

Abstract
Blood levels of extracellular nucleotides (e.g. ATP) are greatly increased during heart ischaemia, but, despite the presence of their specific receptors on cardiomyocytes (both P2X and P2Y subtypes), their effects on the subsequent myocardial damage are still unknown. In this study, we aimed at investigating the role of ATP and specific P2 receptors in the appearance of cell injury in a cardiac model of ischaemic/hypoxic stress. Cells were maintained in a modular incubator chamber in a controlled humidified atmosphere of 95% N2 for 16 hrs in a glucose-free medium. In this condition, we detected an early increase in the release of ATP in the culture medium, which was followed by a massive increase in the release of cytoplasmic histone-associated-DNA-fragments, a marker of apoptosis. Addition of either apyrase, which degrades extracellular ATP, or various inhibitors of ATP release via connexin hemichannels fully abolished ischaemic/hypoxic stress-associated apoptosis. To dissect the role of specific P2 receptor subtypes, we used a combined approach: (i) non-selective and, when available, subtype-selective P2 antagonists, were added to cardiomyocytes before ischaemic/hypoxic stress; (ii) selected P2 receptors genes were silenced via specific small interfering RNAs. Both approaches indicated that the P2Y2 and P2χ7 receptor subtypes are directly involved in the induction of cell death during ischaemic/hypoxic stress, whereas the P2Y4 receptor has a protective effect. Overall, these findings indicate a role for ATP and its receptors in modulating cardiomyocyte damage during ischaemic/hypoxic stress.
Keywords: cardiomyocytes, P2 receptors, ischaemic/hypoxic stress, apoptosis
Introduction
It is now widely accepted that, in addition to functioning as an intracellular energy source, extracellular adenine and uracil nucleotides initiate a wide range of intracellular signalling cascades through the activation of a plethora of purinergic receptors (the P2 receptors). The purines and pyrimidines ATP, adenosine diphosphate (ADP), uridine triphosphate (UTP), uridine diphosphate (UDP), and, as more recently recognized, sugar nucleotides such as UDP-glucose and UDP-galactose [1-4] indeed act on various P2X homo- and heteromultimeric ionotropic and P2Y metabotropic receptor subtypes [3].
In the cardiovascular system, evidence is accumulating to indicate that P2 receptors mediate important physiologic effects, including vasoconstriction and vasodilatation, growth of vascular smooth muscle and endothelial cells, angiogenesis, vascular remodelling, coagulation, platelet aggregation, inflammation and several aspects of cardiac function. The purinergic system and its receptors are also involved in development of myocardial infarction (MI), heart failure and xenograft rejection [5, 6].
However, although the heart has been the first organ to be tested for the physiological actions of nucleotides in early work using adenylic acid [7], the effects mediated by specific myocardial P2 receptors are just now beginning to be elucidated. P2 receptors are abundantly expressed in the foetal human heart [8] as well as in the adult human heart [9-11]. In cardiomyocytes, ATP acting through P2Y and P2X receptors stimulates a pronounced positive inotropic effect and may also act in synergy with β-adrenergic agonists to augment myocyte contractility (Vassort, summarized in 12). Besides adenine nucleotides, uracil nucleotides have been also recently reported to act as positive inotropic agents in rat and mouse cardiomyocytes [10]. In addition, potent effects may be exerted by nucleotides on cardiomyocytes viability, under both physiological conditions and after hypoxia, when cardiomyocytes themselves release massive amounts of ATP. Given its importance as an energy substrate, ATP release is tightly controlled and occurs by several mechanisms such as secretory vesicles, ABC transporter cassette proteins, connexin hemichannels [13], pannexin channels [14] as well as through other channels with large unitary conductances [15]. In line with this hypothesis, short-term activation of UTP receptors protects cardiomyocytes against hypoxic damage and significantly reduces the cell death caused by hypoxia via activation of the P2Y2 and P2Y4 receptors [16]. In a previous study, we have investigated the effects of both adenine and uracil nucleotides on the viability of HL-1 cardiomyocytes, the only available cell line that spontaneously contracts in vitro and maintains a differentiated cardiac phenotype [17]. We showed that murine HL-1 cardiomyocytes express a wide panel of P2X and P2Y receptors known to either exclusively respond to adenine nucleotides (P2X receptors), to both adenine and uracil nucleotides (P2Y2, P2Y4, P2Y6) or to sugar nucleotides (P2Y14 receptor) [18]. Such a large heterogeneity of P2 receptor expression is consistent with previous studies [8, 11], and suggests involvement of these receptors in multiple functional effects. We further demonstrated that the exposure of cardiomyocytes to high concentrations of adenine nucleotides (ATP, ADP or BzATP) induces cardiomyocyte cell death through a mechanism involving both P2Y and P2X receptors [18]. Thus, besides influencing cardiac contractility, P2 receptors may also directly regulate the viability of myocardial cells.
In this study, we set up and characterized an hypoxia/ ischaemia protocol in HL-1 cardiomyocytes to evaluate (a) whether ATP is endogenously released by these cells and possibly plays a role in induction of cell death under these conditions; (b) whether ischaemia-associated cardiomyocyte death is influenced by pharmacological agents known to act on either ATP release/availability or on P2 receptors, with the final aim to; (c) identify the specific P2 receptor subtypes involved in regulation of cardiomyocyte viability. Because apoptosis has a central role in MI, we focused our attention on this type of cell death. Results may have important therapeutic implications and set the basis for the development of novel cardioprotective agents that target-specific P2 receptor subtypes.
Materials and methods
Reagents
Pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonate (PPADS, 100 μmol/l); suramin (100 μmol/l); gadolinium(III) chloride (GdCl3 100 μmol/l); 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate monolithium trisodium salt (TNP-ATP, 10 μmol/l); 4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenylisoquinolinesulfonic acid ester (KN-62, 1 μmol/l); N,N″-1,4-butanediylbis[N’-(3-isothiocyanatophenyl)thiourea (MRS2578 1–10 μmol/l); apyrase (30 U/ml); pertussis toxin (PTX, 100 nmol/l); GF 109203X (1–2 μmol/l); Guanosine 5′-[β-thio]diphosphate trilithium salt (GDP β-S 250–500 μmol/l); and 18aGA (5–10 μmol/l) were from Sigma-Aldrich (St. Louis, MO, USA); Gap 26 (VCYDKSFPISHVR, 300 μmol/L) was from Tocris (Ellisville, MO, USA). 5-[[5-{2,8-Dimethyl-5H-dibenzo[a,d]cyclohepten-5-yl}-3,4-dihydro-2-oxo-4-thioxo-1(2H)-pyrimidinyl]methyl]-N-[1H-tetrazol-5-yl]-2-furancarboxamide (AR-C11892510 μmol/l) was a kind gift from Prof. Dr. C.E. Müller.
