

Abstract
In the relatively short period of time since their discovery, microRNAs have been shown to control many important cellular functions such as cell differentiation, growth, proliferation and apoptosis. In addition, microRNAs have been demonstrated as key drivers of many malignancies and can function as either tumour suppressors or oncogenes. The haematopoietic system is not outside the realm of microRNA control with microRNAs controlling aspects of stem cell and progenitor self-renewal and differentiation, with many, if not all, haematological disorders associated with aberrant microRNA expression and function. In this review, we focus on the current understanding of microRNA control of haematopoiesis and detail the evidence for the contribution and clinical relevance of aberrant microRNA function to the characteristic block of differentiation in acute myeloid leukaemia.
Keywords: microRNA, acute myeloid leukaemia, haematopoiesis
Introduction
Acute myeloid leukaemia (AML) is a heterogeneous disease in which haematopoietic progenitor cells exhibit an increase in their rate of proliferation in concert with a halt in their differentiation programme, resulting in a population of proliferating immature cells. The heterogeneity of AML lies in the numerous recurring chromosomal translocations and gene mutations and their resulting impact on cellular phenotype. These molecular abnormalities result in the generation of fusion proteins or irregular proteins, which disrupt the normal cellular biology of these haematopoietic progenitors. Not surprisingly, these molecular aberrations influence prognosis, wherein the recurrent chromosomal translocations [inv(16), t(8;21) and t(15;17)] confer a favourable prognosis, while the chromosomal deletions [del(5q), -5 and -7], a poorer prognosis 2004. Similarly, gene mutations in CEBPA and NPM1 suggest a favourable prognosis 2 while mutations in FLT3 or WT1 leading to an adverse prognosis [3,4].
Molecular abnormalities in AML cells will conceivably result in an altered gene expression profile that will be somewhat specific to a given abnormality. This appears to be an accurate assumption as several large scale gene expression studies have been performed on AML blasts. The seminal mRNA microarray study of Valk et al. 2004 reports that AML can be clustered into discernable groups based on their mRNA profiles. Similar results have been reported using microRNA microarray (see recent review by Marcucci et al. 2010), suggesting that the expression of microRNAs is at least as informative as mRNA expression when clustering patients based on gene expression. The integration of microRNA and mRNA microarray data confers an additional layer of information regarding microRNA function in AML [7].
With approximately 50% of AML patients presenting with no gross chromosomal abnormalities and a further 23% of those with no mutations in commonly mutated genes [8], focus is now turning to the role of microRNAs in the initiation of AML. With the prognostic and diagnostic significance of microRNA expression in AML well documented, the field is now turning to the influence of microRNAs on normal and malignant cellular function. In this review, we aim to discuss the role of microRNAs in normal myelopoiesis by way of introduction to a discussion on the interplay between microRNA dysregulation and the differentiation block observed in AML.
MicroRNA control of normal haematopoiesis
Haematopoietic stem cell (HSC)
The progression of a cell through the haematopoietic differentiation pathway involves several different intermediaries with progressively reducing capacity to form terminally differentiated blood cells. The HSC is able to re-populate all terminal cell lineages while retaining its self-renewal capacity, wherein multi-potent progenitor the (MPP) has reduced self-renewal capacity.
With regard to human cells, the expression of the microRNA processing protein Dicer has been shown to be essential for HSC maintenance, highlighting the role microRNAs play in HSC biology [9]. An absence of a set of microRNAs directing HSC self-renewal has been reported, although the CD34+ cell population was not segregated into discrete HSC and MPP populations [10]. A subsequent study reported that miR-29a is highly expressed in human HSC compared with the expression observed in more committed progenitors, with miR-29a playing a role in murine stem-cell self-renewal, myeloid differentiation and proliferation of the MPP [11]. Furthermore, miR-125a has been reported to control HSC number primarily through the protection of HSCs from apoptosis via its inhibition of Bak1 expression [9].
It appears that in the mouse HSC, the microRNA expression profiles show a degree of overlap, while upon commitment to a specific lineage, the microRNA expression profile is dramatically reprogrammed [12]. Murine bone marrow cells have been shown to require expression of miR-124, which is regulated by EVI1, to ensure normal cell cycling and self-renewal [13].
As could be expected, microRNAs do not act in isolation, with microRNAs possibly present that oppose the function of other microRNAs, and with different microRNA families shown to oppose one another in the regulation of embryonic stem-cell renewal and differentiation [14]. HSCs are not an exception to this observation, as in the background of opposing microRNA signals, miR-125b was identified as being sufficient to increase engraftment in transplanted mice and to trigger myeloid leukaemia [15].
Partly due to technical issues surrounding the ability to acquire sufficient numbers of well-defined progenitor populations, there is a significantly larger body of knowledge surrounding those cells that are lymphoid- or myeloid-restricted.
Common myeloid progenitor (CMP) and initiation of myelopoiesis
The CMP represents the cellular population that has become myeloid lineage rather than lymphoid lineage restricted and is the point at which myelopoiesis begins. The functional role that microRNAs play in myelopoiesis is implicated primarily via the manipulation of specific microRNA expression levels in HSCs, with a bias towards certain terminal cell lineages a useful readout. Overexpression of miR-29a in HSCs was shown to positively influence myeloid differentiation by increasing the rate of proliferation in myeloid progenitors, causing a myeloproliferative disorder in mice that progressed to AML [11]. miR-146a is found at higher levels in murine CMPs than levels observed in common lymphoid progenitors, with engraftment of miR-146a overexpressing HSCs resulting in facilitation of short-term myeloid differentiation and a reduction in lymphoid and erythroid differentiation [15]. Intriguingly, reduction in miR-146a expression in HSCs resulted in an increase in circulating platelets and variable neutropenia [16].
During the transition from the CMP to granulocyte–monocyte precursors (GMP), the expression of miR-21 and miR-196b significantly decreases, possibly due to inhibition by growth factor-independent 1(GFI1) [17]. Further investigation of the impact of these microRNAs using colony forming assays places miR-21 as a monopoietic effector and miR-196b as antagonist of granulopoiesis [17].
Monocyte/macrophage differentiation
At the point of the GMP, a haematopoietic progenitor cell reaches a bifurcation in the myelopoiesis pathway where cells follow either a monocytic or granulocytic fate. A key characteristic of monocytic lineage is an increase in the expression of the monocytic cellular markers, including CD11b and CD14, a decrease in the stem-cell markers CD117 and CD71 and the granulocytic marker CD15 [18]. Gain- and loss-of-function experiments provide a mechanism by which individual microRNAs can be assessed as to their role in monocytopoiesis, with one such study observing a bias towards megakaryocytic-granulocytic differentiation of CD34+ progenitors at the expense of erythroid-monocytic differentiation following miR-299-5p overexpression [19]. During monocytic differentiation, the transcription factor AML1 is up-regulated, possibly as a result of a simultaneous down-regulation of miR-17-5p, -20a and -106a; resulting in monocyte-colony stimulating factor receptor (M-CSFR) down-regulation, enhanced blast proliferation and inhibition of monocytic differentiation [20].
