

Abstract
It has been shown that ingestion of glucose, amino acids, protein or mixed meals tends to increase serum and salivary cortisol concentrations in healthy adults. Recently, it has been demonstrated that morning glucose ingestion stimulates pulsatile cortisol and adrenocorticotropic hormone (ACTH) secretion, thus elevating their mean concentrations. In light of the above, a question arises: could the frequent food – and specifically glucose – consumption lead to hypercortisolism with possible clinical implications? And can the human body, under normal conditions raise defence mechanisms against the transient hypercortisolism caused by the frequent glucose consumption? Studies have revealed novel mechanisms, which are implicated in the glucocorticoid receptor (GR)-mediated action, providing a kind of glucocorticoid resistance. This glucocorticoid resistance could be mediated through both enhancing acetylation (via, among others, regulation of essential clock genes such as Per) and inhibiting deacetylation of GR (via possible regulation of sirtuin activity). Interestingly, the acetylation/deacetylation processes seem to be regulated by glucose. Thus, glucose apart from causing increased cortisol secretion can, simultaneously, counter-regulate this hypercortisolism, by promoting directly and/or indirectly a glucocorticoid resistance state. Undoubtedly, before extracting conclusions regarding the clinical significance of the increased cortisol secretion following glucose ingestion, we should first thoroughly investigate the ‘defence’ mechanisms provided by ‘nature’ to handle this hypercortisolism.
Keywords: CLOCK system, cortisol secretion, glucocorticoid resistance, glucose, sirtuins
Introduction
Glucocorticoids (GCs) are the cornerstone in the treatment of numerous chronic autoimmune and inflammatory diseases due to their potent anti-inflammatory and immunosuppressive properties. However, GC treatment is accompanied by significant metabolic adverse effects, including insulin resistance, glucose intolerance and diabetes. Notably, even low GC doses just above cortisol replacement levels, as well as endogenous subclinical hypercortisolism, may impair glucose tolerance by reducing hepatic and peripheral insulin sensitivity [1,2].
On the other hand, ingestion of glucose – as well as amino acids, protein, or mixed meals – has been found to increase serum and salivary cortisol levels in healthy adults [3,4]. Both ACTH-dependent and ACTH-independent mechanisms of food-induced cortisol secretion have been postulated under physiological conditions [5,6], although the exact mechanisms still remain elusive. Feeding increases noradrenaline turnover in some parts of the rat hypothalamus [7] and it is possible that this may be the stimulus to postprandial ACTH secretion via α-1 adrenoceptors activated also in humans [8]. In support of these findings, Iranmanesh et al. have recently demonstrated that morning glucose ingestion stimulates pulsatile cortisol and ACTH secretion 2011. The mechanism entailed selective augmentation of ACTH and cortisol secretory-burst mass. Indeed, ACTH and cortisol burst size rose comparably by 27–31% after 75 g oral glucose administration [9].
In view of all the above, a question arises: does the frequent food consumption, and specifically the frequent consumption of glucose-containing foods lead to hypercortisolism with possible clinical implications?
The human body, under normal conditions, can raise defence mechanisms against the transient hypercortisolism caused by the frequent glucose consumption. Indeed, recent studies have unravelled interesting mechanisms that are implicated in the GR-mediated action, providing a kind of glucocorticoid resistance. These mechanisms seem to be regulated, among others, by glucose. Thus, glucose apart from causing increased cortisol secretion can, simultaneously, counter-regulate this hypercortisolism, by promoting directly and/or indirectly a glucocorticoid resistance state (Fig. 1).
Fig 1.
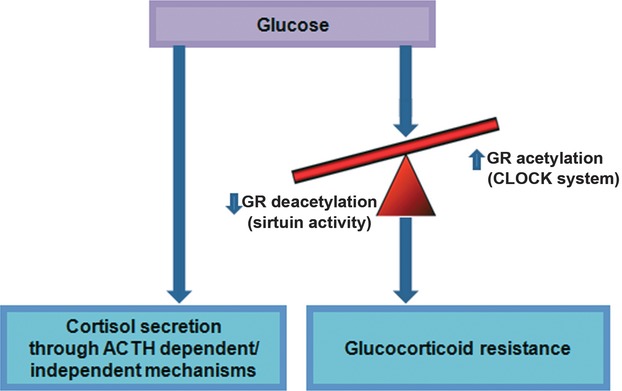
We consider that from one side glucose consumption triggers cortisol secretion, thereby elevating its concentration, while from the other side, it can promote a kind of glucocorticoid resistance. This glucocorticoid resistance could be mediated through both enhancing acetylation (via, among others, regulation of essential clock genes such as Per) and hindering deacetylation of GR (through possible regulation of sirtuin activity).