Cell culture
HL-1 cells, a cardiac muscle cell line derived from the AT-1 mouse atrial myocyte tumour lineage, were a gift from William C. Claycomb, and maintained according to described protocols [17, 19]. In separate experimental groups, cells received no intervention (normoxia control, 95% air and 5% CO2) or were exposed to ischaemic/hypoxic stress. Hypoxia was produced by exposure to 5% CO2 and 95% N2 in a modular incubator chamber for 16 hrs in the presence of serum- and glucose-free DMEM medium. Control cells received vehicle or the indicated compounds.
Real-time RT-PCR
Total cell RNA was extracted using TRIzol Reagent (Invitrogen Life Technologies, Milano, Italy), and reverse transcribed as described [20]. Real-time quantitative PCR was then carried out to detect P2Y2, P2Y4, P2Y6 and P2χ7 mRNA. 18S rRNA was used for sample normalization. The sequences of the primers used were: mP2Y6 sense: 5′- CCC AAC CTG CCT TGA AAA CA-3′, antisense: 5′-TCG GAG AGT CTG TCT CAT GCA A-3′; 18S sense: 5′-CGGCTACCACATCCAAGGAA-3′; 18S antisense: 5′-CCTGTATTGTTATTTTTCGTCACTACCT-3′. Primers for the detection of P2Y2 (QT00097202), P2Y4 (QT00266686) and P2χ7 (QT00130900) were from Qiagen (Milan, Italy). A total of 2.5 μl of cDNAs were incubated in 25 μl IQ Supermix containing P2Y2, P2Y6, P2χ7 or 18S primers and SYBRGreen fluorescence dye (Bio-Rad Laboratories, Milano, Italy). Real-time RT-PCR was carried out in triplicate for each sample on an iCycler Optical System, Bio-Rad Laboratories. Amplifications of P2Y4 were performed as described [11].
RNA interference and cell transfection
Validated high-performance purity grade small interfering RNAs (siRNA) against mouse P2Y2, P2Y4, P2Y6, and P2χ7 were synthesized by Qiagen. Control siRNA, with a non-silencing oligonucleotide sequence that does not recognize any known homology to mammalian genes, was also generated as a negative control, as described in Ref. [20].
Nuclear staining of adhering cells
After cell treatment, cells were fixed in 4% paraformaldehyde in phosphate buffered saline and nuclear chromatin stained by using the fluorescent dye Hoechst 33258 (Sigma-Aldrich), as described in Ref. [21].
Flow cytometric evaluation of apoptosis
The percentage of necrotic cells in the adhering cells was evaluated by means of propidium iodide (PI) staining of DNA followed by flow cytometric analysis as described in Refs. [11, 22]. Induction of apoptosis was also confirmed by measuring Annexin V, as described in [18].
Nucleosome immunoassay
Quantification of histone-complexed DNA fragments (mono- and oligonucleosomes) was performed by one-step sandwich immunoassay (Roche Diagnostics, Mannheim Germany). Data are expressed as absorbance/mg of cell protein determined by Bradford assay.
ATP release assay
ATP release was assayed by means of the ATP Bioluminescence Assay Kit HS II (Roche Diagnostics).
Statistical analysis
Results are expressed as mean ± S.E.M. and were statistically analysed by means of ANOVA followed by Tukey’s post-hoc test. A P-value less than 0.05 was considered significant.
Results
Ischaemic/hypoxic stress induces ATP release in HL-1 cardiomyocytes
Released cellular ATP can serve as a paracrine signalling molecule for intercellular communication, or can act in an autocrine manner to regulate cellular functions, including those of cardiomyocytes. Cultured HL-1 cardiomyocytes exhibited a basal release of ATP (approximately equal to 1.25 nmol/l) that was significantly increased at 30 min. after the induction of an ischaemic/hypoxic stress (approximately 2.28 nmol/l; Fig. 1A). To ensure that, at this time point, membrane damage did not contribute to ATP release, we analysed the conditioned media from ischaemic/hypoxic cells for the presence of the cytosolic enzyme lactate dehydrogenase (LDH), a marker of cellular damage. We consistently found that ischaemic/hypoxic-induced ATP release occurred in the absence of LDH release, hence with no significant plasmalemmal damage compared with normoxic controls (data not shown), suggesting that, at this early stage of hypoxia, ATP does not derive from cell necrosis but from release via transmembrane channels/pumps.
Fig 1.
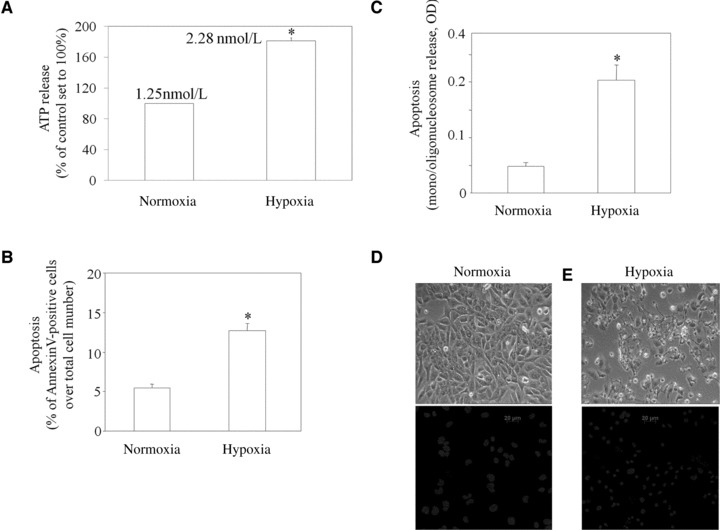
ATP is released by HL-1 cells during hypoxia, which induces apoptosis. (A) ATP was measured in the conditioned medium of cells exposed to normoxia or hypoxia for 30 min. (B, C) Apoptosis of cells exposed to normoxia or hypoxia for 16 hrs were quantified by flow cytometric analysis as annexin V positive cells (B) or by nucleosome immunoassay (C). (D, E) Representative images of HL-1 cells exposed to either normoxia or hypoxia for 16 hrs. Upper images are phase-contrast micrographs; lower images refer to cultures stained with Hoechst 33,258. Results are the mean ± S.E.M., n = 4, *P < 0.05 versus normoxic controls.