Cell lines derived from AML patients provide a convenient reproducible model of monocytic differentiation via exposure to 1, 25-dihydroxyvitaminD3 (VitD3) and VitD3 ± phorbol 12-myristate 13-acetate (PMA). We recently used this model to show a decrease in the expression of miR-181a, -181b, -181d, -130a, -135b and -146a during VitD3/PMA-induced monocytic differentiation of AML cell lines (HL60 and NB4), with several of these microRNAs able to target key myelomonocytic transcription factors [21]. Dysregulated microRNAs do not act in isolation. It is likely that the additive effects of microRNA dysregulation and their individual phenotypes can combine to produce a specific cell phenotype. Although miR-155, -222, -424 and -503 were up-regulated during PMA-induced monocytic differentiation, they all have differing effects, where individual overexpression of these microRNAs had divergent outcomes. miR-155 and miR-222 induce G2 arrest and apoptosis, respectively, and miR-424 and miR-503 target cell cycle regulators, thereby inducing G1 cell-cycle arrest, whereby overexpression of all four microRNAs produces a PMA-like response [22].
miR-155 is able to restrict the commitment of RAW264.7 monocyte progenitors towards a macrophage phenotype, with a concurrent drop in osteoclast differentiation [23]. During human macrophage differentiation, a decrease in miR-223, -15a and -16 occurs with a corresponding increase in IKKalpha expression; when considered with a concurrent stabilization of the NF-κB-inducing kinase, this suggests that the decrease in these microRNAs may prevent macrophage hyperactivation while priming for future responses to pro-inflammatory stimuli [24].
Transcriptional networks involving coding and non-coding RNAs that control key cellular processes are well established; in addition, networks involved in monocyte/macrophage differentiation is not different. Using a bioinformatics approach to interpret time course expression data, Schmeier et al. 2009 identified 12 transcription factors (ATF2, E2F3, HOXA4, NFE2L1, SP3 and YY1) that putatively control microRNA expression during monocyte to macrophage differentiation with six microRNAs previously not described. Furthermore, using this approach, several microRNAs previously linked to myeloid differentiation (including miR-21) and monocyte/macrophage differentiation (miR-155, -424 and -17-92) were identified.
A similar transcriptional network driving macrophage differentiation centres on the myeloid transcription factor PU.1 and its control of miR-424 [26]. miR-424 up-regulation by PU.1 in turn represses NFI-A expression, which activates macrophage differentiation genes such as M-CSFR. Ectopic expression of miR-424 (and similar knock-down of NFI-A) augments monocytic differentiation in human myeloid cell lines and CD34+ progenitors [26]. Via the occupation of binding sites within regulatory regions neighbouring their genomic loci, PU.1 was shown to control the expression of four microRNAs: miR-146a, -342, -338 and -155 [27]. miR-146a increase in HSCs resulted in their differentiation into functional peritoneal macrophages, while inhibition of miR-146a in a zebrafish model of haematopoiesis halted the development of macrophages [27].
Granulocytic differentiation
Unlike monocytic differentiation, granulocytic differentiation influenced by microRNAs is well established in the literature. One of the earliest studies ascertaining a functional role for microRNAs in granulocytic differentiation involved the now well-characterized miR-223, NFI-A and CCAAT/enhancer binding protein alpha (C/EBPα). Both NFI-A and C/EBPα competitively inhibit each other's binding to the miR-223 promoter, with NFI-A and C/EBPα down- and up-regulating miR-223 expression, respectively [28]. Thus, miR-223 overexpression in acute promyelocytic leukaemia (APL) cell lines enhanced differentiation into granulocytes; in contrast, inhibiting miR-223 reduced differentiation. miR-223 has subsequently been shown to be up-regulated in APL cell lines treated with all-trans retinoic acid (ATRA) [21,29,,30], down- and up-regulated in erythroid and megakaryocytic differentiation of K562 cells, respectively [31]. Transgenic mice with a miR-223 loss-of-function have an enlarged granulocytic compartment resulting from a cell-autonomous increase in granulocytic progenitors, and surprisingly show that miR-223 negatively regulates granulocyte differentiation [32].
CCAAT/enhancer binding protein alpha also regulates the expression of miR-34a during granulocytic differentiation, which in turn targets E2F3 and blocks myeloid cell proliferation [33]. Transcription networks involving microRNAs and important myeloid transcription factors are not limited to C/EBPα, with RUNX1 also forming networks that affect granulocytic differentiation. RUNX1 has been shown to control miR-24 expression, an increase in miR-24 blocks granulocyte differentiation [34], while miR-27 directly inhibits RUNX1 translation resulting in increased granulocytic differentiation [35]. Similarly, PU.1 and IRF-9 are both up-regulated, and IRF-1 is down-regulated via miR-342 overexpression during ATRA-induced granulocytic differentiation; in addition, an increase in miR-342 is associated with an increased ATRA-induced granulocytic differentiation [36].
During granulocyte colony-stimulating factor (G-CSF) treatment, miR-125b blocks granulocytic differentiation by repressing a number of mRNAs including STAT3, JUND and BAK1 [37]. Growth factor-independent 1 (GFI-1) has been shown to control the expression of several microRNAs. Growth factor-independent 1 knockout mice have a block in G-CSF-stimulated granulocytic differentiation and a concurrent dysregulation expression of miR-21 and miR-196b in bone marrow cells. Co-expression of miR-21 and miR-196b in lineage negative bone marrow cells recapitulates the dampening in granulocytic differentiation observed in GFI1 knockout mice [17].
Erythroid/megakaryocytic differentiation
The leukaemic cell line K562 provides an experimental model for the study of erythroid and megakaryocytic differentiation. During megakaryocyte differentiation, miR-181 represses Lin28 expression leading to an increase in let-7 expression, resulting in an increase in megakaryocytic differentiation, with no appreciable role in haemin-induced erythrocytic differentiation 2011. Similarly, miR-451 has been shown to promote erythroid differentiation of K562 cells [39], while during megakaryocytic differentiation, miR-34a inhibits cellular proliferation via the repression of mitogen-activated protein kinase kinase 1 (MEK1) [40]. The role of miR-34a in promoting megakaryocytic differentiation is explained partly via its regulation of MYB and is independent of p53 [41].
Increasing miR-126 expression in human embryonic stem cells inhibited the generation of erythroid colonies [42], with a reciprocal experiment in zebrafish indicating that inhibition of miR-126 promotes the generation of erythrocytes at the expense of generating thrombocytes [43]. Similarly, mice lacking miR-451 exhibits defective erythroid differentiation [44,45] via the repression of 14-3-3zeta [44], and thalassemic CD34+ cells undergoing erythroid differentiation exhibit dysregulation of miR-451 [46].
