

Abstract
Loss-of-function mutations in human adenomatous polyposis coli (APC) lead to multiple colonic adenomatous polyps eventually resulting in colonic carcinoma. Similarly, heterozygous mice carrying defective APC (apcMin/+) suffer from intestinal tumours. The animals further suffer from anaemia, which in theory could result from accelerated eryptosis, a suicidal erythrocyte death triggered by enhanced cytosolic Ca2+ activity and characterized by cell membrane scrambling and cell shrinkage. To explore, whether APC-deficiency enhances eryptosis, we estimated cell membrane scrambling from annexin V binding, cell size from forward scatter and cytosolic ATP utilizing luciferin–luciferase in isolated erythrocytes from apcMin/+ mice and wild-type mice (apc+/+). Clearance of circulating erythrocytes was estimated by carboxyfluorescein-diacetate-succinimidyl-ester labelling. As a result, apcMin/+ mice were anaemic despite reticulocytosis. Cytosolic ATP was significantly lower and annexin V binding significantly higher in apcMin/+ erythrocytes than in apc+/+ erythrocytes. Glucose depletion enhanced annexin V binding, an effect significantly more pronounced in apcMin/+ erythrocytes than in apc+/+ erythrocytes. Extracellular Ca2+ removal or inhibition of Ca2+ entry with amiloride (1 mM) blunted the increase but did not abrogate the genotype differences of annexin V binding following glucose depletion. Stimulation of Ca2+-entry by treatment with Ca2+-ionophore ionomycin (10 μM) increased annexin V binding, an effect again significantly more pronounced in apcMin/+ erythrocytes than in apc+/+ erythrocytes. Following retrieval and injection into the circulation of the same mice, apcMin/+ erythrocytes were more rapidly cleared from circulating blood than apc+/+ erythrocytes. Most labelled erythrocytes were trapped in the spleen, which was significantly enlarged in apcMin/+ mice. The observations point to accelerated eryptosis and subsequent clearance of apcMin/+ erythrocytes, which contributes to or even accounts for the enhanced erythrocyte turnover, anaemia and splenomegaly in those mice.
Keywords: phosphatidylserine, cell membrane scrambling, calcium, cell volume, eryptosis, APC
Introduction
The APC protein binds the oncogenic protein β-catenin and favours its degradation [1-4]. Lack of APC is followed by accumulation of β-catenin, which enters the nucleus and stimulates the expression of several genes involved in the regulation of cell proliferation [5, 6]. Loss-of-function mutations affecting APC lead to the development of multiple colonic adenomatous polyps, which eventually results in colonic carcinoma [7, 8]. Mice carrying a mutation in the APC gene (apcMin/+), which leads to truncation of the APC protein at amino acid 850, develop multiple intestinal tumours [9]. Beyond that the mice suffer from enhanced gastric acid secretion [10], hyperaldosteronism and increased blood pressure [11]. Moreover, the animals were shown to suffer from anaemia, a disorder considered to be secondary to blood loss [12].
A blood count of those mice revealed an anaemia despite excessive reticulocyte numbers. The observation points to enhanced erythrocyte turnover, which may, at least in theory, result from enhanced eryptosis, a suicidal erythrocyte death characterized by cell shrinkage and cell membrane scrambling [13].
Eryptosis is triggered by activation of Ca2+-permeable cation channels [14-21]. Cytosolic Ca2+ activates Ca2+-sensitive K+ channels [22, 23] leading to exit of KCl with osmotically obliged water and thus to cell shrinkage [24]. Enhanced cytosolic Ca2+ further stimulates scrambling of the erythrocyte membrane with exposure of phosphatidylserine at the cell surface [30-32]. Accordingly, accelerated eryptosis may lead to anaemia [13]. On the other hand, eryptosis is a physiological mechanism preventing haemolysis of defective erythrocytes [13].
This study was performed to elucidate whether the anaemia and reticulocytosis in apcMin/+ mice is secondary to increased eryptosis. Thus, eryptosis was determined in erythrocytes from apcMin/+ mice and from wild-type mice (apc+/+).
Materials and methods
Mice
Mice with mutated APC (apcMin/+) and wild-type mice (apc+/+) were generated by breeding of apcMin/+ mice initially obtained from the Jackson Laboratory. The mice (four to eight per experiment, sex-matched, age as indicated) were fed a control diet (C1314; Altromin, Heidenau, Germany) and had access to drinking water ad libitum. Unless otherwise stated, 9- to 26-week-old mice were used.
Blood count and reticulocyte estimation
Blood was withdrawn into heparinized capillaries by puncturing the retrobulbar plexus. To this end, the mice were anaesthetized with diethylether (Roth, Karlsruhe, Germany). Anaesthesia was verified by testing the hind limb reflex. Then, 50 μl of blood was taken by puncturing the retrobulbar plexus. For all experiments except for the blood count heparin blood was obtained. For the blood count, EDTA blood was analysed using an electronic haematology counter (scil VET abc, Weinheim, Germany). Relative reticulocyte numbers were determined using the Retic-COUNT reagent (BD, Heidelberg, Germany) according to the manufacturer’s instructions.
Incubation, chemicals and solutions
The erythrocytes were isolated by washing two times with Ringer solution containing (in mM) 125 NaCl, 5 KCl, 1 MgSO4, 32 HEPES/NaOH (pH 7.4), 5 glucose, 1 CaCl2. Erythrocytes were incubated in vitro at a haematocrit of 0.4% in Ringer solution at 37°C for 8 hrs unless otherwise stated. Where indicated, amiloride or ionomycin (both Sigma-Aldrich, Schnelldorf, Germany) were added, glucose removed or 1 mM CaCl2 substituted with 4 mM ethylene glycol tetraacetic acid (EGTA; Sigma-Aldrich).