Ischaemic/hypoxic stress promotes apoptosis of HL-1 cardiomyocytes
Ischaemic/hypoxic stress induced significant increases in the appearance of HL-1 cardiomyocytes apoptosis, as assessed by two complementary assays including annexin V and nucleosomal immunoassay. An increased number of annexin-positive cardiomyocytes was registered after 16 hrs of exposure to ischaemic/hypoxic stress (Fig. 1B). We found that, at this time after ischaemic/hypoxic stress induction, the number of PI-positive cells was also increased compared to control value (+72.2% ± 9.9, P < 0.05, data not shown). To confirm the membrane-related changes associated with apoptosis, we performed nucleosomal ELISA assay, which detect later stages of apoptosis. ELISA analysis showed a strong increase in the apoptotic index, up to 320.9% ± 15.8 of control value at 16 hrs (Fig. 1C). Because one important sign of apoptosis is the appearance of specific morphological changes, including nuclear condensation and chromatin fragmentation, we used Hoechst 33258, a fluorescent DNA-binding dye, to stain nuclear chromatin in HL-1 cardiomyocytes, and examined cell morphology by phase-contrast microscopy. Figure 1D and E shows the phase contrast morphology of HL-1 cardiomyocytes exposed to ischaemic/hypoxic stress or to normoxia. Control cultures have normal cell bodies and intact features with nuclei homogeneous in size and shape and very lightly stained with Hoechst 33258 (Fig. 1D). However, when HL-1 cardiomyocytes were exposed to ischaemia/hypoxia for 16 hrs, we noticed morphologic alterations typical of cells undergoing apoptosis, which became rounded and condensed and lost adhesion from the monolayer. Furthermore, HL-1 cells displayed condensed chromatin that was brightly and uniformly stained by Hoechst 33258 and ranged in shape from a single uniform sphere to a collection of multiple chromatin dots typical of fragmented apoptotic nuclei (Fig. 1E).
Role of ATP in the ischaemic/hypoxic stress-induced apoptosis in cardiomyocytes
Endogenously released ATP plays an important role in mediating HL-1 cardiomyocytes apoptosis, as treatment with apyrase, a nucleotidase that degrades nucleotide triphosphates into nucleotide monophosphates and thus completely abolishes the amount of ATP released in the medium during ischaemia/hypoxia stress (Fig. 2A), significantly prevented the induction of apoptosis (Fig. 2B). Because there is, as expected, a constitutive basal release of ATP even in the absence of hypoxia, apyrase also significantly reduced basal ATP levels during normoxia (Fig. 2A). However, this was not associated to any change in cell viability, to confirm that, under control conditions, ATP does not mediate cell death. To investigate the role of connexin hemichannels in the induction of apoptosis following ischaemic/hypoxic stress experiments were carried out in the presence of established inhibitors of these channels. Gap26, a highly specific inhibitor of connexin hemichannels, significantly prevented the induction of apoptosis induced by ischaemic/hypoxic stress (Fig. 2C). A further less specific inhibitor of connexin channels, 18aGA, was also protective (Fig. 2D). The maxi-anion channel blocker GdCl3, which has been previously involved in ATP release [15], also inhibited ischaemic/hypoxic stress-associated apoptosis (data not shown). To further confirm the role of released ATP in the induction of apoptosis by ischaemic/hypoxic stress in HL-1 cells, we found that stimulation of HL-1 cardiomyocytes by ATP itself for 24 hrs at 250–500 μmol/l increased apoptosis up to 254% ± 37 of control values set to 100% (Fig. 2E).
Fig 2.
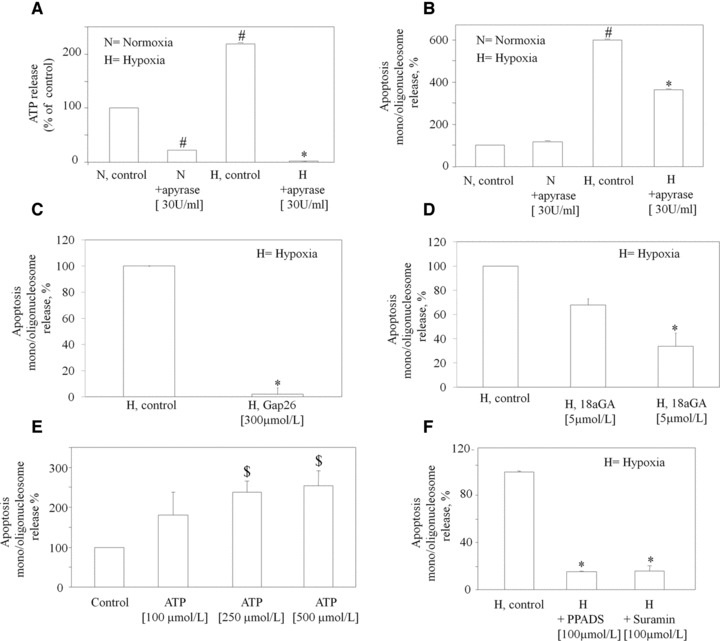
Ischaemia/hypoxia-induced release of ATP is required for the induction of apoptosis. (A) Apyrase was added to cells immediately before normoxia (N) or hypoxia (H). After 30 min., ATP was measured in the medium. Cells were treated with Apyrase (B), Gap 26 (C) and 18aGA (D) 1 hr before hypoxia and apoptosis determined by nucleosome immunoassay. Histogram reports apoptosis under hypoxic conditions after subtracting the apoptosis value detected under normoxic conditions. (E) Cells were treated with either vehicle (Control) or ATP at the indicated concentrations for 24 hrs and apoptosis determined by nucleosome immunoassay. (F) PPADS and suramin were added to cells 1 hr before hypoxia and apoptosis determined by nucleosome immunoassay. Values represent changes with respect to apoptosis found in control (vehicle) hypoxic cells set to 100% and are the mean ± S.E.M. of three independent experiments run in triplicate. *P < 0.05 versus hypoxia control cells; #P < 0.05 versus normoxia control cells; $P < 0.05 versus corresponding control value (vehicle). H is the apoptosis value under hypoxic conditions after subtraction of the apoptosis value detected under normoxic conditions.