Megakaryopoiesis requires a decrease in the levels of miR-155 with a maintenance of expression inhibiting this differentiation, mediated in part via the removal of the repression of Ets-1 and Meis1 [47]. Runx1 is an important transcription factor controlling many aspects of haematopoiesis and its regulation of miR-27a and the repression of Runx1 by miR-27a forms an important feedback loop regulating megakaryocytic differentiation [48]. A similar transcription factor-based regulatory pathway controlling megakaryocytic differentiation is observed where PLZF suppresses miR-146a expression, which leads to the increase in expression of the bona fide miR-146a mRNA target of CXCR4 [49].
MicroRNA control of malignant differentiation
miR-155 is found in the non-coding B cell integration cluster transcript and is elevated in many solid tumours and leukaemias. miR-155 has been found to be up-regulated in patients with FTL3-ITD mutations [50,51,,52]; however, miR-155 has not been found to rely on FTL3 signalling [50], and blocking or stimulating FTL3 signalling in mouse myeloid cells led to no variations in miR-155 levels. Functionally, overexpression of miR-155 in murine HSCs that were transplanted into lethally irradiated mice found that miR-155 caused a myeloproliferative disorder, which was characterized by the bone marrow dominated by cells at a variety of myeloid developmental stages. These mice also exhibited splenomegally and extramedullary haematopoiesis, although no overt AML development was observed up to 2 months after transplant [53], indicating that additional mutations may be necessary for bona fide leukaemogenesis. Interestingly, miR-155 perturbed mature peripheral blood cell populations, with a decrease in numbers of erythrocytes, leucocytes and platelets, possibly due to the abnormal development of progenitors. AML patient blasts were found to have elevated levels of miR-155, especially those suffering from AML-M4 and AML-M5 [53]. Ectopic overexpression of miR-155 followed by treatment with a differentiation agent demonstrated that miR-155 decreased erythroid and megakaryocytic differentiation in the human leukaemia cell line K562. Overexpression of miR-155 in human CD34+ cells demonstrated a decrease in clonogenicity, with a decreased number of myeloid colonies, which were also smaller than their control-transfected counterpart [10]. Several targets of miR-155 were identified and confirmed by luciferase assay, including PU.1, PICALM, CUTL1, CSF1R, ARNTL, JARID2, HIF1a [53], C/EBP, CREBBP, JUN, MEIS1, AGTR1/2 and FOS [10]. However, no further functional assays were conducted to decipher the potential mechanism of miR-155 involvement in AML. In the lymphoid system, miR-155 transgenic mice were found to accumulate large pre-B cells and develop leukaemia/high-grade lymphoma [54]. In this system, miR-155 was found to target Src homology 2 domain-containing iositol-5-phosphatase (SHIP), a negative regulator of cell signalling by potentially down-regulating MAPK and PI3K/AKT pathways [54,55,,56]. Src homology 2 domain-containing iositol-5-phosphatase knockout mice were found to develop a myeloproliferative disease similar to that seen in the study by O'Connell et al., with an increased number of GMPs at different stages of myeloid development, and decreased erythroid development 2009.
The miR-29 family is composed of three isoforms arranged in two clusters: miR-29b-1/and miR-29b-2/miR-29c [57]. miR-29b has been found to have a potentially important effect in AML development, high expression being correlated with an increase in apoptosis in leukaemic cell lines K562 and Kasumi-1, and a decrease in tumour size in a xenograft model [57]. The molecular basis of miR-29b tumour suppression was characterized by mRNA microarray analysis followed by supervised analysis and use of DAVID analysis software, which yielded genes involved in apoptosis (MCL-1, TRAF4 and MYBl2) and cell cycle regulation (CDK4, CDK6 and CCND2) as targets of miR-29b regulation; the levels of these genes, especially MCL-1, were found to be largely inversely correlated with low miR-29b expression in 45 AML samples [57]. Further studies to ascertain the regulatory role of miR-29b found that it was overexpressed in AML cell lines, resulting in a reduction in DNA methyltransferases DNMT1, DNMT3A and DNMT3B, leading to a global decrease in DNA methylation and an overexpression of p15INK4b and ESR1 tumour-suppressor genes [58]. Functionally, miR-29b overexpression was found to induce partial differentiation of the Kasumi-1 cell lines, with a 38% increase in myeloid marker (CD11b) expression [58]. This indicated an important hypomethylating role, which is typically silenced in AML. Interestingly, analysis of miR-29b levels in older AML patients who underwent treatment with a single-agent decitabine found a relationship between miR-29b pre-treatment levels and clinical response, with higher levels associated with a clinical response to treatment (P = 0.02) [59].
The analysis of changes in transcript mRNA expression in AML cell lines after transfection of miR-29a/b identified several targets including oncogenes MCL1, CDK6, IGFR and JAK2 [57]. Furthermore, miR-29b is involved in a protein-microRNA network, which included SP1/NFkB/HDAC whose regulation results in KIT expression in core binding factor-AML (CBF-AML) and predicts poor outcome [60]. KIT is a tyrosine kinase receptor that regulates survival, differentiation and proliferation. KIT gain of function mutations lead to MYC-dependent miR-29b repression and increased levels of Sp1 (miR-29b target). Sp1 enhances its own expression by participating in an NFkB/HDAC complex that further represses miR-29b transcription. This up-regulated Sp1 binds to NFkB and transactivates KIT. miR-29b overexpression (through transfection or treatment with bortezomin or mithramycin A) leads to a down-regulation in KIT expression [60], supporting the tumour suppressor function of miR-29b, and thus supports the use of synthetic miR-29b as a therapeutic option for AML.
miR-29a has demonstrated its importance in leukaemogenesis as part of the important miR-29 family. miR-29a is expressed at its highest in primary human HSCs, and then progressively decreases as cells mature through the haematopoietic progenitor stages: MPPs, CMPs and GMPs. In AML, CD34+ and CD34− cells express similar levels of miR-29a, which is close to that of normal MPPs. Overexpression of miR-29a in mouse bone marrow progenitors and transplant into lethally irradiated mice found increasing levels of donor-derived myeloid (Mac-1+) chimerism 8–12 weeks after transplantation, at the expense of B cell development, and significantly increased proportion of granulocytes (Mac-1+, Gr-1+) and monocytes (Mac-1+, Gr-1lo). This myeloproliferative effect was found to act on MPPs and not GMPs or CMPs, with an increase in colony-forming potential in MPPs: a simultaneous decreased megakaryocytic potential and increased monocytic development. MPP also exhibit uncharacteristic self-renewal potential seen in vivo. Transplant of these chimeric cells into secondary mice found that these mice began to expire four months following transplantation. The primary transplanted mice demonstrated hepatosplenomegaly, aberrant splenic and bone marrow architecture overrun with immature mononuclear population, typical of AML. Interestingly, miR-29b did not demonstrate a similar effect when bone marrow cells were transplanted into mice [11].
The loss of miR-29a in AML with monosomy 7 (-7) or deletion of 7q (del7q) has been found to increase the nucleoprotein SKI, which is thought to repress retinoic acid-induced myeloid differentiation. Therefore, the loss of miR-29a may contribute to leukaemogenesis by repressing the nuclear oncogene SKI [61].