FACS analysis of annexin V-binding and forward scatter
For FACS analysis of annexin V-binding and forward scatter 50 μl cell suspensions were washed in Ringer solution containing 5 mM CaCl2 and then stained with Annexin-V-FITC (1:250 dilution; Immunotools, Friesoythe, Germany) in this solution for 20 min. at 37°C under protection from light. In the stained erythrocyte cell suspensions, forward scatter of the cells was determined, and annexin V fluorescence intensity was measured in FL-1 with an excitation wavelength of 488 nm and an emission wavelength of 530 nm on a FACS calibur (BD, Heidelberg, Germany).
Measurement of the in vivo clearance of fluorescence-labelled erythrocytes
The in vivo clearance of fluorescence-labelled erythrocytes was determined as described previously [33]. Briefly, erythrocytes (obtained from 200 μl blood) were fluorescence labelled by staining the cells with 5 μM carboxyfluorescein-diacetate-succinimidyl-ester (CFSE; Molecular Probes, Leiden, The Netherlands) in PBS and incubated for 30 min. at 37°C. After washing twice in PBS containing 1% FCS, the pellet was resuspended in Ringer solution (37°C), and 100 μl of the CFSE-labelled erythrocytes were injected into the tail vein of the recipient mouse. As indicated, blood was retrieved from the tail veins of the mice, and CFSE-dependent fluorescence intensity of the erythrocytes was measured in FL-1 as described earlier. The percentage of CFSE-positive erythrocytes was calculated in percentage of the total labelled fraction determined 10 min. after injection.
Confocal microscopy and immunofluorescence
For the detection of annexin V-binding and CFSE-dependent fluorescence of erythrocytes in splenic tissue, the mice were deeply anaesthetized with diethyleter. Then, they were killed by cervical dislocation. After laparotomy, the spleens of apcMin/+ and of apc+/+ mice were removed, weighed and mechanically homogenized in 1 ml cold PBS. The suspension was then centrifuged at 500 χ g for 10 min. at 4°C. The cell pellet was resuspended in 200 μl cold PBS. Five microlitres of Annexin V-APC (BD) were added, and incubation was carried out for 20 min. at 37°C protected from light. The suspension was then transferred onto a glass slide and mounted with Prolong® Gold antifade reagent (Invitrogen). Images were taken on a Zeiss LSM 5 EXCITER Confocal Laser Scanning Microscope (Carl Zeiss MicroImaging GmbH, Germany) with a water immersion Plan-Neofluar 63/1.3 NA DIC.
Estimation of intracellular ATP concentration
For determination of erythrocyte ATP, 80 μl of erythrocyte pellets were incubated for 12 hrs at 37°C in Ringer solution with or without glucose (final haematocrit 5%). All manipulations were then performed at 4°C to avoid ATP degradation. Cells were lysed in distilled water, and proteins were precipitated by addition of HClO4 (5%). After centrifugation, an aliquot of the supernatant (400 μl) was adjusted to pH 7.7 by addition of saturated KHCO3 solution. After dilution of the supernatant, the ATP concentration of the aliquots was determined utilizing the luciferin–luciferase assay kit (Roche Diagnostics, Mannheim, Germany) on a luminometer (Berthold Biolumat LB9500, Bad Wildbad, Germany) according to the manufacturer’s protocol. ATP concentrations refer to the cystol of erythrocytes.
Statistics
Data are expressed as arithmetic means ± S.E.M., and statistical analysis was made by non-parametric Mann–Whitney test as indicated in the figure legends using GraphPad InStat Version 3.06 (San Diego, CA, USA); n denotes the number of different erythrocyte specimens studied.
Results
Blood count and percentage of reticulocytes
A blood count revealed moderate anaemia of the apcMin/+ mice. Erythrocyte count, haemoglobin concentration and haematocrit were significantly smaller in apcMin/+ than in apc+/+ mice (Table 1). The mean corpuscular volume was, however, significantly increased. According to FACS analysis, the reticulocyte number was significantly higher in apcMin/+ than in apc+/+ mice at different ages (4, 6, 8 and 12 weeks), pointing to enhanced erythrocyte formation (Table 1).
Table 1.
Anaemia in APC-deficient mice
| Parameter | apc+/+ | apcMin/+ | Normal range |
|---|---|---|---|
| RBC (106/μl) | 10.7 ± 0.6 | 5.5 ± 0.1* | 5.0–9.5 |
| HGB (g/dl) | 16.4 ± 0.7 | 10.0 ± 0.4* | 10.9–16.3 |
| HCT (%) | 55.2 ± 2.0 | 34.1 ± 1.2* | 38.5–45.1 |
| MCV (fl) | 51.8 ± 1.8 | 62.3 ± 1.7* | 48.0–56.0 |
| MCHC (g/dl) | 29.8 ± 0.5 | 29.3 ± 0.5 | 25.9–35.1 |
| MCH (pg) | 15.5 ± 0.3 | 18.4 ± 0.7* | 11.9–19.0 |
| Reticulocytes (%) | |||
| 4 weeks (age) | 3.7 ± 0.3 | 7.0 ± 0.7* | 1.0–6.0 |
| 6 weeks (age) | 5.3 ± 0.4 | 12.6 ± 0.7* | |
| 8 weeks (age) | 5.3 ± 1.3 | 20.7 ± 4.6* | |
| 12 weeks (age) | 4.1 ± 0.4 | 17.8 ± 6.0* |
Arithmetic mean ± S.E.M. (n = 4) of erythrocyte count (RBC), haemoglobin concentration (HGB), haematocrit (HCT), mean corpuscular volume (MCV), mean corpuscular haemoglobin (MCH), mean corpuscular haemoglobin concentration (MCHC) of 15- to 16-week-old APC-deficient mice (apcMin/+) and wild-type mice (apc+/+). Reticulocyte number (n = 4) of APC-deficient mice (apcMin/+) and wild-type mice (apc+/+) as a function of age. The data are compared to the normal range in mice [67, 68]. * indicates significant differences between genotypes (Mann–Whitney test; P = 0.0286).