P2 receptors antagonists prevent the appearance of HL-1 cardiomyocyte apoptosis induced by ischaemia/hypoxia
We have previously shown that HL-1 cardiomyocytes express all known P2X receptors (except for P2χ2), as well as the P2Y2,4,6,14 subtypes [18]. To verify if the apoptotic effect of the ischaemic/ hypoxic stress was actually due to activation of P2 receptors by ATP or other nucleotides released during hypoxia, HL-1 cardiomyocytes were exposed to ischaemic/hypoxic stress in the presence of non-selective P2 receptor antagonists, such as PPADS and suramin [2, 23]. These treatments almost completely prevented the appearance of apoptosis (−82.7% ± 7.7 after PPADS; −68% ± 12 after suramin; Fig. 2F).
Because PPADS antagonises P2χ1,2,3,5 and P2Y4,6, whereas suramin effectively blocks P2χ1,2,3,5 and P2Y2,6,11 [24, 25], these data suggest that both P2X and P2Y receptors contribute to the cardiomyocytic apoptosis induced by ischaemic/hypoxic stress. However, because P2X receptors can form heteromers characterized by different and unpredictable pharmacological profiles [24], a definite conclusion regarding the specific receptor subtypes involved cannot be drawn from these data.
Inhibition of P2×7 partially prevents the ischaemia/hypoxia-induced apoptosis
Our previous data are in support of a role of the P2X receptors in the induction of apoptosis mediated by ATP in HL-1 cardiomyocytes [18]. Indeed, a 24 hr-exposure of HL-1 cardiomyocytes to the selective P2χ7 agonist BzATP resulted in a dramatic reduction of the number of cells adhering to the culture substrate [18]. In this study, pre-treatment with TNP-ATP, an antagonist for P2χ1–4 receptors [26], failed to significantly prevent the appearance of apoptosis induced by ischaemic/hypoxic stress (Fig. 3A), thus ruling out a role for these P2X receptor subtypes in our model. By contrast, when the specific P2χ7 antagonist KN-62 was tested, a significant reduction of apoptosis in the ischaemic/hypoxic condition was monitored (Fig. 3A). To validate the effect of the P2χ7 antagonist, we silenced the expression of P2χ7 receptor using small interfering (si) RNAs. Treatment of HL-1 cells with a P2χ7 receptor siRNA construct reduced the P2χ7 receptor mRNA levels by 54% ± 3.4 (Fig. 3B). The decrease in P2χ7 receptor expression resulted in a partial, albeit statistically significant, inhibition of apoptosis in response to the ischaemic/hypoxic stress (−34% ± 2.7; Fig. 3C). Taken together, these results point to a possible contribution of the P2χ7 receptor in the apoptosis in HL-1 cardiomyocytes induced by ischaemic/hypoxic stress.
Fig 3.
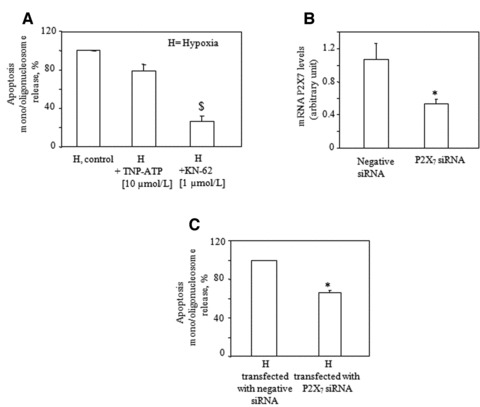
Inhibition of P2×7, but not of P2×1–4 receptors, prevents cardiomyocyte apoptosis induced by hypoxia. (A) TNP-ATP or KN-62 were added to cells 1 hr before hypoxia. After 16 hrs, apoptosis was determined by nucleosome immunoassay. Values refer to apoptosis values in hypoxic cells set to 100% and are the mean ± S.E.M. from four independent experiments run in triplicate; $P < 0.01 versus corresponding control values. (B) mRNA analysis of P2×7 by qRT-PCR in HL-1 cells transfected with ineffective (negative) siRNA or with P2×7 siRNA, and then exposed to normoxia or hypoxia for 16 hrs for detection of apoptosis (C). Apoptosis values were calculated in hypoxic cells and set to 100%. Values are the mean ± S.E.M. from three independent experiments run in triplicate. *P < 0.05 versus negative siRNA-transfected cells. H is the apoptosis value under hypoxic conditions after subtraction of the apoptosis value detected under normoxic conditions.
Inhibition of P2Y2 prevents ischaemia/hypoxia-induced apoptosis
To evaluate the role of P2Y receptor subtypes in the induction of apoptosis by the ischaemic/hypoxic stress, we used selective P2Y2 and P2Y6 antagonists. AR-C118925, the only known selective, heterocyclic antagonist of the P2Y2 receptor [27], significantly prevented the appearance of apoptosis by −50.8% ± 2 (Fig. 4). By contrast, the potent antagonist of P2Y6 nucleotide receptor diisothiocyanate derivative MRS2578 [28] did not affect hypoxia-induced apoptosis in HL-1 cardiomyocytes (Fig. 4). This pharmacological approach could not be adopted for the P2Y4 receptor, because no selective antagonists are currently available for this receptor subtype. Globally, these pharmacological data support a role for P2Y2, but not P2Y6, in ischaemic/hypoxic stress-induced apoptosis. To validate these results, we adopted a second approach based on the use of specific siRNAs against the various P2Y receptors. Analysis of P2Y2, and P2Y6 mRNAs after transfection of HL-1 cardiomyocytes with specific siRNAs showed a reduction of the corresponding receptor mRNAs of 70% ± 6.8 and 89% ± 3.6, for the P2Y2 and P2Y6 receptors, respectively (Fig. 5A and B). Semi-quantitative real-time analysis could not be performed for the P2Y4 receptor in silenced cells, due to the lack of efficacious oligonucleotide primers for this type of analysis [29]. However, standard RT-PCR in cardiomyocytes transfected with P2Y4 siRNA showed a dramatic reduction of P2Y4 amplification product at the expected 447 bp (Fig. 5C) with respect to cells transfected with corresponding negative siRNAs.
Fig 4.
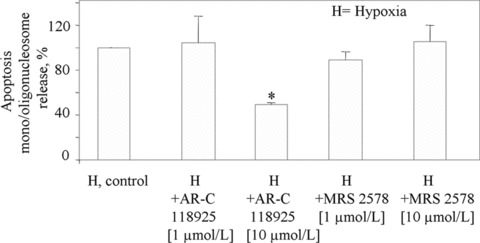
Effect of the selective P2Y2 receptor antagonist AR-C118925 and the selective P2Y6 receptor antagonist MRS2578 on the induction of cardiomyocyte apoptosis. AR-C118925 and MRS2578 were added to cells 1 hr before either normoxia or hypoxia. At the end of the 16 hrs of incubation, apoptosis values were determined in hypoxic cells by ELISA assay and set to 100%. Values are the mean ± S.E.M. from three individual experiments run in triplicate, *P < 0.05 versus corresponding control values. H is the apoptosis value under hypoxic conditions after subtraction of the apoptosis value detected under normoxic conditions.