Up-regulation of miR-221 has been observed in AML blasts when compared with normal bone marrow and CD34+ haematopoietic progenitor cells [62,63]. Up-regulation of miR-221 in CD34+ progenitors resulted in reduced proliferation and hastened erythroid differentiation with a simultaneous down-regulation of c-kit protein. Transplantation studies reveal that modulation of miR-221 impairs the engraftment and stem-cell activity of CD34+ progenitors. These results suggest that the inhibition of c-kit protein expression is removed during erythroid cell growth via the down-regulation of miR-221 [64].
Functional loss of C/EBPα, a master regulatory transcription factor in the haematopoietic system, can result in a differentiation block in granulopoiesis and thus contribute to leukeamic transformation. Analysis of the DNA methylation profile of the CpG island of C/EBPα identified a densely methylated upstream promoter region in over 50% of AML cases. Aberrant DNA methylation was strongly associated with AML subtypes: those with cytogenetic inv(16) and t(15:17). miR-124a is typically silenced by epigenetic mechanisms in leukaemia cell lines and, upon demethylation treatment with DAC, becomes activated and further inhibits C/EBPα transcription [65].
CEBPα has been shown to be crucial for granulopoiesis, up-regulating myeloid-specific genes necessary for granulocytic maturation and inhibiting myeloid cell proliferation. Inhibition of E2F1 activity, a cell cycle protein, by CEBPα is a key step towards inhibiting cell cycling during granulopoiesis. This occurs via the CEBPα activation of miR-223, which in turn targets E2F1 via translational inhibition. Furthermore, E2F1 binds to the miR-223 promoter and inhibits its transcription, generating a negative feedback loop [66]. miR-223 has also been found to be down-regulated in AML with CEBPα mutations, compared with AML with normal cytogenetics [66], thereby having its anti-proliferation role decreased.
In AML, CEBPα function is frequently disrupted, with 10% of patients showing dominant negative mutations in the CEBPα gene, and with suppression by the fusion proteins AML1-ETO, AML1-MDS1-EVI1 or CBFB-SMMHC from patients with t(8;21), t(3;21) or inv(16), respectively. CEBPα has been found to correlate with miR-29b expression in myeloid cell lines, and 21 AML patients with a CEBPα mutation also expressed lower levels of miR-29b compared with normal granulocytes; these levels were similar to those found in the immature CD34+ fraction of normal bone marrow [67]. The regulation of miR-29b by CEBPα was confirmed by luciferase assay and ChIP analysis, which found that CEBPα bound to the upstream promoter of the pri-miR-29a/b coding site [67]. The gene MLLT11, a mixed-lineage leukaemia (MLL) fusion partner in AML, is highly regulated in HSCs and is found at high expression levels in AML, plus MLLT11 has been reported to be an AML prognostic marker [68]. miR-29b represses MLLT11, and at its low expression levels, miR-29b correlates with poorer prognosis for AML patients [69].
miR-34a is a widely expressed microRNA that is regulated by p53 and functions as a tumour suppressor in other cancers [70]. miR-34a expression has been found to correlate with CEBPα mutations in AML, with low CEBPα levels equating with low miR-34a levels. CEBPα directly regulates miR-34a expression during granulopoiesis, with high microRNA levels leading to cell cycle block at G0/G1 [33]. Furthermore, miR-34a blocks myeloid cell cycle progression by inhibiting E2F3, and its overexpression in primary AML samples induces partial differentiation with increased levels of CD11b and G-CSFR, but no change in CD14 (monocytic) levels was observed [33].
miR-34b targets cyclic AMP-responsive element binding protein (CREB), which has been documented to be overexpressed in leukaemia, causing aberrant cell proliferation, cell cycle progression and higher clonogenic potential. AML patients with increased CREB expression were found to have a significantly lower expression of miR-34b than normal cases [71].
Increased levels of miR-17-5p-20a-106a have been found to be involved in myelopoiesis, with high levels increasing blast cell proliferation and blocking monocytic differentiation [20]. These microRNAs appear to function through the AML1 gene, which transactivates the M-CSF receptor to induce monocyte differentiation. Therefore, overexpression of miR17-5p-20a-106a down-regulates AML1 and mimics the effects of the AML1/ETO fusion protein on monopoiesis.
The miR-17-92 polycistron cluster of microRNAs contains mRNA-17, -17*, -18a, -19a, -20a, -19b-1 and -92a-1, and these are thought to function as oncogenes in other cancers including lymphomas [72]. They are usually correlated with the amplification of the genomic locus, indicating a wide involvement in oncogenesis [73,74]. This cluster is frequently overexpressed in MLL-rearranged AMLs [75]. In fact, fusion proteins of MLL exhibit stronger binding to the locus of the miR cluster than wild-type MLL, which are associated with increased histone H3 acetylation and H3K4 trimethylation levels, along with increased miR cluster expression [76]. Ectopic expression of this microRNA cluster caused increase in proliferation, inhibited apoptosis in human cell lines, and increased the clonogenicity of mouse bone marrow cells, especially when combined with MLL fusion proteins [76]. Overexpression of this cluster can also inhibit monocyte and megakaryocyte development, with monocyte development inhibited by suppression of the AML1 proto-oncogene, which goes on to suppress M-CSFR transactivation and decrease monopoiesis [20,77]. This miR-17 polycistron has been found to regulate the leukaemic stem-cell (LSC, c-kit+) potential in a mouse model of MLL-AML, by repressing expression of the cyclin-dependent kinase inhibitor p21. miR-17-92 is highly expressed in malignant LSCs, once LSCs leave the self-renewing compartment; the miR-17-92 polycistron is down-regulated, with the lowest levels in cells displaying the most mature myeloid phenotype (c-kit—Gr-1+, Mac-1+). Forced overexpression leads to the block in myeloid differentiation, with an increase in myeloid blasts and decreased number of mature neutrophils and macrophages, enhances LSC clonogenic potential and decreases leukaemia latency [78].
miR-196b, located between HOXA9 and HOXA10 genes, is typically expressed at higher levels in HSCs and decreases as cells differentiate; however, miR-196b is up-regulated in MLL-AML patients. Its overexpression in bone marrow stem cells leads to a partial block in their differentiation ability, increased proliferation ability and increased survival. MLL fusion proteins induced miR-196b. Knockdown of miR-196b levels in cells containing the MLL-AF9 fusion gene decreased the clonogenic proliferative capacity of these cells [79].
MicroRNA associated with abnormal monocytic differentiation
An excellent model of monocytic differentiation in vitro is the exposure of myeloid cells such as HL60, NB4 and U937 to VitD3, which induces a monocytic phenotype (with CD11b and CD14 expression) and arrest cell proliferation [80]. This cell cycle arrest in G1 phase, has been found to occur due to the stabilization and accumulation of p27kip1, due to reduced expression of subunits of S-phase kinase protein 2 (Skp2). P27kip degradation is also controlled by the cyclin-dependent sub-unit 1 (Csk1) and by Spy1. miR-181a is down-regulated during VitD3 treatment, and targets p27kip1, causing its down-regulation when miR-181a is overexpressed. Furthermore, overexpression of miR-181a in HL60s and U937s enhanced monocytic differentiation, increasing CD14 expression by 24% [81] (Fig. 1).