In vivo clearance of CFSE-labelled erythrocytes
The enhanced reticulocyte number accompanied by moderate anaemia points to enhanced erythrocyte turnover. To determine the life span of circulating erythrocytes blood was drawn from apcMin/+ and apc+/+ (control) mice, erythrocytes were labelled with CFSE and the labelled apcMin/+ erythrocytes and apc+/+ erythrocytes were injected into the same mice. As illustrated in Figure 1, labelled apcMin/+ erythrocytes disappeared from circulating blood of apcMin/+ mice significantly more rapidly than labelled apc+/+ erythrocytes. Thus, the life span of apcMin/+ erythrocytes in apcMin/+ mice was significantly shorter than the life span of apc+/+ erythrocytes in apc+/+ mice. The percentage of apcMin/+ erythrocytes remaining in circulating blood of apc+/+ mice within 24 hrs tended to be higher (62.1 ± 7.4, n = 3) than the respective percentage of apcMin/+ erythrocytes in apcMin/+ mice (50.3 ± 4.9, n = 4), a difference, however, not reaching statistical significance. The percentage of apc+/+ erythrocytes remaining within 24 hrs in circulating blood of apcMin/+ mice tended to be lower (84.2 ± 3.2, n = 3) as the respective percentage of apc+/+ erythrocytes in apc+/+ mice (89.4 ± 1.2, n = 4), a difference, however, again not statistically significant.
Fig 1.
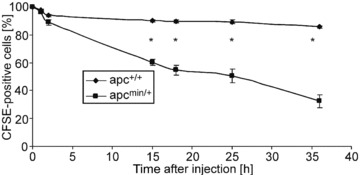
Accelerated clearance of erythrocytes in APC-deficient mice. Time-dependent decay of CFSE-labelled circulating erythrocytes drawn from 20-week-old APC-deficient mice (black squares) and wild-type mice (black diamonds) and injected into the same mice. The percentage of CFSE-labelled circulating cells is plotted against time after injection. Values are normalized arithmetic mean ± S.E.M. (n =4) of the percentages of CFSE-labelled erythrocytes. * indicates significant difference between genotypes (Mann–Whitney test; P < 0.05).
Analysis of the spleen and splenic erythrocytes
As evident from Figure 2A and B, the labelled erythrocytes were mainly trapped in the spleen, which was significantly larger in apcMin/+ mice than in apc+/+ mice. A detailed non-quantitative analysis revealed that the number of fluorescent annexin V binding and thus phosphatidylserine-exposing erythrocytes was higherin the spleens from apcMin/+ mice than from apc+/+ mice (control; Fig. 2C). CFSE accumulates in the cytosol, whereas annexin V binds to phosphatidylserine in the cell membrane.
Fig 2.
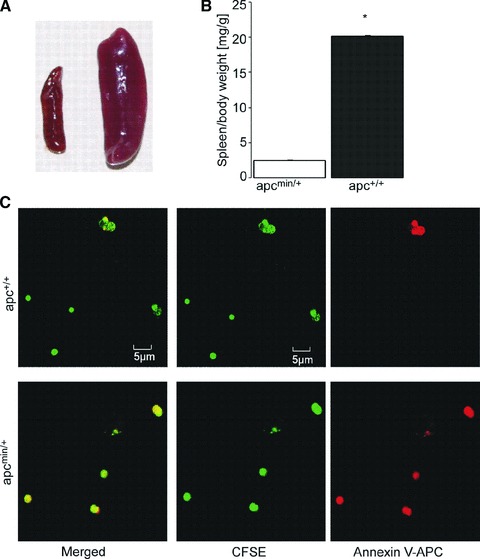
Splenomegaly associated with increased number of annexin V binding erythrocytes in APC-deficient mice. (A) Photograph of spleens from 20-week-old APC-deficient mice (right) and wild-type mice (left). (B) Arithmetic mean ± S.E.M. (n = 4) of the spleen/body weight ratios of 20-week-old APC-deficient mice (apcMin/+, black bar) and wild-type mice (apc+/+, white bar), * indicates significant difference between genotypes (Mann-Whitney test; P < 0.05;). (C) Confocal microscopy of CFSE dependent (middle panels), annexin V-APC (right panels) dependent and merged fluorescence (left panels) of erythrocytes from the spleens of APC-deficient (apcMin/+, lower panels) mice and wild-type mice (apc+/+, upper).
Phosphatidylserine exposure of apc+/+ and apcMin/+ erythrocytes
In view of the accelerated clearance of circulating erythrocytes in the spleen of apcMin/+ mice and their enhanced phosphatidylserine exposure at the cell surface, additional experiments were performed to determine annexin V binding of apcMin/+ erythrocytes and apc+/+ erythrocytes in FACS analysis. The experiments were performed in the presence and absence of glucose, as energy depletion is known to foster eryptosis [34]. As shown in Figure 3A and B, annexin V binding reflecting phosphatidylserine exposure at the erythrocyte surface was significantly higher in apcMin/+ erythrocytes than in apc+/+ erythrocytes following energy depletion.
Fig 3.