Fig 5.
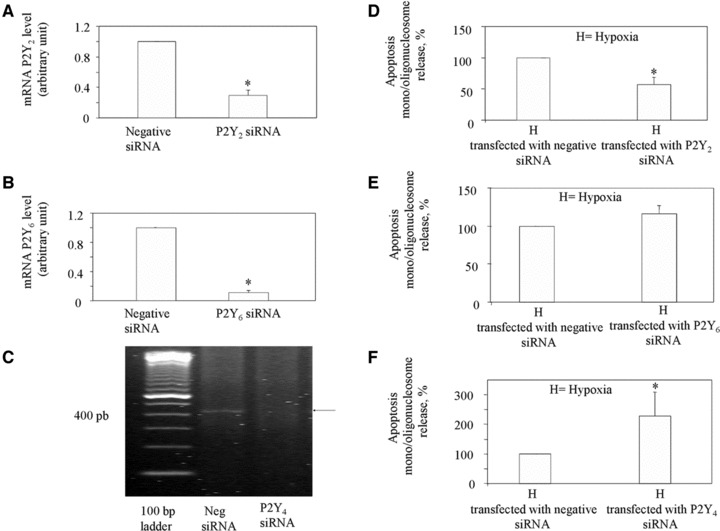
Effect of P2Y2, P2Y4 and P2Y6 gene silencing on the induction of cardiomyocyte apoptosis after ischaemia/hypoxia. mRNA analysis of P2Y2 (A) and P2Y6 (B) by quantitative qRT-PCR in HL-1 cells transfected with either negative siRNA or with P2Y2 and P2Y6 siRNA. (C) Micrograph showing amplification of P2Y4 mRNA by standard RT-PCR in HL-1 cells transfected with either negative siRNA or with specific P2Y4 siRNA. Similar data have been obtained in three independent experiments analysis. (D–F) HL-1 cardiomyocytes transfected with either negative siRNA or with specific P2Y2, P2Y6, P2Y4 siRNA were exposed to normoxia or hypoxia. After 16 hrs, apoptosis was determined in hypoxic cells and set to 100%. Values are the mean ± S.E.M. from three individual experiments run in triplicate *P < 0.05 versus control siRNA-negative treated cells. H is the apoptosis value under hypoxic conditions after subtraction of the apoptosis value detected under normoxic conditions.
In line with the pharmacological data of Figure 5, silencing of P2Y2 resulted in a marked prevention of apoptosis following ischaemic/hypoxic stress (Fig. 5D). No effect was instead detected after silencing of P2Y6 (Fig. 5E). Of interest, gene silencing of P2Y4 resulted in a significant increase of cell apoptosis during ischaemic/hypoxic stress, thus suggesting a potential protective role for this receptor (Fig. 5F).
Both G proteins and protein kinase C are involved in the cardiomyocyte apoptosis induced by hypoxia and ATP
To shed some light on the post-receptor mechanisms involved in ischaemic/hypoxic stress and ATP-induced apoptosis of HL1 cardiomyocytes, we also performed experiments with either the selective Gi protein inhibitor PTX, the pan-G protein inhibitor GDP-β-S or the protein kinase C inhibitor GF. Both G-protein inhibitors partially but significantly inhibited hypoxia-associated apoptosis, whereas the protein kinase C inhibitor showed a non-statistically significant trend to a reduction (Fig. 6A). When apoptosis was induced by exposure to high ATP concentrations, a highly significant reduction of cell death was observed in the presence of either PTX or GF (Fig. 6B). No significant effects were induced by these agents on normoxic cells.
Fig 6.
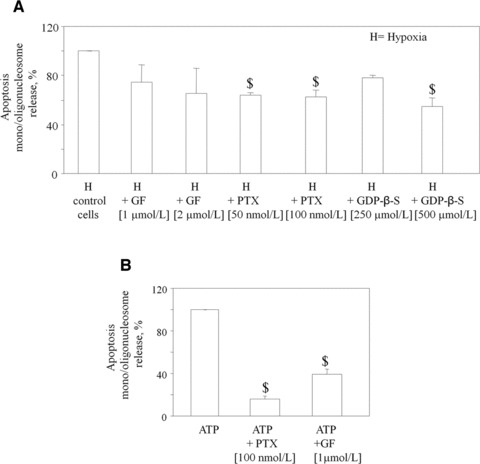
Effect of various inhibitors of signalling cascades on cardiomyocyte apoptosis induced by ischaemic hypoxia or by ATP. (A) GF, PTX and GDP-β-S were added to cells 1 hr before hypoxia. Parallel samples receiving these same inhibitors were maintained under normoxic conditions. At the end of the incubation, apoptosis was determined by nucleosome immunoassay, and set to 100%. Values are the mean ± S.E.M. from three individual experiments, run in triplicate $P < 0.01 versus corresponding values in control cells. H is the apoptosis value under hypoxic conditions after subtraction of the apoptosis value detected under normoxic conditions. (B) HL-1 cells were treated with ATP at the indicated concentrations. PTX and GF were added to cells 1 hr before ATP addition (250 μmol/l). At the end of a 24 hr incubation, apoptosis was determined by nucleosome immunoassay and set to 100%. Values are the mean ± S.E.M. from three individual experiments, run in triplicate $P < 0.01 versus corresponding control values.
Discussion
Here we show that ATP and P2 receptors play a key role in ischaemic/hypoxic stress-induced apoptosis of HL-1 cardiomyocytes, that express various P2X and P2Y receptors (Ref. 18 and this issue). Specifically, we demonstrate that (i) ischaemic/ hypoxic stress induces rapid ATP release from cultured cardiomyocytes, and that (ii) distinct P2 receptors regulate cardiomyocyte death, with both pro- and anti-apoptotic effects. These actions are likely mediated by direct stimulation of P2 receptors by ATP, although we cannot exclude that other concomitantly released nucleotides (e.g. uracil nucleotides, see also below) are also involved. Moreover, by using pharmacological inhibitors and gene silencing of selected P2 receptors, we provide evidence for a specific role of P2χ7 and P2Y2 receptors in ischaemic/hypoxic stress-induced cardiomyocyte apoptosis. Our data also suggest a protective role for P2Y4 receptors; conversely, no effects are apparently induced by co-expressed P2Y6 receptors. Finally, we provide initial hints involving G-proteins and PKC in the post-receptor mechanisms at the basis of hypoxia- and ATP-induced cardiomyocyte apoptosis.