Fig 1.
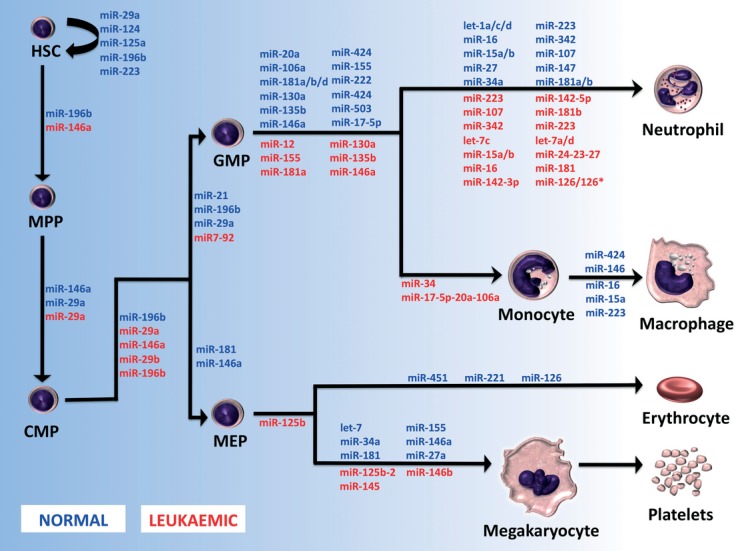
MicroRNAs associated with normal myelopoiesis and myeloid leukaemia. A model for microRNA activity at specific stages of normal development is represented in BLUE. microRNAs postulated to be dysregulated in various leukaemic subtypes are represented in RED and are located at the stage where they are dysregulated. HSC: haematopoietic stem cell; MPP: multi-potent progenitor cell; GMP: granulocyte–monocyte progenitor; MEP: megakaryocytic–erythroid progenitor.
Overexpression of miR-125b, up to 90 fold, was found to be present in patients with myelodysplastic syndrome (MDS) and AML with a relatively rare chromosomal translocation t(2;11). In vitro studies overexpressing miR-125b in the AML cell lines HL60 and NB4 found that cells overexpressing miR-125b had a decreased differentiation capacity as they progress down the myelomonocytic lineage [82]. Indeed, miR-125b negatively regulates many proteins in the p53 pathway, and has been found to regulate stem-cell pool size and confer a competitive advantage to engrafting HSCs [83]. In addition, miR-125b increases the proliferation and self-renewal ability of megakaryocyte progenitors (MP) and MEP [84]. Transplantation of miR-125b overexpressing haematopoietic progenitors into lethally irradiated mice demonstrated heightened proliferation of myeloid cells, and the occurrence of macrocytic anaemia with decreased erythrocyte, haemoglobin and haematocrit levels. Of the transplanted mice, half of them succumbed to myeloproliferative neoplasm or acute lymphoblastic leukaemia within 12–29 weeks, demonstrating that miR-125b acts as an oncogene, promoting malignant transformation [85].
MicroRNA associated with abnormal granulocytic differentiation
Acute promyelocytic leukaemia (APL) is characterized by the differentiation arrest of granulopoiesis at the promyelocyte stage. It is associated with chromosomal translocations that typically implicated the gene encoding the retinoic acid receptor α (RARA), which frequently fuses with the promyelocytic leukaemia protein (PML) gene. Although APL is an exceedingly malignant form of AML, with a fatal course of only weeks, pharmaceutical doses of ATRA overcome PML-RARA-mediated repression, restore normal gene transcription and alleviate the granulocytic maturation block, leading to increased patient survival. It has been found that let-1a/c/d, miR-16, -15a/b, -107, -147,-223 and -342 are up-regulated by ATRA, whereas miR-181a and -181b are down-regulated in NB4 cells [29,36,,86]. The up-regulation of miR-223 is consistent with its role in normal granulopoiesis [28], where its expression increases once suppression by NFI-A ceases. Interestingly, Garzon et al. reported that miR-107 targets NFI-A, describing the regulatory loop leading to granulocyte development. PML-RARA fusion protein represses miR-210, the miR-23a/24-2 cluster, miR-342 and let-7c, which is alleviated upon ATRA treatment, seen in cell lines and primary patient samples [86,87]. Assessment of these microRNA levels in patients at diagnosis (APL blasts) and after treatment confirmed these results seen in vitro by Careccia et al. 2009, and suggests a role for these microRNAs in the prognosis of APL patients.
Comparison of APL blasts from newly diagnosed patients, with normal promyelocytes differentiated in vitro from umbilical cord blood CD34+ cells, found that nine microRNAs were aberrantly up-regulated: miR-15a/b, -16, -142-3p, -142-5p, -181b, -223 and let-7a/d; while three were down-regulated: miR-107, -342 and let-7c [87]. This is the only study, which was found to date, that attempted to compare stage-matched normal cells with their leukaemic counterpart. Unfortunately, the authors did not compare the microRNA levels of promyelocytes and granulocytes with ATRA-treated APL patient cells. This would have determined if the microRNA profile after ATRA treatment was comparable to normal levels.
RUNX1 (or AML1 gene) is a key haematopoietic transcription factor, which is frequently rearranged in myeloid leukaemia, disrupting the transcriptional function of this protein. The AML1-ETO fusion protein resulting from t(8;21) lacks the RUNX1 carboxyl terminus and thus associated functions such as transcriptional activation and sub-nuclear targeting. Although it is known that AML1-ETO occupies RUNX1 target gene promoters, resulting in gene suppression, the exact leukaemogenic mechanism of this chimeric protein remains unknown. The miR-24-23-27 cluster and miR-181 family are significantly up-regulated in patients and cell lines with abnormal RUNX1, while remaining at a lower level with wild- type RUNX1. Of these microRNAs, miR-24 was found to down-regulate MKP-7 (which abrogates myeloid cell proliferation by negatively regulating p38 and JNK) and activate MAPK signalling. miR-24 overexpression was found to block granulocytic differentiation of 32D cells with up to 80% of transfected cells remaining immature, and enhance growth factor-independent proliferation of these cells [34].
miR-223 is expressed in low levels in HSCs and increases during granulocytic differentiation [28]; it was found to be transcriptionally repressed in cells harbouring AML1/ETO. This fusion protein targets miR-223 by interacting with the AML1 site at the upstream region of miR-223, where it recruits HDAC and DNMT deacetylating enzymes, which deacetylate histone proteins and methylate CpGs [88]. These CpGs serve as binding sites for MeCP2 (a DNA-methyl CpG-binding protein). This changes the chromatin conformation, rendering miR-223 silent. This is thought to contribute to the maturation block evident in this AML subtype, with miR-223 overexpression, demethylation and knockdown of AML1/ETO, all of which improve the cell differentiation capacity [88].
