endothelial FAK is required for tumour angiogenesis. EMBO Mol Med. 2010;2:516–28. doi: 10.1002/emmm.201000106. This report provides genetic evidence for a causative role for endothelial cell FAK expression in promoting tumor angiogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kostourou V, et al. FAK-heterozygous mice display enhanced tumour angiogenesis. Nat Commun. 2013;4:2020. doi: 10.1038/ncomms3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19:777–88. doi: 10.1517/13543784.2010.489548. [DOI] [PubMed] [Google Scholar]
- 95.Halder J, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67:10976–83. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- 96.Bagi CM, et al. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol Ther. 2009;8:856–65. doi: 10.4161/cbt.8.9.8246. [DOI] [PubMed] [Google Scholar]
- 97.Arnold KM, Goeckeler ZM, Wysolmerski RB. Loss of focal adhesion kinase enhances endothelial barrier function and increases focal adhesions. Microcirculation. 2013;20:637–49. doi: 10.1111/micc.12063. [DOI] [PubMed] [Google Scholar]
- 98.Schober M, et al. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol. 2007;176:667–80. doi: 10.1083/jcb.200608010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hiratsuka S, et al. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci U S A. 2011;108:3725–30. doi: 10.1073/pnas.1100446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kasorn A, et al. Focal adhesion kinase regulates pathogen-killing capability and life span of neutrophils via mediating both adhesion-dependent and -independent cellular signals. J Immunol. 2009;183:1032–43. doi: 10.4049/jimmunol.0802984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Owen KA, et al. Regulation of lamellipodial persistence, adhesion turnover, and motility in macrophages by focal adhesion kinase. J Cell Biol. 2007;179:1275–87. doi: 10.1083/jcb.200708093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stokes JB, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10:2135–45. doi: 10.1158/1535-7163.MCT-11-0261. Shows that FAK inhibition blocks pancreatic tumor proliferation either directly by inhibiting tumor cell proliferation or indirectly by impairing the recruitment and/or proliferation of stromal cells to the tumor microenvironment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11:R68. doi: 10.1186/bcr2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lu J, et al. Fak depletion in both hematopoietic and nonhematopoietic niche cells leads to hematopoietic stem cell expansion. Exp Hematol. 2012;40:307–17 e3. doi: 10.1016/j.exphem.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vemula S, et al. Essential role for focal adhesion kinase in regulating stress hematopoiesis. Blood. 2010;116:4103–15. doi: 10.1182/blood-2010-01-262790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hitchcock IS, et al. Roles of focal adhesion kinase (FAK) in megakaryopoiesis and platelet function: studies using a megakaryocyte lineage specific FAK knockout. Blood. 2008;111:596–604. doi: 10.1182/blood-2007-05-089680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roh ME, Cosgrove M, Gorski K, Hitchcock IS. Off-targets effects underlie the inhibitory effect of FAK inhibitors on platelet activation: studies using Fak-deficient mice. J Thromb Haemost. 2013;11:1776–8. doi: 10.1111/jth.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barker HE, Bird D, Lang G, Erler JT. Tumor-secreted LOXL2 activates fibroblasts through FAK signaling. Mol Cancer Res. 2013;11:1425–36. doi: 10.1158/1541-7786.MCR-13-0033-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lagares D, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–64. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Greenberg RS, et al. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 2006;20:1006–8. doi: 10.1096/fj.05-4838fje. [DOI] [PubMed] [Google Scholar]
- 111.Mitra SK, et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–84. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- 112.Despeaux M, et al. Critical features of FAK-expressing AML bone marrow microenvironment through leukemia stem cell hijacking of mesenchymal stromal cells. Leukemia. 2011;25:1789–93. doi: 10.1038/leu.2011.145. Supports the idea that the ability of bone marrow mesenchymal stromal cells to organize a pro-tumoral niche (favoring quiescence and resistance of hematopoietic stem cells) is dependent on FAK expression within acute myeloid leukemia. [DOI] [PubMed] [Google Scholar]
- 113.Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14:627–32. doi: 10.1158/1078-0432.CCR-07-2220. [DOI] [PubMed] [Google Scholar]
- 114.McLean GW, et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Slack-Davis JK, Hershey ED, Theodorescu D, Frierson HF, Parsons JT. Differential requirement for focal adhesion kinase signaling in cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol Cancer Ther. 2009;8:2470–7. doi: 10.1158/1535-7163.MCT-09-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bolos V, Gasent JM, Lopez-Tarruella S, Grande E. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco Targets Ther. 2010;3:83–97. doi: 10.2147/ott.s6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Serrels A, et al. The role of focal adhesion kinase catalytic activity on the proliferation and migration of squamous cell carcinoma cells. Int J Cancer. 2012;131:287–97. doi: 10.1002/ijc.26351. [DOI] [PubMed] [Google Scholar]