These data are relevant to the pathogenic mechanisms of myocardial damage during hypoxia/ischaemia in patients. Under physiological conditions, in both rat and human heart and in human plasma, extracellular ATP levels are around 20–40 nmol/l [30]. Upon hypoxia/ischaemia, due to both increased exocytosis from cells’ vesicular pool and to cytoplasmic release, these levels are markedly increased to micromolar concentrations [31]. More recently, during MI, human venous plasma levels of UTP have been also demonstrated to increase to about 10% of the ATP concentrations [10]. It has been postulated that, under these conditions, nucleotides release is originally meant at augmenting heart function via a positive inotropic effect mainly exerted through myocardial P2Y2 receptors (although P2Y4 and P2Y6 receptors may also contribute) (ibidem). However, if heart reperfusion is impaired or prevented and nucleotides are not removed, their local concentrations (especially those of ATP) increase to toxic levels and contribute to cardiomyocyte death. This hypothesis is consistent with our data demonstrating that ATP is released from cardiomyocytes very early after hypoxia/ischaemia, and that this release is later associated to apoptosis. It has to be underlined that, in our experiments, after ischaemic/hypoxic stress, the ATP concentration was found to double to 2.28 nM, a value that may seem still low to activate P2Y receptors (which are generally responsive to microMolar nucleotide concentrations, e.g. http://www.iuphardb.org/DATABASE/ObjectDisplayForward?objectId=324&familyId=52).
However, it may well be that the local ATP concentration at membrane receptors is much higher than that measured in the culture medium. It has been indeed previously recognized that, after being released, the ATP concentration in the vicinity of the cell surface is often different from the bulk concentration, being influenced by both delayed diffusion (due to unstirred layer effects), and by immediate degradation by membrane bound ectoATPAses [32].
Regarding the threshold exogenous ATP concentration needed to induce apoptosis, the micromolar values found in our experimental model are in line with previous literature data reporting apoptosis induction between 10 and 50 μM to mM concentrations, depending on the cell type and specific conditions [33-36].
In our model, early release of ATP does not involve cell lysis; moreover, subsequent ATP-mediated apoptosis is prevented by the blockage of connexin hemichannels through which ATP is released. To confirm a causal relationship between ATP release and cardiomyocyte death, both the connexin hemichannels blockers (Gap26 and 18aGa), apyrase and P2 blockers inhibited cardiomyocyte apoptosis (the present data; Ref. 37). In line with these findings, apyrase expression was increased in ischaemic heart samples of patients [5, 38]. Moreover, it has been recently shown that connexin hemichannels inhibitors abolished the release of ATP induced by simulated ischaemia also in rat neonatal cardiomyocytes [39]. As highlighted earlier, our data point to differential roles of various P2 receptors in cardiomyocyte viability during ischaemic/hypoxic stress. The pro-apoptotic role of P2Y2 receptors under hypoxic conditions is in line with previous data demonstrating this as the most expressed P2 receptor undergoing up-regulation in the human failing heart [40]. Moreover, three single nucleotide polymorphisms of the human P2Y2 receptor gene have been associated with MI and proposed as genetic markers for MI in Japanese men [41]. P2Y2 receptors also promote a profibrotic response in isolated myocardial fibroblasts; in mice lacking this receptor, extracellular nucleotides are no longer able to increase the expression of pro-fibrotic plasminogen activator inhibitor type 1 and α-smooth muscle actin [42]. Regarding P2χ7 receptor, despite abundant information on its role in induction of apoptosis in macrophages, monocytes, microglia and other cell types [43], very little is known on its presence and role in MI. Although no direct evidence for P2χ7 presence in rodent heart is available, in dog, this receptor has been originally cloned from heart cDNA [44]. Significant levels of P2χ7 have been found in human myocardial samples from normal subjects and patients with chronic heart failure [11]. Moreover, a loss-of-function P2χ7 polymorphism has been reported in heart failure patients [45], but the relevance of this finding is unknown. More recently, pannexin-1/P2χ7 channels have been shown to mediate the release of cardioprotectants upon ischaemic pre- and post-conditioning [46]. This is the first time that, based on both pharmacological and gene-silencing data, P2χ7 is directly implicated in ischaemia-associated cardiomyocyte death.
Our data also highlight a protective role for P2Y4. In cultured hypoxic cardiomyocytes, activation of some P2Y receptors by UTP reduced LDH release [16], prevented intracellular calcium elevation, maintained ATP intracellular levels [47] and preserved mitochondrial activity [48]. In line with these findings, in vivo experiments showed that UTP administration reduced infarct size and improved myocardial function [49]. However, the exact P2 receptor involved was not univocally identified in these studies. UTP can activate both P2Y2 and P2Y4 receptors [2]; this data suggest, for the first time, that it is the latter that mediates UTP-induced protection. Regarding the P2Y6 receptor that has been proposed to mediate positive inotropic effects [10], recent papers have involved this receptor in pressure overload-induced cardiac fibrosis [5, 50]. However, our data suggest that, despite playing a role in heart remodelling during chronic heart failure, P2Y6 is not directly involved in regulation of cardiomyocyte viability during acute hypoxic stress. Although our study focused on the role of P2 receptors it is known that, upon addition to cells, ATP is quickly degraded to adenosine, which can then activate the P1 receptors (subdivided into the A1, A2A, A2B and A3 subtypes [51]). However in our experimental model, ATP-induced cytotoxicity was not counteracted by either 8-cyclopentyl-1,3-dipropylxanthine (CPX, which primarily antagonizes the A1 and A2B receptors [52-54]), or by the selective A2A antagonist SCH58261, or by the selective A3 antagonist MRS1220 [51] (data not shown), thus ruling out a role for these receptors in the detected effects. Finally, this data shed some light on the post-receptor mechanisms involved in P2 receptor regulation of cardiomyocyte viability. Inhibition of both G-proteins and PKC resulted in apoptosis reduction, in line with our proposal that the G-protein coupled P2Y2 receptor participates to cardiomyocyte death. Globally, our results suggest that the pharmacological modulation of P2Y2 and P2χ7 receptors by mean of selective antagonists may represent a new therapeutic strategy in ischaemic heart disease.