Li et al. found that some AML subtypes clustered together after unsupervised analysis of microarray data, including miR-126/126*, which was overexpressed in t(8:21) and inv(16) samples. Study of the function of miR-126 found that it increases cell viability and inhibits apoptosis in AML cell lines THP1 and ME-1 2008. It was also found that miR-126 functions in cooperation with the AML-ETO. This protein has been found to be insufficient for leukaemogenesis by itself; however, studies of clonogenicity of mouse bone marrow progenitor cells found a synergistic effect between miR-126 and AML1-ETO leading to a significant increase in colonies of up to 36 fold. Interestingly, the authors attempted to clarify the mechanisms underlying the up-regulation of miR-126 in CBF-AML, finding that the overexpression was not a consequence of amplification or mutation of the genomic locus, but an association with partial de-methylation of the CpG island in which miR-126 is embedded [75].
MicroRNAs associated with abnormal erythroid and megakaryocytic differentiation
MicroRNAs have been observed to contribute to MDSs and leukaemia, which particularly relate to the erythroid and megakaryocytic compartments. In megakaryoblastic leukaemia, miR-125b-2 has been identified as an oncogenic microRNA by increasing the proliferation and self-renewal of human and mouse megakaryocytic progenitors and megakaryocytic/erythroid progenitors [84]. The -5q syndrome phenotype is characterized in part by dysplastic megakaryocytes with the deletion of the 5q region correlating with the loss of expression of miR-145 and -146a, where functional studies indicate that reducing the expression of these microRNAs in mouse HSCs results in thrombocytosis and megakaryocytic dysplasia [16]. Pure erythroid leukaemia is extremely rare and erythroleukemia comprises <5% of cases of AML [15], perhaps explaining the deficiency in data pertaining to microRNA control of these neoplasms.
Conclusion
The studies detailed in this review highlight the important role of microRNAs in normal and malignant haematopoiesis, primarily via the observations made by several groups through the use of common techniques such as gene expression profiling. This field is reaching the stage where gene expression profiles have been reported for the major progenitor cell populations, particularly in the case of AML. Profiles have been reported for the key subtypes of AML and, in many cases, profiles for specific AML gene mutations have been defined.
The challenge that the myeloid biology field now faces is translating these gene expression profiles into functional studies to link an aberrant expression with abnormal function. Only when these functional studies have been completed can the huge potential of microRNAs leave the laboratory and enter the clinic to aid the treatment of haematological malignancies.
Acknowledgments
This work was supported by grants from the Arrow Bone Marrow Transplant Foundation, St Vincent's Hospital Haematology Research Fund, St Vincent's Clinic Foundation and the Sydney Foundation for Medical Research. M.L. conceived the article and M.L., C.P., E.T. and D.M. wrote the paper.
Conflict of interest
The authors wish to declare no conflicts of interest.
References
- Mrozek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukaemia. Blood Rev. 2004;18:115–36. doi: 10.1016/S0268-960X(03)00040-7. [DOI] [PubMed] [Google Scholar]
- Mrozek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukaemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–48. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukaemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–6. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- Paschka P, Marcucci G, Ruppert AS, et al. Wilms' tumour 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukaemia: a cancer and leukaemia group B study. J Clin Oncol. 2008;26:4595–602. doi: 10.1200/JCO.2007.15.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukaemia. N Engl J Med. 2004;350:1617–28. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Marcucci G, Mrozek K, Radmacher MD, et al. The prognostic and functional role of microRNAs in acute myeloid leukaemia. Blood. 2010;117:1121–9. doi: 10.1182/blood-2010-09-191312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havelange V, Stauffer N, Heaphy CC, et al. Functional implications of microRNAs in acute myeloid leukaemia by integrating microRNA and messenger RNA expression profiling. Cancer. 2011 doi: 10.1002/cncr.26096. ; in press: doi: 10.1002/cncr.26096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow S, Campo S, Harris N, et al. WHO classification of tumours of haematopoietic and lymphoid tissue. 4th ed. Lyon, France: IARC; 2008. [Google Scholar]
- Guo S, Lu J, Schlanger R, et al. MicroRNA miR-125a controls hematopoietic stem cell number. Proc Natl Acad Sci USA. 2010;107:14229–34. doi: 10.1073/pnas.0913574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgantas RW, 3rd, Hildreth R, Morisot S, et al. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci USA. 2007;104:2750–5. doi: 10.1073/pnas.0610983104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YC, Park CY, Bhagat G, et al. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukaemia. J Exp Med. 2010;207:475–89. doi: 10.1084/jem.20090831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriv OI, Kuchenbauer F, Delaney AD, et al. Comprehensive microRNA expression profiling of the hematopoietic hierarchy. Proc Natl Acad Sci USA. 2010;107:15443–8. doi: 10.1073/pnas.1009320107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein J, Senyuk V, Premanand K, et al. Methylation and silencing of miRNA-124 by EVI1 and self-renewal exhaustion of hematopoietic stem cells in murine myelodysplastic syndrome. Proc Natl Acad Sci USA. 2010;107:9783–8. doi: 10.1073/pnas.1004297107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–6. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starczynowski DT, Kuchenbauer F, Wegrzyn J, et al. MicroRNA-146a disrupts hematopoietic differentiation and survival. Exp Hematol. 2011;39:167–78 e4. doi: 10.1016/j.exphem.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Starczynowski DT, Kuchenbauer F, Argiropoulos B, et al. Identification of miR-145 and miR-146a as mediators of the 5q-syndrome phenotype. Nat Med. 2010;16:49–58. doi: 10.1038/nm.2054. [DOI] [PubMed] [Google Scholar]
- Velu CS, Baktula AM, Grimes HL. Gfi1 regulates miR-21 and miR-196b to control myelopoiesis. Blood. 2009;113:4720–8. doi: 10.1182/blood-2008-11-190215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SL, Belov L, Barber N, et al. Immunophenotypic changes induced on human HL60 leukaemia cells by 1alpha,25-dihydroxyvitamin D3 and 12-O-tetradecanoyl phorbol-13-acetate. Leuk Res. 2005;29:1141–51. doi: 10.1016/j.leukres.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Tenedini E, Roncaglia E, Ferrari F, et al. Integrated analysis of microRNA and mRNA expression profiles in physiological myelopoiesis: role of hsa-mir-299-5p in CD34+ progenitor cells commitment. Cell Death Dis. 2010;1:e28. doi: 10.1038/cddis.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Pelosi E, Greco P, et al. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol. 2007;9:775–87. doi: 10.1038/ncb1613. [DOI] [PubMed] [Google Scholar]