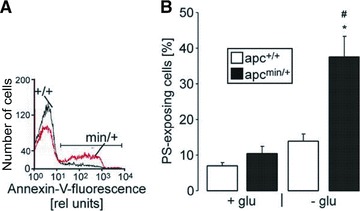
Enhanced phosphatidylserine abundance at the surface of erythrocytes from APC-deficient mice. (A) Histogram of annexin V binding reflecting phosphatidylserine exposure in a representative experiment of erythrocytes from APC-deficient mice (apcMin/+) and their wild-type littermates (apc+/+) exposed for 8 hrs to glucose-depleted Ringer. (B) Arithmetic mean ± S.E.M. (n = 4) of the percentage of annexin V-binding erythrocytes from 8-week-old APC-deficient mice (apcMin/+, black bars) and wild-type mice (apc+/+, white bars) exposed for 8 hrs to glucose-containing (left bars) or glucose-depleted (right bars) Ringer. Annexin V bound to the cell membrane whereas CFSE accumulated in the cytosol. * indicates significant (P < 0.05) difference from glucose-containing Ringer; # indicates significant difference (P < 0.05) between genotypes (Mann–Whitney test).
Role of Ca2+ for cell membrane scrambling of apc+/+ and apcMin/+ erythrocytes
As cytosolic Ca2+ is important for triggering of eryptosis, the Ca2+ sensitivity of annexin V binding was tested by exposing apcMin/+ and apc+/+ erythrocytes to the Ca2+ ionophore ionomycin (10 μM). As illustrated in Figure 4A, the ionomycin effect on annexin V binding was significantly stronger in apcMin/+ erythrocytes than in apc+/+ erythrocytes pointing to higher Ca2+ sensitivity of apcMin/+ erythrocytes. To define the role of Ca2+ entry for the triggering of energy depletion-induced eryptosis, apcMin/+ and apc+/+ erythrocytes were incubated with glucose-free Ringer in the presence and absence of amiloride (1 mM), an inhibitor of the non-specific cation conductance in erythrocytes. As a result, amiloride significantly attenuated the increase in annexin V binding following energy depletion in both apcMin/+ erythrocytes and apc+/+ erythrocytes (Fig. 4B). However, amiloride did not abolish the differences between apcMin/+ erythrocytes and apc+/+ erythrocytes of the annexin V binding following energy depletion. Similar observations were made in the absence of extracellular Ca2+. As shown in Figure 4C, removal of extracellular Ca2+ tended to attenuate the increase in annexin V binding following energy depletion in both apcMin/+ erythrocytes and apc+/+ erythrocytes. However, similar to amiloride administration, Ca2+ removal did not abolish the differences between apcMin/+ erythrocytes and apc+/+ erythrocytes of the annexin V binding following energy depletion. Collectively, these observations point to enhanced susceptibility of apcMin/+ erythrocytes to the cell membrane scrambling effect of energy depletion and enhanced cytosolic Ca2+ activity.
Fig 4.
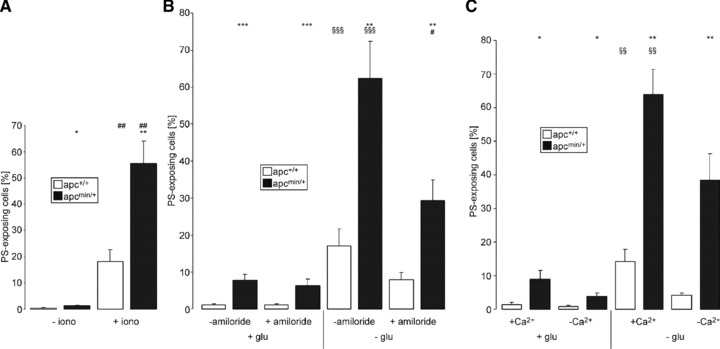
Ca2+ sensitivity of erythrocytes from APC-deficient and wild-type mice. (A) Arithmetic mean ± S.E.M. (n = 5) of the percentage of annexin V-binding erythrocytes from APC-deficient mice (apcMin/+, black bars) and wild-type mice (apc+/+, white bars) exposed for 30 min. to Ringer solution without (left bars) or with (right bars) 10 μM ionomycin. *, ** indicate (P < 0.05, P < 0.01) significant difference between genotypes; ## indicates (P < 0.01) significant difference from absence of ionomycin (Mann–Whitney test). (B) Arithmetic mean ± S.E.M. (n = 7 experiments with samples from four different mice) of the percentage of annexin V-binding erythrocytes from APC-deficient mice (apcMin/+, black bars) and wild-type mice (apc+/+, white bars) exposed for 10 hrs to Ringer solution with (left bars) or without (right bars) glucose in the absence and presence of 1 mM amiloride. **, *** indicate significant (P < 0.01, P < 0.001) difference between genotypes, # significant (P < 0.05) difference from absence of amiloride, §§§ indicates significant (P < 0.001) difference from the presence of glucose (Mann–Whitney test). (C) Arithmetic mean ± S.E.M. (n = 5) of the percentage of annexin V-binding erythrocytes from APC-deficient mice (apcMin/+, black bars) and wild-type mice (apc+/+, white bars) exposed for 10 hrs to Ringer solution with (left bars) or without (right bars) glucose in the absence (−Ca2+) and presence (+Ca2+) of 1 mM extracellular Ca2+. *, ** indicate significant (P < 0.05, P < 0.001) difference between genotypes; §§ indicates significant (P < 0.01) difference from the presence of glucose (Mann–Whitney test).