Acknowledgments
Authors warmly thank Professor William Claycomb, LSU Health Sciences Center, New Orleans, LA, USA, for the kind gift of HL-1 cells and Prof. Dr. Christa E. Müller, PharmaCenter Bonn, Pharmaceutical Institute, Pharmaceutical Chemistry I, An der Immenburg 4, D-53121 Bonn, Germany for the kind gift of AR-C118925.
This work was supported by a grant from Italian Ministry of Health, Rome, Italy (Ricerca Corrente 2008 BIO 25) and by Ministry of Health Progetto di Ricerca Finalizzata 2006 ‘Identificazione di markers patogenetico/prognostici nello scompenso cardiaco (SC)’ to MPA.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Burnstock G. The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology. 1997;36:1127–39. doi: 10.1016/s0028-3908(97)00125-1. [DOI] [PubMed] [Google Scholar]
- 2.Abbracchio MP, Burnstock G, Boeynaems JM, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–92. [PubMed] [Google Scholar]
- 4.Brunschweiger A, Mller CE. P2 receptors activated by uracil nucleotides-an update. Curr Med Chem. 2006;13:289–312. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 5.Erlinge D, Burnstock G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008;4:1–20. doi: 10.1007/s11302-007-9078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pelleg A, Vassort G P2 receptors in the cardiovascular system. In: Purinergic and pyrimidergic signalling II. Abbracchio MP, Williams M, editors. Berlin/Heidelberg: Springer-Verlag; 2001. pp. 73–100. [Google Scholar]
- 7.Drury AN, Szent-Györgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol. 1929;68:213–37. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bogdanov Y, Rubino A, Burnstock G. Characterisation of subtypes of the P2X and P2Y families of ATP receptors in the foetal human heart. Life Sci. 1998;62:697–703. doi: 10.1016/s0024-3205(97)01168-5. [DOI] [PubMed] [Google Scholar]
- 9.Vassort G. Adenosine 5′-triphosphate: a P2-purinergic agonist in the myocardium. Physiol Rev. 2001;81:767–806. doi: 10.1152/physrev.2001.81.2.767. [DOI] [PubMed] [Google Scholar]
- 10.Wihlborg AK, Balogh J, Wang L, et al. Positive inotropic effects by uridine triphosphate (UTP) and uridine diphosphate (UDP) via P2Y2 and P2Y6 receptors on cardiomyocytes and release of UTP in man during myocardial infarction. Circ Res. 2006;98:970–6. doi: 10.1161/01.RES.0000217402.73402.cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banfi C, Ferrario S, De Vincenti O, et al. P2 receptors in human heart: upregulation of P2 χ 6 in patients undergoing heart transplantation, interaction with TNFalpha and potential role in myocardial cell death. J Mol Cell Cardiol. 2005;39:929–39. doi: 10.1016/j.yjmcc.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- 13.Leybaert L, Braet K, Vandamme W, et al. Connexin channels, connexin mimetic peptides and ATP release. Cell Commun Adhes. 2003;10:251–7. doi: 10.1080/cac.10.4-6.251.257. [DOI] [PubMed] [Google Scholar]
- 14.Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;572:65–8. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Dutta AK, Sabirov RZ, Uramoto H, et al. Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J Physiol. 2004;559:799–812. doi: 10.1113/jphysiol.2004.069245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yitzhaki S, Shneyvays V, Jacobson KA, et al. Involvement of uracil nucleotides in protection of cardiomyocytes from hypoxic stress. Biochem Pharmacol. 2005;69:1215–23. doi: 10.1016/j.bcp.2005.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–9. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 18.Mazzola A, Amoruso E, Beltrami E, et al. Opposite effects of uracil and adenine nucleotides on the survival of murine cardiomyocytes. J Cell Mol Med. 2008;12:522–36. doi: 10.1111/j.1582-4934.2007.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claycomb WC, Lanson NA, Jr, Stallworth BS, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–84. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banfi C, Brioschi M, Barcella S, et al. Tissue factor induction by protease-activated receptor 1 requires intact caveolin-enriched membrane microdomains in human endothelial cells. J Thromb Haemost. 2007;5:2437–44. doi: 10.1111/j.1538-7836.2007.02759.x. [DOI] [PubMed] [Google Scholar]
- 21.Ceruti S, Franceschi C, Barbieri D, et al. Apoptosis induced by 2-chloro-adenosine and 2-chloro-2’-deoxy-adenosine in a human astrocytoma cell line: differential mechanisms and possible clinical relevance. J Neurosci Res. 2000;60:388–400. doi: 10.1002/(SICI)1097-4547(20000501)60:3<388::AID-JNR14>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 22.Ceruti S, Beltrami E, Matarrese P, et al. A key role for caspase-2 and caspase-3 in the apoptosis induced by 2-chloro-2′-deoxy-adenosine (cladribine) and 2-chloro-adenosine in human astrocytoma cells. Mol Pharmacol. 2003;63:1437–47. doi: 10.1124/mol.63.6.1437. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson KA, Costanzi S, Joshi BV, et al. Agonists and antagonists for P2 receptors. Novartis Found Symp. 2006;276:58–68. doi: 10.1002/9780470032244.ch6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–67. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 25.Abbracchio MP, Verderio C. Pathophysiological roles of P2 receptors in glial cells. Novartis Found Symp. 2006;276:91–103. [PubMed] [Google Scholar]
- 26.Jarvis MF, Khakh BS. ATP-gated P2X cation-channels. Neuropharmacology. 2009;56:208–15. doi: 10.1016/j.neuropharm.2008.06.067. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson KA, Ivanov AA, de Castro S, et al. Development of selective agonists and antagonists of P2Y receptors. Purinergic Signal. 2009;5:75–89. doi: 10.1007/s11302-008-9106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mamedova LK, Joshi BV, Gao ZG, et al. Diisothiocyanate derivatives as potent, insurmountable antagonists of P2Y6 nucleotide receptors. Biochem Pharmacol. 2004;67:1763–70. doi: 10.1016/j.bcp.2004.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Govindaraju V, Martin JG, Maghni K, et al. The effects of extracellular purines and pyrimidines on human airway smooth muscle cells. J Pharmacol Exp Ther. 2005;315:941–8. doi: 10.1124/jpet.105.089698. [DOI] [PubMed] [Google Scholar]