- Lutherborrow M, Bryant A, Jayaswal V, et al. Expression profiling of cytogenetically normal acute myeloid leukaemia identifies microRNAs that target genes involved in monocytic differentiation. Am J Hematol. 2011;86:2–11. doi: 10.1002/ajh.21864. [DOI] [PubMed] [Google Scholar]
- Forrest AR, Kanamori-Katayama M, Tomaru Y, et al. Induction of microRNAs, mir-155, mir-222, mir-424 and mir-503, promotes monocytic differentiation through combinatorial regulation. Leukemia. 2010;24:460–6. doi: 10.1038/leu.2009.246. [DOI] [PubMed] [Google Scholar]
- Mann M, Barad O, Agami R, et al. miRNA-based mechanism for the commitment of multipotent progenitors to a single cellular fate. Proc Natl Acad Sci USA. 2010;107:15804–9. doi: 10.1073/pnas.0915022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Morgan MJ, Choksi S, et al. MicroRNAs modulate the noncanonical transcription factor NF-kappaB pathway by regulating expression of the kinase IKKalpha during macrophage differentiation. Nat Immunol. 2010;11:799–805. doi: 10.1038/ni.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeier S, MacPherson CR, Essack M, et al. Deciphering the transcriptional circuitry of microRNA genes expressed during human monocytic differentiation. BMC Genomics. 2009;10:595. doi: 10.1186/1471-2164-10-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A, Ballarino M, Sorrentino A, et al. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc Natl Acad Sci USA. 2007;104:19849–54. doi: 10.1073/pnas.0706963104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghani S, Riemke P, Schonheit J, et al. Macrophage development from HSCs requires PU.1-coordinated microRNA expression. Blood. 2011;118:2275–84. doi: 10.1182/blood-2011-02-335141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazi F, Rosa A, Fatica A, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123:819–31. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Garzon R, Pichiorri F, Palumbo T, et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukaemia. Oncogene. 2007;26:4148–57. doi: 10.1038/sj.onc.1210186. [DOI] [PubMed] [Google Scholar]
- Jian P, Li ZW, Fang TY, et al. Retinoic acid induces HL-60 cell differentiation via the upregulation of miR-663. J Hematol Oncol. 2011;4:20. doi: 10.1186/1756-8722-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JY, Wang F, Yu J, et al. MicroRNA-223 reversibly regulates erythroid and megakaryocytic differentiation of K562 cells. J Cell Mol Med. 2009;13:4551–9. doi: 10.1111/j.1582-4934.2008.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–9. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- Pulikkan JA, Peramangalam PS, Dengler V, et al. C/EBPalpha regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood. 2010;116:5638–49. doi: 10.1182/blood-2010-04-281600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Dowdy CR, van Wijnen AJ, et al. Altered Runx1 subnuclear targeting enhances myeloid cell proliferation and blocks differentiation by activating a miR-24/MKP-7/MAPK network. Cancer Res. 2009;69:8249–55. doi: 10.1158/0008-5472.CAN-09-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Iwama A, Satake M, et al. MicroRNA-27 enhances differentiation of myeloblasts into granulocytes by post-transcriptionally downregulating Runx1. Br J Haematol. 2009;145:412–23. doi: 10.1111/j.1365-2141.2009.07632.x. [DOI] [PubMed] [Google Scholar]
- De Marchis ML, Ballarino M, Salvatori B, et al. A new molecular network comprising PU.1, interferon regulatory factor proteins and miR-342 stimulates ATRA-mediated granulocytic differentiation of acute promyelocytic leukaemia cells. Leukemia. 2009;23:856–62. doi: 10.1038/leu.2008.372. [DOI] [PubMed] [Google Scholar]
- Surdziel E, Cabanski M, Dallmann I, et al. Enforced expression of miR-125b affects myelopoiesis by targeting multiple signaling pathways. Blood. 2011;117:4338–48. doi: 10.1182/blood-2010-06-289058. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang J, Gao L, et al. MiR-181 mediates cell differentiation by interrupting the Lin28 and let-7 feedback circuit. Cell Death Differ. 2011 doi: 10.1038/cdd.2011.127. ; in press: doi: 10.1038/cdd.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchova-Votavova H, Yoon D, Prchal JT. miR-451 enhances erythroid differentiation in K562 cells. Leuk Lymphoma. 2010;51:686–93. doi: 10.3109/10428191003629362. [DOI] [PubMed] [Google Scholar]
- Ichimura A, Ruike Y, Terasawa K, et al. MicroRNA-34a inhibits cell proliferation by repressing mitogen-activated protein kinase kinase 1 during megakaryocytic differentiation of K562 cells. Mol Pharmacol. 2010;77:1016–24. doi: 10.1124/mol.109.063321. [DOI] [PubMed] [Google Scholar]
- Navarro F, Gutman D, Meire E, et al. miR-34a contributes to megakaryocytic differentiation of K562 cells independently of p53. Blood. 2009;114:2181–92. doi: 10.1182/blood-2009-02-205062. [DOI] [PubMed] [Google Scholar]
- Huang X, Gschweng E, Van Handel B, et al. Regulated expression of microRNAs-126/126* inhibits erythropoiesis from human embryonic stem cells. Blood. 2011;117:2157–65. doi: 10.1182/blood-2010-08-302711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabher C, Payne EM, Johnston AB, et al. Zebrafish microRNA-126 determines hematopoietic cell fate through c-Myb. Leukemia. 2011;25:506–14. doi: 10.1038/leu.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick DM, Zhang CC, Tao Y, et al. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3zeta. Genes Dev. 2010;24:1614–9. doi: 10.1101/gad.1942810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Simmini S, Abreu-Goodger C, et al. The miR-144/451 locus is required for erythroid homeostasis. J Exp Med. 2010;207:1351–8. doi: 10.1084/jem.20100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svasti S, Masaki S, Penglong T, et al. Expression of microRNA-451 in normal and thalassemic erythropoiesis. Ann Hematol. 2010;89:953–8. doi: 10.1007/s00277-010-0980-7. [DOI] [PubMed] [Google Scholar]
- Romania P, Lulli V, Pelosi E, et al. MicroRNA 155 modulates megakaryopoiesis at progenitor and precursor level by targeting Ets-1 and Meis1 transcription factors. Br J Haematol. 2008;143:570–80. doi: 10.1111/j.1365-2141.2008.07382.x. [DOI] [PubMed] [Google Scholar]
- Ben-Ami O, Pencovich N, Lotem J, et al. A regulatory interplay between miR-27a and Runx1 during megakaryopoiesis. Proc Natl Acad Sci USA. 2009;106:238–43. doi: 10.1073/pnas.0811466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbaye C, Spinello I, Quaranta MT, et al. A three-step pathway comprising PLZF/miR-146a/CXCR4 controls megakaryopoiesis. Nat Cell Biol. 2008;10:788–801. doi: 10.1038/ncb1741. [DOI] [PubMed] [Google Scholar]
- Garzon R, Garofalo M, Martelli MP, et al. Distinctive microRNA signature of acute myeloid leukaemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA. 2008;105:3945–50. doi: 10.1073/pnas.0800135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R, Volinia S, Liu CG, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukaemia. Blood. 2008;111:3183–9. doi: 10.1182/blood-2007-07-098749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongen-Lavrencic M, Sun SM, Dijkstra MK, et al. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukaemia. Blood. 2008;111:5078–85. doi: 10.1182/blood-2008-01-133355. [DOI] [PubMed] [Google Scholar]