Cytosolic ATP concentration in apc+/+ and apcMin/+ erythrocytes
Glucose deprivation is likely to affect the intracellular ATP content. Hence, the intracellular ATP concentration of erythrocytes incubated in the presence or absence of glucose for 12 hrs was determined. As shown in Figure 5, glucose depletion indeed decreased the intracellular ATP concentration of erythrocytes from both genotypes. In the presence of glucose, cytosolic ATP content was significantly lower in apcMin/+ erythrocytes than in apc+/+ erythrocytes. Following glucose depletion cytosolic ATP content tended to be lower in apcMin/+ erythrocytes than in apc+/+ erythrocytes, a difference, however, not reaching statistical significance (Fig. 5).
Fig 5.
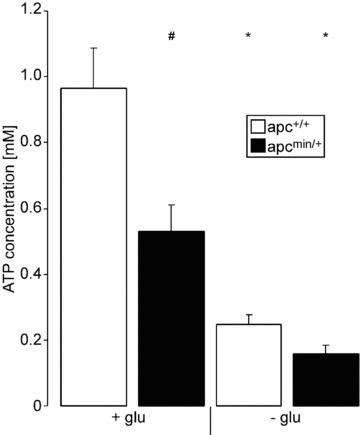
Intracellular ATP content in erythrocytes from APC-deficient and wild-type mice. Arithmetic mean ± S.E.M. (n = 4) of the cytosolic ATP concentration in erythrocytes from APC-deficient mice (apcMin/+, black bars) and wild-type mice (apc+/+, white bars) exposed for 12 hrs to Ringer solution without (left bars) or with (right bars) glucose. # indicates significant (P < 0.05) difference between genotypes; * indicates significant (P < 0.05) difference from presence of glucose (Mann–Whitney test).
Cell volume of apc+/+ and apcMin/+ erythrocytes
To depict cell shrinkage, another hallmark of eryptosis, forward scatter of apcMin/+ erythrocytes and apc+/+ erythrocytes was determined in FACS analysis. As shown in Figure 6, the forward scatter was significantly reduced by energy depletion in erythrocytes from both genotypes, an effect not significantly different between apcMin/+ erythrocytes and apc+/+ erythrocytes.
Fig 6.
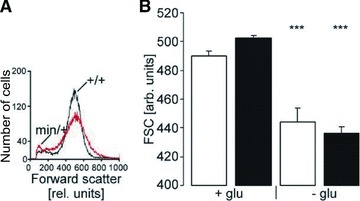
Forward scatter in erythrocytes from APC-deficient and wild-type mice. (A) Histogram of forward scatter as a measure of cell volume in a representative experiment of erythrocytes from APC- deficient mice (Min/+) and wild-type mice (+/+) exposed for 8 hrs to glucose-depleted Ringer. (B) Arithmetic mean ± S.E.M. (n = 8) of forward scatter of erythrocytes from 8-week-old APC-deficient mice (apcMin/+, black bars) and wild-type mice (apc+/+, white bars) exposed for 8 hrs to glucose-containing (left bars) or glucose-depleted (right bars) Ringer. *** indicate significant (P < 0.001) difference from glucose-containing Ringer (Mann–Whitney test).
Discussion
According to the present observations, heterozygous mice carrying defective APC (apcMin/+) suffer from mild anaemia with decreased erythrocyte count, haemoglobin concentration and haematocrit. The anaemia occurs despite significantly higher reticulocyte count in apcMin/+ mice, pointing to enhanced formation of new erythrocytes. Accordingly, the anaemia is secondary to enhanced turnover of apcMin/+ erythrocytes, which is further apparent from accelerated in vivo clearance of CFSE-labelled erythrocytes. The erythrocytes are to a large part trapped in the spleen. The splenic accumulation of eryptotic erythrocytes presumably accounts for the splenomegaly of those mice. Conversely, splenomegaly may foster splenic trapping of erythrocytes.
The accelerated clearance of the apcMin/+ erythrocytes is presumably due to enhanced phosphatidylserine exposure at their surface. Phosphatidylserine-exposing cells are trapped by macrophages [35], engulfed and degraded [36]. Phosphatidylserine-exposing erythrocytes are thus rapidly cleared from circulating blood [32]. To the extent that the accelerated loss of circulating erythrocytes is not matched by a similarly enhanced formation of new erythrocytes, the accelerated eryptosis leads to anaemia.
The present observations do not allow safe conclusions as to the mechanism linking APC deficiency to eryptosis. Clearly, erythrocytes from APC-deficient mice are more susceptible to the eryptotic effects of increased cytosolic Ca2+ activity, a property unmasked by the enhanced eryptosis of those erythrocytes following treatment with the Ca2+ ionophore ionomycin. The Ca2+ ionophore should increase cytosolic Ca2+ levels to similarly high values in APC-deficient and wild-type erythrocytes. Thus, at least part of the defect must be downstream of cytosolic Ca2+. Along those lines, even in the absence of extracellular Ca2+, eryptosis was enhanced in APC-deficient erythrocytes.
Erythrocytes from APC-deficient mice have lower cytosolic ATP levels, which should render them indeed more vulnerable to eryptosis. Energy depletion is known to stimulate protein kinase C, which in turn has been shown to trigger cell shrinkage and cell membrane scrambling [34].
Eryptosis is typically paralleled by decrease of cell volume [24]. However, no significant differences were observed in forward scatter between apcMin/+ and apc+/+ erythrocytes. Possibly, the energy depletion of apcMin/+ erythrocytes impairs the activity of the Na+/K+ ATPase leading to cellular loss of K+ and cellular gain of Na+. K+ depletion expectedly blunts the K+ exit following activation of K+ channels and thus compromises the cellular KCl loss and cell shrinkage following Ca2+ entry.