Glucose could promote directly and/or indirectly a glucocorticoid resistance state
GCs mediate their effects via their intracellular receptor GRα (the classical GR), a member of the nuclear receptor superfamily (GRβ, albeit expressed widely, does not bind GCs). The GC–GR complex can act as a transcription factor that regulates transcription of many GC-responsive genes via several mechanisms. These include trans-activation through binding of GC–GR complex to consensus glucocorticoid response elements (GREs), as well as trans-repression via interaction with other transcription factors such as activator protein-1 (AP-1) and nuclear factor κB (NF-κB) [10].
This transcriptional activity is modulated not only by various co-regulators (co-activators and co-repressors) and other transcription factors, but also via post-translational modifications of the GR protein, such as methylation, phosphorylation, acetylation etc [11]. Indeed, acetylation is a general epigenetic alteration that controls activity of, among others, GR protein. The human GR was first shown to be acetylated at lysines 494 and 495 located in its hinge region [12]; this acetylation of GR was found to attenuate the binding of the receptor to GREs hence repressing GR-induced trans-activation of GRE-driven promoters, while it is also possible that GR acetylation alters nuclear translocation of the receptor, with both the above mechanisms leading to a kind of glucocorticoid resistance at the cellular level. According to a recent work by Charmandari et al., GR acetylation is higher in the morning than in the evening in human peripheral blood mononuclear cells (PBMCs), mirroring the fluctuations of circulating cortisol in reverse phase and serving as a counter regulatory mechanism that effectively decreases tissue sensitivity to GCs in the morning and increases it at night 2011. Interestingly, GR is deacetylated by histone deacetylase 2 (HDAC2) [13]. It should be noted that sirtuin 1 (Sirt1), a class III HDAC and one of the seven human sirtuins, deacetylates several nuclear receptors; this HDAC might also deacetylate the GR.
Apparently, changes in the glucocorticoid resistance – which represents a counter-regulatory mechanism against increased cortisol secretion – could result from changes in the balance between acetylation and deacetylation of GR. For example, increased acetylation and/or reduced deacetylation of GR could sustain a state of glucocorticoid resistance.
Could glucose affect this acetylation/deacetylation process in a manner that counter-regulates the increased cortisol secretion caused by itself?
We know that glycolysis is employed by all body cells to obtain part of the chemical energy entrapped in the glucose molecule. Glycolytic pathway is known to lead to the net production of ATP, consuming NAD during the metabolism of glyceraldehyde-3-phosphate to 1,3-diphosphoglycerate and NADH as products. Sirtuins – which may deacetylate the GR – are unique in that they require NAD as a cofactor [14,15]. In a complicated reaction, sirtuins couple lysine deacetylation to NAD hydrolysis, yielding O-acetyl-ADPribose and nicotinamide [15]. As such, sirtuin activity seems to be governed by cellular [NAD]/[NADH] ratios and responds to changes in cellular metabolism [16,17]. An increased glycolytic activity would tend to provoke an accumulation of NADH and lower NAD availability, resulting directly in a decreased sirtuin (Sirt1) activity [18]. Moreover, a glucose challenge, by potentiation of insulin–insulin receptor signalling cascade could down-regulate sirtuin activity via activation of mTOR signalling, an effect mediated through repression of the nicotinamidase gene PNC1 expression. In both cases, glucose, through decreasing the sirtuin activity, could allow GR to remain acetylated, thus sustaining a glucocorticoid resistance state [18].
Is this the only possible mechanism for glucose to regulate acetylation/deacetylation of GR, promoting glucocorticoid resistance at the cellular level?
Humans, like most other organisms, have an endogenous pacemaker, the circadian locomotor output cycle kaput (CLOCK) system, which generates a circadian (‘about a day’) rhythm in several physiologic processes, including hypothalamic–pituitary–adrenal (HPA) secretion [19]. Both central and peripheral CLOCKs utilize the same transcription regulatory machinery for generating intrinsic circadian rhythms [20]. A central role in this machinery is played by the Clock and its heterodimer partner brain-muscle-arnt-like protein 1 (Bmal1) transcription factors. Interaction between Clock and Bmal1 stimulates the transcription of other essential clock genes, such as Periods (Per1, Per2 and Per3) and Cryptochromes (Cry1 and Cry2). The resulting PER and CRY proteins heterodimerize, translocate to the nucleus and interact with the Clock–Bmal1 complex to inhibit their transcription. Of note, degradation of the PER–CRY repressor complex allows the Clock–Bmal1 to become activated again [21].