- 30.Kuzmin AI, Lakomkin VL, Kapelko VI, et al. Interstitial ATP level and degradation in control and postmyocardial infarcted rats. Am J Physiol. 1998;275:C766–71. doi: 10.1152/ajpcell.1998.275.3.C766. [DOI] [PubMed] [Google Scholar]
- 31.Dutta AK, Sabirov RZ, Uramoto H, et al. Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J Physiol. 2004;559:799–812. doi: 10.1113/jphysiol.2004.069245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi S, Hazama A, Dutta AK, et al. Detecting ATP release by a biosensor method. Sci STKE. 2004;258:pl14. doi: 10.1126/stke.2582004pl14. [DOI] [PubMed] [Google Scholar]
- 33.Schrier SM, Florea BI, Mulder GJ, et al. Apoptosis induced by extracellular ATP in the mouse neuroblastoma cell line N1E-115: studies on involvement of P2 receptors and adenosine. Biochem Pharmacol. 2002;63:1119–26. doi: 10.1016/s0006-2952(01)00939-x. [DOI] [PubMed] [Google Scholar]
- 34.Delarasse C, Gonnord P, Galante M, et al. Neural progenitor cell death is induced by extracellular ATP via ligation of P2χ7 receptor. J Neurochem. 2009;109:846–57. doi: 10.1111/j.1471-4159.2009.06008.x. [DOI] [PubMed] [Google Scholar]
- 35.Placido R, Auricchio G, Falzoni S, et al. P2X(7) purinergic receptors and extracellular ATP mediate apoptosis of human monocytes/macrophages infected with Mycobacterium tuberculosis reducing the intracellular bacterial viability. Cell Immunol. 2006;244:10–8. doi: 10.1016/j.cellimm.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Lepine S, Le Stunff H, Lakatos B, et al. ATP-induced apoptosis of thymocytes is mediated by activation of P2χ7 receptor and involves de novo ceramide synthesis and mitochondria. Biochim Biophys Acta. 2006;1761:73–82. doi: 10.1016/j.bbalip.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Sugimoto S, Lin X, Lai J, et al. Apyrase treatment prevents ischaemia-reperfusion injury in rat lung isografts. J Thorac Cardiovasc Surg. 2009;138:752–9. doi: 10.1016/j.jtcvs.2009.04.049. [DOI] [PubMed] [Google Scholar]
- 38.Kittel A, Kiss AL, Müllner N, et al. Expression of NTPDase1 and caveolins in human cardiovascular disease. Histochem Cell Biol. 2005;124:51–9. doi: 10.1007/s00418-005-0018-8. [DOI] [PubMed] [Google Scholar]
- 39.Clarke TC, Williams OJ, Martin PE, et al. ATP release by cardiac myocytes in a simulated ischaemia model: inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur J Pharmacol. 2009;605:9–14. doi: 10.1016/j.ejphar.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 40.Hou M, Malmsjö M, Möller S, et al. Increase in cardiac P2χ1- and P2Y2-receptor mRNA levels in congestive heart failure. Life Sci. 1999;65:1195–206. doi: 10.1016/s0024-3205(99)00353-7. [DOI] [PubMed] [Google Scholar]
- 41.Wang ZX, Nakayama T, Sato N, et al. Association of the purinergic receptor P2Y, G-protein coupled, 2 (P2RY2) gene with myocardial infarction in Japanese men. Circ J. 2009;73:2322–9. doi: 10.1253/circj.cj-08-1198. [DOI] [PubMed] [Google Scholar]
- 42.Braun OO, Lu D, Aroonsakool N, et al. Uridine triphosphate (UTP) induces profibrotic responses in cardiac fibroblasts by activation of P2Y2 receptors. J Mol Cell Cardiol. 2010;49:362–9. doi: 10.1016/j.yjmcc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baraldi PG, Di Virgilio F, Romagnoli R. Agonists and antagonists acting at P2χ7 receptor. Curr Top Med Chem. 2004;4:1707–17. doi: 10.2174/1568026043387223. [DOI] [PubMed] [Google Scholar]
- 44.Roman S, Cusdin FS, Fonfria E, et al. Cloning and pharmacological characterization of the dog P2χ7 receptor. Br J Pharmacol. 2009;158:1513–26. doi: 10.1111/j.1476-5381.2009.00425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eslick GD, Thampan BV, Nalos M, et al. Circulating interleukin-18 concentrations and a loss-of-function P2χ7 polymorphism in heart failure. Int J Cardiol. 2009;137:81–3. doi: 10.1016/j.ijcard.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 46.Vessey DA, Li L, Kelley M. Pannexin-I/P2X 7 purinergic receptor channels mediate the release of cardioprotectants induced by ischaemic pre- and postconditioning. J Cardiovasc Pharmacol Ther. 2010;15:190–5. doi: 10.1177/1074248409360356. [DOI] [PubMed] [Google Scholar]
- 47.Yitzhaki S, Shainberg A, Cheporko Y, et al. Uridine-5’-triphosphate (UTP) reduces infarct size and improves rat heart function after myocardial infarct. Biochem Pharmacol. 2006;72:949–55. doi: 10.1016/j.bcp.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yitzhaki S, Hochhauser E, Porat E, et al. Uridine-5’-triphosphate (UTP) maintains cardiac mitochondrial function following chemical and hypoxic stress. J Mol Cell Cardiol. 2007;43:653–62. doi: 10.1016/j.yjmcc.2007.07.060. [DOI] [PubMed] [Google Scholar]
- 49.Shainberg A, Yitzhaki S, Golan O, et al. Involvement of UTP in protection of cardiomyocytes from hypoxic stress. Can J Physiol Pharmacol. 2009;87:287–99. doi: 10.1139/Y09-010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishida M, Sato Y, Uemura A, et al. P2Y6 receptor-Galpha12/13 signalling in cardiomyocytes triggers pressure overload-induced cardiac fibrosis. EMBO J. 2008;27:3104–15. doi: 10.1038/emboj.2008.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–64. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Golembiowska K, Dziubina A. Striatal adenosine A(2A) receptor blockade increases extracellular dopamine release following l-DOPA administration in intact and dopamine-denervated rats. Neuropharmacology. 2004;47:414–26. doi: 10.1016/j.neuropharm.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 53.Goldenberg I, Shainberg A, Jacobson KA, et al. Adenosine protects against angiotensin II-induced apoptosis in rat cardiocyte cultures. Mol Cell Biochem. 2003;252:133–9. doi: 10.1023/a:1025551229566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ji X, Kim YC, Ahern DG, et al. [3H]MRS 1754, a selective antagonist radioligand for A(2B) adenosine receptors. Biochem Pharmacol. 2001;61:657–63. doi: 10.1016/s0006-2952(01)00531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]