Eryptosis has been determined in Ringer, indicating that the enhanced eryptosis was a property of the erythrocytes rather than a result from direct effects of plasma components on erythrocyte survival. Moreover, the clearance of CSFE-labelled erythrocytes from apcMin/+ mice is enhanced even in wild-type mice. The present observation could be explained by a role of APC in the maintenance of cytosolic ATP levels and survival of erythrocytes. Alternatively, the erythrocytes have been rendered more vulnerable to eryptotic stimuli by some component in circulating blood prior to the experiments. It is noteworthy that reticulocytosis increases with age of the animals and may at least in part be related to tumour growth. Anaemia is a well-known complication of malignancy [37, 38] including familial adenomatous polyposis [12, 39]. In view of the present observations, tumour anaemia may at least in part be due to enhanced eryptosis followed by accelerated clearance of eryptotic cells from circulating blood. In patients with malignancy, the eryptosis may be further triggered by cytostatic treatment, as several cytotoxic drugs have been shown to stimulate eryptosis [13]. Eryptosis is triggered by a wide variety of further anaemia-causing xenobiotics and endogeneous substances [40-47], and accelerated eryptosis has been observed in anaemia of several clinical disorders [13], including iron deficiency [32], phosphate depletion [48], haemolytic uraemic syndrome [49], sepsis [50], malaria [51-54] or Wilson’s disease [55]. It is considered likely that APC deficiency enhances the susceptibility to the eryptotic effect of those xenobiotics, endogeneous substances and clinical disorders. In view of the rapid clearance of erythrocytes, the splenomegaly and the profound anaemia despite reticulocytosis in apcMin/+ mice, confounding triggers of eryptosis may be present in the blood of those mice.
Phosphatidylserine-exposing erythrocytes have been shown to adhere to the vascular wall [56-60], and to stimulate blood clotting [56, 61, 62]. Accordingly, excessive eryptosis due to oxidative stress may lead to derangements of microcirculation and enhanced eryptosis has been suggested to participate in the vascular injury of metabolic syndrome [63], a chronic clinical condition consisting of the clustering of cardiovascular risk factors including hypertension, that in humans relates also to colo-rectal cancer, and other forms of malignancies [64]. Intriguingly, the apcMin/+ mice suffer from hyperaldosteronism and hypertension [11], an observation similarly made in APC patients [65]. Oxidative stress has further been shown to be relevant for ageing of stored red blood cells [66].
In conclusion, lack of APC leads to enhanced suicidal erythrocyte death or eryptosis. The effect contributes to the anaemia in APC-deficient mice and presumably in patients carrying a loss-of-function mutation of the gene encoding the APC protein. Future studies may explore whether eryptosis is similarly enhanced in human patients suffering from APC.
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (La 315/13-3 to FL) and the Carl-Zeiss-Stiftung (to MF).
Conflict of interest
The authors state that they have no conflict of interest to disclose.
References
- 1.Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 2.Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 3.Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 4.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–51. [PubMed] [Google Scholar]
- 5.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 6.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 8.Smith KJ, Johnson KA, Bryan TM, et al. The APC gene product in normal and tumour cells. Proc Natl Acad Sci U S A. 1993;90:2846–50. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 10.Rotte A, Bhandaru M, Foller M, et al. APC sensitive gastric acid secretion. Cell Physiol Biochem. 2009;23:133–42. doi: 10.1159/000204102. [DOI] [PubMed] [Google Scholar]
- 11.Bhandaru M, Kempe DS, Rotte A, et al. Hyperaldosteronism, hypervolemia, and increased blood pressure in mice expressing defective APC. Am J Physiol Regul Integr Comp Physiol. 2009;297:R571–75. doi: 10.1152/ajpregu.00070.2009. [DOI] [PubMed] [Google Scholar]
- 12.Ousingsawat J, Spitzner M, Schreiber R, et al. Upregulation of colonic ion channels in APC (Min/+) mice. Pflugers Arch. 2008;456:847–55. doi: 10.1007/s00424-008-0451-3. [DOI] [PubMed] [Google Scholar]
- 13.Lang F, Gulbins E, Lerche H, et al. Eryptosis, a window to systemic disease. Cell Physiol Biochem. 2008;22:373–80. doi: 10.1159/000185448. [DOI] [PubMed] [Google Scholar]
- 14.Foller M, Mahmud H, Gu S, et al. Modulation of suicidal erythrocyte cation channels by an AMPA antagonist. J Cell Mol Med. 2009;13:3680–6. doi: 10.1111/j.1582-4934.2009.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernhardt I, Weiss E, Robinson HC, et al. Differential effect of HOE642 on two separate monovalent cation transporters in the human red cell membrane. Cell Physiol Biochem. 2007;20:601–6. doi: 10.1159/000107543. [DOI] [PubMed] [Google Scholar]