Interestingly, it has been recently found that the Clock transcription factor acetylates a lysine cluster located in the hinge region of the human GR, including lysines at amino acid positions 480, 492, 494 and 495. These acetylations reduce GR's affinity for its cognate GREs, thus repressing the GR-induced transcription of several GC-responsive genes (glucocorticoid resistance state) [22].
Could glucose regulate the CLOCK system, therefore controlling the acetylation of GR?
A study by Hirota et al. identified glucose as a key molecule that can directly reset the peripheral clock by down-regulating Per1 and Per2 mRNA levels in rat fibroblasts 2002. In the same study, TIEG1 and VDUP1 were suggested as candidates for Clock-associated molecule(s) mediating the glucose signal. It could be hypothesized that this down-regulation of Per1 and Per2 liberates the Clock transcription factor, allowing it to acetylate the GR.
Nevertheless, the finding of clock genes regulated by glucose suggests the activation of several transcriptional regulators and cellular pathways that have not been known to respond to glucose and that remain to be investigated.
Discussion and considerations
It could be suggested that from one side, glucose consumption stimulates cortisol secretion, thereby elevating its concentration, while from the other side, it can promote a kind of glucocorticoid resistance. This glucocorticoid resistance could be mediated through both enhancing acetylation (via, among others, regulation of essential clock genes such as Per) and hindering deacetylation of GR (through possible regulation of sirtuin activity) (Fig. 1). Any dysregulation in this acetylation/deacetylation process could lead to an excess cortisol action, following carbohydrates consumption. Nonetheless, this is not the only mechanism since, according to a recent study by Nader et al., AMP-activated protein kinase (AMPK), the fuel sensor which is regulated by AMP/ATP levels and consequently by glycolysis rate, regulates GC actions by phosphorylating the GR hence modifying transcription of GC-responsive genes 2010.
It is conceivable that many points of this speculation remain to be explored, such as: is this feedback loop which avoids an increased cortisol action a generalized mechanism or a tissue-, or even cell-specific one, serving special purposes? Could glucose-induced effect on cortisol secretion and GR-mediated action through receptor acetylation/deacetylation be expanded to other food ingredients (amino acids, fatty acids)? And, finally, how aberrant regulation of this balance (secretion versus action) could contribute to a possible glucose-induced or even food-induced hypercortisolism?
Noteworthy, the response of the HPA axis to meals containing different macronutrient proportions has been recently investigated; a higher cortisol secretion after ingestion of large amounts of carbohydrates compared with high-lipid/protein meal was shown, suggesting a possible pathophysiological relevance in obesity [25,26], albeit there being still inconsistencies in the literature [27,28].
Undoubtedly, before extracting conclusions regarding the clinical significance of the increased cortisol secretion following glucose ingestion, the ‘defence’ mechanisms provided by ‘nature’ to handle this hypercortisolism should be first thoroughly explored.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- Rossi R, Tauchmanova L, Luciano A, et al. Subclinical Cushing's syndrome in patients with adrenal incidentaloma: clinical and biochemical features. J Clin Endocrinol Metab. 2000;85:1440–8. doi: 10.1210/jcem.85.4.6515. [DOI] [PubMed] [Google Scholar]
- Van Raalte DH, Brands M, van der Zijl NJ, et al. Low-dose glucocorticoid treatment affects multiple aspects of intermediary metabolism in healthy humans: a randomised controlled trial. Diabetologia. 2011;54:2103–12. doi: 10.1007/s00125-011-2174-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slag MF, Ahmad M, Gannon MC, et al. Meal stimulation of cortisol secretion: a protein induced effect. Metabolism. 1981;30:1104–8. doi: 10.1016/0026-0495(81)90055-x. [DOI] [PubMed] [Google Scholar]
- Follenius M, Brandenberger G, Hietter B. Diurnal cortisol peaks and their relationships to meals. J Clin Endocrinol Metab. 1982;55:757–61. doi: 10.1210/jcem-55-4-757. [DOI] [PubMed] [Google Scholar]
- Fehm HL, Holl R, Klein E, et al. The meal-related peak in plasma cortisol is not mediated by radioimmunoassayable ACTH. Clin Physiol Biochem. 1983;1:329–33. [PubMed] [Google Scholar]