- O'Connell RM, Rao DS, Chaudhuri AA, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–94. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costinean S, Sandhu SK, Pedersen IM, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009;114:1374–82. doi: 10.1182/blood-2009-05-220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Chaudhuri AA, Rao DS, et al. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci USA. 2009;106:7113–8. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen IM, Otero D, Kao E, et al. Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of B cell lymphomas. EMBO Mol Med. 2009;1:288–95. doi: 10.1002/emmm.200900028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R, Heaphy CE, Havelange V, et al. MicroRNA 29b functions in acute myeloid leukaemia. Blood. 2009;114:5331–41. doi: 10.1182/blood-2009-03-211938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R, Liu S, Fabbri M, et al. MicroRNA-29b induces global DNA hypomethylation and tumour suppressor gene reexpression in acute myeloid leukaemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–8. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum W, Garzon R, Klisovic RB, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci USA. 2010;107:7473–8. doi: 10.1073/pnas.1002650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wu LC, Pang J, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukaemia. Cancer Cell. 2010;17:333–47. doi: 10.1016/j.ccr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichler S, Illmer T, Roemhild J, et al. MicroRNA29a regulates the expression of the nuclear oncogene Ski. Blood. 2011;118:1899–902. doi: 10.1182/blood-2010-09-306258. [DOI] [PubMed] [Google Scholar]
- Cammarata G, Augugliaro L, Salemi D, et al. Differential expression of specific microRNA and their targets in acute myeloid leukaemia. Am J Hematol. 2010;85:331–9. doi: 10.1002/ajh.21667. [DOI] [PubMed] [Google Scholar]
- Isken F, Steffen B, Merk S, et al. Identification of acute myeloid leukaemia associated microRNA expression patterns. Br J Haematol. 2008;140:153–61. doi: 10.1111/j.1365-2141.2007.06915.x. [DOI] [PubMed] [Google Scholar]
- Felli N, Fontana L, Pelosi E, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci USA. 2005;102:18081–6. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackanson B, Bennett KL, Brena RM, et al. Epigenetic modification of CCAAT/enhancer binding protein alpha expression in acute myeloid leukaemia. Cancer Res. 2008;68:3142–51. doi: 10.1158/0008-5472.CAN-08-0483. [DOI] [PubMed] [Google Scholar]
- Pulikkan JA, Dengler V, Peramangalam PS, et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukaemia. Blood. 2010;115:1768–78. doi: 10.1182/blood-2009-08-240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyholzer M, Schmid S, Wilkens L, et al. The tumour-suppressive miR-29a/b1 cluster is regulated by CEBPA and blocked in human AML. Br J Cancer. 2010;103:275–84. doi: 10.1038/sj.bjc.6605751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse W, Meshinchi S, Alonzo TA, et al. Elevated expression of the AF1q gene, an MLL fusion partner, is an independent adverse prognostic factor in pediatric acute myeloid leukaemia. Blood. 2004;104:3058–63. doi: 10.1182/blood-2003-12-4347. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Li Z, Ji M, et al. MIR29B regulates expression of MLLT11 (AF1Q), an MLL fusion partner, and low MIR29B expression associates with adverse cytogenetics and poor overall survival in AML. Br J Haematol. 2011;153:753–7. doi: 10.1111/j.1365-2141.2011.08662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–9. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- Pigazzi M, Manara E, Baron E, et al. miR-34b targets cyclic AMP-responsive element binding protein in acute myeloid leukaemia. Cancer Res. 2009;69:2471–8. doi: 10.1158/0008-5472.CAN-08-3404. [DOI] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–32. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- Venturini L, Battmer K, Castoldi M, et al. Expression of the miR-17-92 polycistron in chronic myeloid leukaemia (CML) CD34+ cells. Blood. 2007;109:4399–405. doi: 10.1182/blood-2006-09-045104. [DOI] [PubMed] [Google Scholar]
- Li Z, Lu J, Sun M, et al. Distinct microRNA expression profiles in acute myeloid leukaemia with common translocations. Proc Natl Acad Sci USA. 2008;105:15535–40. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Li Z, Chen P, et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukaemia. Proc Natl Acad Sci USA. 2010;107:3710–5. doi: 10.1073/pnas.0914900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R, Pichiorri F, Palumbo T, et al. MicroRNA fingerprints during human megakaryocytopoiesis. Proc Natl Acad Sci USA. 2006;103:5078–83. doi: 10.1073/pnas.0600587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Iwasaki M, Somervaille TC, et al. The miR-17-92 microRNA polycistron regulates MLL leukaemia stem cell potential by modulating p21 expression. Cancer Res. 2010;70:3833–42. doi: 10.1158/0008-5472.CAN-09-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic R, Riesbeck LE, Velu CS, et al. Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood. 2009;113:3314–22. doi: 10.1182/blood-2008-04-154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackman D, Lund-Johansen F, Aarskog D. Expression of cell surface antigens during the differentiation of HL-60 cells induced by 1,25-dihydroxyvitamin D3, retinoic acid and DMSO. Leuk Res. 1995;19:57–64. doi: 10.1016/0145-2126(94)00061-e. [DOI] [PubMed] [Google Scholar]
- Wang X, Gocek E, Liu CG, et al. MicroRNAs181 regulate the expression of p27Kip1 in human myeloid leukaemia cells induced to differentiate by 1,25-dihydroxyvitamin D3. Cell Cycle. 2009;8:736–41. doi: 10.4161/cc.8.5.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet M, Quelen C, Rosati R, et al. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukaemia with the t(2;11)(p21;q23) translocation. J Exp Med. 2008;205:2499–506. doi: 10.1084/jem.20080285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Chaudhuri AA, Rao DS, et al. MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc Natl Acad Sci USA. 2010;107:14235–40. doi: 10.1073/pnas.1009798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klusmann JH, Li Z, Bohmer K, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukaemia. Genes Dev. 2010;24:478–90. doi: 10.1101/gad.1856210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet M, Harris MH, Zhou B, et al. MicroRNA miR-125b causes leukaemia. Proc Natl Acad Sci USA. 2010;107:21558–63. doi: 10.1073/pnas.1016611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saumet A, Vetter G, Bouttier M, et al. Transcriptional repression of microRNA genes by PML-RARA increases expression of key cancer proteins in acute promyelocytic leukaemia. Blood. 2009;113:412–21. doi: 10.1182/blood-2008-05-158139. [DOI] [PubMed] [Google Scholar]
- Careccia S, Mainardi S, Pelosi A, et al. A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene. 2009;28:4034–40. doi: 10.1038/onc.2009.255. [DOI] [PubMed] [Google Scholar]
- Fazi F, Racanicchi S, Zardo G, et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell. 2007;12:457–66. doi: 10.1016/j.ccr.2007.09.020. [DOI] [PubMed] [Google Scholar]