- 16.Duranton C, Huber SM, Lang F. Oxidation induces a Cl(-)-dependent cation conductance in human red blood cells. J Physiol. 2002;539:847–55. doi: 10.1113/jphysiol.2001.013040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duranton C, Huber S, Tanneur V, et al. Electrophysiological properties of the Plasmodium Falciparum-induced cation conductance of human erythrocytes. Cell Physiol Biochem. 2003;13:189–98. doi: 10.1159/000072421. [DOI] [PubMed] [Google Scholar]
- 18.Huber SM, Gamper N, Lang F. Chloride conductance and volume-regulatory nonselective cation conductance in human red blood cell ghosts. Pflugers Arch. 2001;441:551–8. doi: 10.1007/s004240000456. [DOI] [PubMed] [Google Scholar]
- 19.Kaestner L, Christophersen P, Bernhardt I, et al. The non-selective voltage-activated cation channel in the human red blood cell membrane: reconciliation between two conflicting reports and further characterisation. Bioelectrochemistry. 2000;52:117–25. doi: 10.1016/s0302-4598(00)00110-0. [DOI] [PubMed] [Google Scholar]
- 20.Kaestner L, Bernhardt I. Ion channels in the human red blood cell membrane: their further investigation and physiological relevance. Bioelectrochemistry. 2002;55:71–4. doi: 10.1016/s1567-5394(01)00164-5. [DOI] [PubMed] [Google Scholar]
- 21.Lang KS, Duranton C, Poehlmann H, et al. Cation channels trigger apoptotic death of erythrocytes. Cell Death and Differentiation. 2003;10:249–56. doi: 10.1038/sj.cdd.4401144. [DOI] [PubMed] [Google Scholar]
- 22.Bookchin RM, Ortiz OE, Lew VL. Activation of calcium-dependent potassium channels in deoxygenated sickled red cells. Prog Clin Biol Res. 1987;240:193–200. [PubMed] [Google Scholar]
- 23.Brugnara C, de Franceschi L, Alper SL. Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J Clin Invest. 1993;92:520–6. doi: 10.1172/JCI116597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang PA, Kaiser S, Myssina S, et al. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am J Physiol Cell Physiol. 2003;285:C1553–60. doi: 10.1152/ajpcell.00186.2003. [DOI] [PubMed] [Google Scholar]
- 25.Berg CP, Engels IH, Rothbart A, et al. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001;8:1197–206. doi: 10.1038/sj.cdd.4400905. [DOI] [PubMed] [Google Scholar]
- 26.Brand VB, Sandu CD, Duranton C, et al. Dependence of Plasmodium falciparum in vitro growth on the cation permeability of the human host erythrocyte. Cell Physiol Biochem. 2003;13:347–56. doi: 10.1159/000075122. [DOI] [PubMed] [Google Scholar]
- 27.Bratosin D, Estaquier J, Petit F, et al. Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ. 2001;8:1143–56. doi: 10.1038/sj.cdd.4400946. [DOI] [PubMed] [Google Scholar]
- 28.Daugas E, Cande C, Kroemer G. Erythrocytes: death of a mummy. Cell Death Differ. 2001;8:1131–3. doi: 10.1038/sj.cdd.4400953. [DOI] [PubMed] [Google Scholar]
- 29.Lang KS, Myssina S, Brand V, et al. Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ. 2004;11:231–43. doi: 10.1038/sj.cdd.4401311. [DOI] [PubMed] [Google Scholar]
- 30.Foller M, Feil S, Ghoreschi K, et al. Anaemia and splenomegaly in cGKI-deficient mice. Proc Natl Acad Sci U S A. 2008;105:6771–6. doi: 10.1073/pnas.0708940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foller M, Sopjani M, Koka S, et al. Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J. 2009;23:1072–80. doi: 10.1096/fj.08-121772. [DOI] [PubMed] [Google Scholar]
- 32.Kempe DS, Lang PA, Duranton C, et al. Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 2006;20:368–70. doi: 10.1096/fj.05-4872fje. [DOI] [PubMed] [Google Scholar]
- 33.Lang PA, Kasinathan RS, Brand VB, et al. Accelerated clearance of Plasmodium-infected erythrocytes in sickle cell trait and annexin-A7 deficiency. Cell Physiol Biochem. 2009;24:415–28. doi: 10.1159/000257529. [DOI] [PubMed] [Google Scholar]
- 34.Klarl BA, Lang PA, Kempe DS, et al. Protein kinase C mediates erythrocyte “programmed cell death” following glucose depletion. Am J Physiol Cell Physiol. 2006;290:C244–53. doi: 10.1152/ajpcell.00283.2005. [DOI] [PubMed] [Google Scholar]
- 35.Fadok VA, Bratton DL, Rose DM, et al. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- 36.Boas FE, Forman L, Beutler E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anaemia. Proc Natl Acad Sci U S A. 1998;95:3077–81. doi: 10.1073/pnas.95.6.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colloca G, Venturino A, Vitucci P, et al. Management of anaemia in prostate cancer. Cancer Invest. 2010;28:280–8. doi: 10.3109/07357900903124480. [DOI] [PubMed] [Google Scholar]
- 38.Spivak JL, Gascon P, Ludwig H. Anaemia management in oncology and hematology. Oncologist. 2009;14:43–56. doi: 10.1634/theoncologist.2009-S1-43. [DOI] [PubMed] [Google Scholar]
- 39.Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. doi: 10.1186/1750-1172-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhavsar SK, Bobbala D, Xuan NT, et al. Stimulation of suicidal erythrocyte death by alpha-lipoic acid. Cell Physiol Biochem. 2010;26:859–68. doi: 10.1159/000323995. [DOI] [PubMed] [Google Scholar]
- 41.Bhavsar SK, Eberhard M, Bobbala D, et al. Monensin induced suicidal erythrocyte death. Cell Physiol Biochem. 2010;25:745–52. doi: 10.1159/000315094. [DOI] [PubMed] [Google Scholar]