- Modlinger RS, Schonmuller JM, Arora SP. Adrenocorticotropin release by tryptophan in man. J Clin Endocrinol Metab. 1980;50:360–3. doi: 10.1210/jcem-50-2-360. [DOI] [PubMed] [Google Scholar]
- Myers RD, McCaleb ML. Feeding satiety signal from intestine triggers brain's noradrenergic mechanism. Science. 1980;209:1035–7. doi: 10.1126/science.7403866. [DOI] [PubMed] [Google Scholar]
- Al-Damluji S, Iveson T, Thomas JM, et al. Food-induced cortisol secretion is mediated by central α-1 adrenoceptor modulation of pituitary ACTH secretion. Clin Endocrinol (Oxf) 1987;26:629–36. doi: 10.1111/j.1365-2265.1987.tb00819.x. [DOI] [PubMed] [Google Scholar]
- Iranmanesh A, Lawson D, Dunn B, et al. Glucose ingestion selectively amplifies ACTH and cortisol secretory-burst mass and enhances their joint synchrony in healthy men. J Clin Endocrinol Metab. 2011;96:2882–8. doi: 10.1210/jc.2011-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronacher K, Hadley K, Avenant C, et al. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. 2009;299:219–31. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102:11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Charmandari E, Chrousos GP, Lambrou GI, et al. Peripheral CLOCK regulates target-tissue glucocorticoid receptor transcriptional activity in a circadian fashion in man. PLoS One. 2011;6:e25612. doi: 10.1371/journal.pone.0025612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Yamamura S, Essilfie-Quaye S, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–35. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Denu JM. Linking chromatin function with metabolic networks: Sir2 family of NAD(+)- dependent deacetylases. Trends Biochem Sci. 2003;28:41–8. doi: 10.1016/s0968-0004(02)00005-1. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Kaeberlein M, Andalis AA, et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–8. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Ford E, Haigis M, et al. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–6. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassi E, Papavassiliou AG. Could glucose be a proaging factor? J Cell Mol Med. 2008;12:1194–8. doi: 10.1111/j.1582-4934.2008.00329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafarczyk A, Ixart G, Malaval F, et al. Effects of lesions of the suprachiasmatic nuclei and of p-chlorophenylalanine on the circadian rhythms of adrenocorticotrophic hormone and corticosterone in the plasma, and on locomotor activity of rats. J Endocrinol. 1979;83:1–16. doi: 10.1677/joe.0.0830001. [DOI] [PubMed] [Google Scholar]
- Takahashi JS, Hong HK, Ko CH, et al. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, et al. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–67. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 2009;23:1572–83. doi: 10.1096/fj.08-117697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Okano T, Kokame K, et al. Glucose down-regulates Per1 and Per2 mRNA levels and induces circadian gene expression in cultured Rat-1 fibroblasts. J Biol Chem. 2002;277:44244–51. doi: 10.1074/jbc.M206233200. [DOI] [PubMed] [Google Scholar]
- Nader N, Ng SS, Lambrou GI, et al. AMPK regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 MAPK. Mol Endocrinol. 2010;24:1748–64. doi: 10.1210/me.2010-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicennati V, Ceroni L, Gagliardi L, et al. Comment: response of the hypothalamic-pituitary-adrenocortical axis to high-protein/fat and high-carbohydrate meals in women with different obesity phenotypes. J Clin Endocrinol Metab. 2002;87:3984–8. doi: 10.1210/jcem.87.8.8718. [DOI] [PubMed] [Google Scholar]
- Martens MJ, Rutters F, Lemmens SG, et al. Effects of single macronutrients on serum cortisol concentrations in normal weight men. Physiol Behav. 2010;101:563–7. doi: 10.1016/j.physbeh.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Gibson EL, Checkley S, Papadopoulos A, et al. Increased salivary cortisol reliably induced by a protein-rich midday meal. Psychosom Med. 1999;61:214–24. doi: 10.1097/00006842-199903000-00014. [DOI] [PubMed] [Google Scholar]
- Lemmens SG, Born JM, Martens EA, et al. Influence of consumption of a high-protein versus high-carbohydrate meal on the physiological cortisol and psychological mood response in men and women. PLoS One. 2011;6:e16826. doi: 10.1371/journal.pone.0016826. [DOI] [PMC free article] [PubMed] [Google Scholar]