- 118.Lu H, et al. IGFBP2/FAK pathway is causally associated with dasatinib resistance in non-small cell lung cancer cells. Mol Cancer Ther. 2013;12:2864–73. doi: 10.1158/1535-7163.MCT-13-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shi Q, et al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinog. 2007;46:488–96. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- 120.Slack-Davis JK, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 121.Roberts WG, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935–44. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 122.Tanjoni I, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9:764–77. doi: 10.4161/cbt.9.10.11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lietha D, Eck MJ. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS One. 2008;3:e3800. doi: 10.1371/journal.pone.0003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 125.Shapiro IM, et al. Merlin Deficiency Predicts FAK Inhibitor Sensitivity: A Synthetic Lethal Relationship. Sci Transl Med. 2014;6:237ra68. doi: 10.1126/scitranslmed.3008639. This study shows that mesothelioma cells with low expression of the tumor suppressor Merlin are very sensitive to pharmacological FAK inhibition, and is therefore the first to identify a potential biomarker for patient stratification for chemotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shah NR, et al. Analyses of merlin/NF2 connection to FAK inhibitor responsiveness in serous ovarian cancer. Gynecol Oncol. 2014 doi: 10.1016/j.ygyno.2014.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tomita N, et al. Structure-based discovery of cellular-active allosteric inhibitors of FAK. Bioorg Med Chem Lett. 2013;23:1779–85. doi: 10.1016/j.bmcl.2013.01.047. [DOI] [PubMed] [Google Scholar]
- 128.Heinrich T, et al. Fragment-based discovery of new highly substituted 1H-pyrrolo[2,3-b]- and 3H-imidazolo[4,5-b]-pyridines as focal adhesion kinase inhibitors. J Med Chem. 2013;56:1160–70. doi: 10.1021/jm3016014. [DOI] [PubMed] [Google Scholar]
- 129.Golubovskaya VM, et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)]decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis. 2012;33:1004–13. doi: 10.1093/carcin/bgs120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci Signal. 2013;6:pe10. doi: 10.1126/scisignal.2004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Golubovskaya VM, et al. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer. 2013;13:342. doi: 10.1186/1471-2407-13-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Infante JR, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30:1527–33. doi: 10.1200/JCO.2011.38.9346. First clinical trial report of a completed phase I study with an ATP-competitive FAK kinase inhibitor shows that it is safe and well-tolerated. [DOI] [PubMed] [Google Scholar]
- 133.Konstantinidou G, et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 2013;3:444–57. doi: 10.1158/2159-8290.CD-12-0388. Shows that RhoA/FAK signaling drives lung cancer tumor progression upon loss of Cdkn2a and expression of mutant KRAS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Crompton BD, et al. High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer Res. 2013;73:2873–83. doi: 10.1158/0008-5472.CAN-12-1944. [DOI] [PubMed] [Google Scholar]
- 135.Tancioni I, et al. FAK inhibition disrupts a beta5 integrin signaling axis controlling anchorage-independent ovarian carcinoma growth. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-13-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lulo J, Yuzawa S, Schlessinger J. Crystal structures of free and ligand-bound focal adhesion targeting domain of Pyk2. Biochem Biophys Res Commun. 2009;383:347–52. doi: 10.1016/j.bbrc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 137.Kuang BH, et al. Proline-rich tyrosine kinase 2 and its phosphorylated form pY881 are novel prognostic markers for non-small-cell lung cancer progression and patients’ overall survival. Br J Cancer. 2013;109:1252–63. doi: 10.1038/bjc.2013.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Loftus JC, et al. miRNA expression profiling in migrating glioblastoma cells: regulation of cell migration and invasion by miR-23b via targeting of Pyk2. PLoS One. 2012;7:e39818. doi: 10.1371/journal.pone.0039818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fan H, Guan JL. Compensatory function of Pyk2 protein in the promotion of focal adhesion kinase (FAK)-null mammary cancer stem cell tumorigenicity and metastatic activity. J Biol Chem. 2011;286:18573–82. doi: 10.1074/jbc.M110.200717. Demonstrates how Pyk2 compensates for FAK loss in mouse tumor models and controls mammary cancer stem cell (MaCSC) renewal, tumor growth and metastasis. [DOI] [PMC free article] [PubMed] [Google Scholar]