- 42.Eberhard M, Ferlinz K, Alizzi K, et al. FTY720-induced suicidal erythrocyte death. Cell Physiol Biochem. 2010;26:761–6. doi: 10.1159/000322343. [DOI] [PubMed] [Google Scholar]
- 43.Lang F, Gulbins E, Lang PA, et al. Ceramide in suicidal death of erythrocytes. Cell Physiol Biochem. 2010;26:21–8. doi: 10.1159/000315102. [DOI] [PubMed] [Google Scholar]
- 44.Braun M, Foller M, Gulbins E, et al. Eryptosis triggered by bismuth. Biometals. 2009;22:453–60. doi: 10.1007/s10534-008-9180-5. [DOI] [PubMed] [Google Scholar]
- 45.Mahmud H, Mauro D, Qadri SM, et al. Triggering of suicidal erythrocyte death by amphotericin B. Cell Physiol Biochem. 2009;24:263–70. doi: 10.1159/000233251. [DOI] [PubMed] [Google Scholar]
- 46.Mahmud H, Mauro D, Foller M, et al. Inhibitory effect of thymol on suicidal erythrocyte death. Cell Physiol Biochem. 2009;24:407–14. doi: 10.1159/000257433. [DOI] [PubMed] [Google Scholar]
- 47.Mahmud H, Foller M, Lang F. Arsenic-induced suicidal erythrocyte death. Arch Toxicol. 2009;83:107–13. doi: 10.1007/s00204-008-0338-2. [DOI] [PubMed] [Google Scholar]
- 48.Birka C, Lang PA, Kempe DS, et al. Enhanced susceptibility to erythrocyte “apoptosis” following phosphate depletion. Pflugers Arch. 2004;448:471–7. doi: 10.1007/s00424-004-1289-y. [DOI] [PubMed] [Google Scholar]
- 49.Lang PA, Beringer O, Nicolay JP, et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J Mol Med. 2006;84:378–88. doi: 10.1007/s00109-006-0058-0. [DOI] [PubMed] [Google Scholar]
- 50.Kempe DS, Akel A, Lang PA, et al. Suicidal erythrocyte death in sepsis. J Mol Med. 2007;85:269–77. doi: 10.1007/s00109-006-0123-8. [DOI] [PubMed] [Google Scholar]
- 51.Bobbala D, Alesutan I, Foller M, et al. Effect of anandamide in Plasmodium Berghei-infected mice. Cell Physiol Biochem. 2010;26:355–62. doi: 10.1159/000320559. [DOI] [PubMed] [Google Scholar]
- 52.Foller M, Bobbala D, Koka S, et al. Suicide for survival—death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol Biochem. 2009;24:133–40. doi: 10.1159/000233238. [DOI] [PubMed] [Google Scholar]
- 53.Siraskar B, Ballal A, Bobbala D, et al. Effect of amphotericin B on parasitemia and survival of plasmodium berghei-infected mice. Cell Physiol Biochem. 2010;26:347–54. doi: 10.1159/000320558. [DOI] [PubMed] [Google Scholar]
- 54.Koka S, Bobbala D, Lang C, et al. Influence of paclitaxel on parasitemia and survival of Plasmodium berghei infected mice. Cell Physiol Biochem. 2009;23:191–8. doi: 10.1159/000204107. [DOI] [PubMed] [Google Scholar]
- 55.Lang PA, Schenck M, Nicolay JP, et al. Liver cell death and anaemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat Med. 2007;13:164–70. doi: 10.1038/nm1539. [DOI] [PubMed] [Google Scholar]
- 56.Andrews DA, Low PS. Role of red blood cells in thrombosis. Curr Opin Hematol. 1999;6:76–82. doi: 10.1097/00062752-199903000-00004. [DOI] [PubMed] [Google Scholar]
- 57.Closse C, Dachary-Prigent J, Boisseau MR. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br J Haematol. 1999;107:300–2. doi: 10.1046/j.1365-2141.1999.01718.x. [DOI] [PubMed] [Google Scholar]
- 58.Gallagher PG, Chang SH, Rettig MP, et al. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood. 2003;101:4625–7. doi: 10.1182/blood-2001-12-0329. [DOI] [PubMed] [Google Scholar]
- 59.Pandolfi A, Di Pietro N, Sirolli V, et al. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J Cell Physiol. 2007;213:699–709. doi: 10.1002/jcp.21138. [DOI] [PubMed] [Google Scholar]
- 60.Wood BL, Gibson DF, Tait JF. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: flow-cytometric measurement and clinical associations. Blood. 1996;88:1873–80. [PubMed] [Google Scholar]
- 61.Chung SM, Bae ON, Lim KM, et al. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler Thromb Vasc Biol. 2007;27:414–21. doi: 10.1161/01.ATV.0000252898.48084.6a. [DOI] [PubMed] [Google Scholar]
- 62.Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–88. doi: 10.1007/s00018-005-4527-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zappulla D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: adaptations to CO2 increases? J Cardiometab Syndr. 2008;3:30–4. doi: 10.1111/j.1559-4572.2008.07263.x. [DOI] [PubMed] [Google Scholar]
- 64.Bloomgarden ZT. Definitions of the insulin resistance syndrome: the 1st World Congress on the Insulin Resistance Syndrome. Diabetes Care. 2004;27:824–30. doi: 10.2337/diacare.27.3.824. [DOI] [PubMed] [Google Scholar]
- 65.Hopkins TG, Salem V, El Gayar H, et al. Familial adenomatous polyposis and hypertension. Lancet. 2010;375:1752. doi: 10.1016/S0140-6736(10)60405-9. [DOI] [PubMed] [Google Scholar]
- 66.Kriebardis AG, Antonelou MH, Stamoulis KE, et al. Progressive oxidation of cytoskeletal proteins and accumulation of denatured hemoglobin in stored red cells. J Cell Mol Med. 2007;11:148–55. doi: 10.1111/j.1582-4934.2007.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.JG Fox, MT Davisson, FW Quimby, et al. The Mouse in Biomedical Research. New York: Academic Press; 2006. [Google Scholar]
- 68.MA Suckow, P Danneman, C Brayton. The laboratory mouse. Boca Raton: CRC Press; 2001. [Google Scholar]