

Abstract
It remains elusive as to what bone marrow (BM) cell types infiltrate into injured and/or diseased tissues and subsequently differentiate to assume the phenotype of residential cells, for example, neurons, cardiac myocytes, keratocytes, etc., to repair damaged tissue. Here, we examined the possibility of whether BM cell invasion via circulation into uninjured and injured corneas could assume a keratocyte phenotype, using chimeric mice generated by transplantation of enhanced green fluorescent protein (EGFP)+ BM cells into keratocan null (Kera−/−) and lumican null (Lum−/−) mice. EGFP+ BM cells assumed dendritic cell morphology, but failed to synthesize corneal-specific keratan sulfate proteoglycans, that is KS-lumican and KS-keratocan. In contrast, some EGFP+ BM cells introduced by intrastromal transplantation assumed keratocyte phenotypes. Furthermore, BM cells were isolated from Kera-Cre/ZEG mice, a double transgenic mouse line in which cells expressing keratocan become EGFP+ due to the synthesis of Cre driven by keratocan promoter. Three days after corneal and conjunctival transplantations of such BM cells into Kera−/− mice, green keratocan positive cells were found in the cornea, but not in conjunctiva. It is worthy to note that transplanted BM cells were rejected in 4 weeks. MSC isolated from BM were used to examine if BM mesenchymal stem cells (BM-MSC) could assume keratocyte phenotype. When BM-MSC were intrastromal-transplanted into Kera−/− mice, they survived in the cornea without any immune and inflammatory responses and expressed keratocan in Kera−/− mice. These observations suggest that corneal intrastromal transplantation of BM-MSC may be an effective treatment regimen for corneal diseases involving dysfunction of keratocytes.
Keywords: bone marrow, mesenchymal stem cells, cell therapy, cornea, keratocytes
Introduction
In the 1970s, circumstantial evidence implicated that BM cells might egress from BM and migrate into injured/diseased tissues; thereafter, further studies suggested that BM cells might contribute to tissue repair [1-3]. These studies imply that injury to a tissue or organ might induce the egression and homing of BM stem cells that are capable of differentiation to assume the phenotype of residential cells and repair damaged tissue [4-12]. Subsequently, numerous studies have suggested that BM-derived stem cells can give rise to various cell phenotypes and transplantation with BM stem cells may be a promising strategy for tissue repair and regeneration. For example, introduction of BM-derived endothelial and haematopoietic progenitors restores tissue vascularization after ischaemic events in retina, myocardium and adult mouse BM cells participate in the reconstruction of skin [13-17]. However, the hypothesis that BM stem cells can transdifferentiate to other cell types has been questioned. For example, genetically marked BM cells transplanted into the areas of induced muscle degeneration do not undergo myogenic differentiation rather they assume a cardiomyocyte phenotype and function via cell fusion, or they actually differentiate to haematopoietic cell lineages instead of cardiomyocytes [18,19]. The variegation may be derived from the fact that there are two major stem cell types in BM, that is MSC and HSC (haematopoietic stem cells). It is very likely that these two stem cell populations may assume different fates when they are transplanted in vivo.
There has been a growing interest in the application of MSC transplantation in regenerative medicine to repair and restore the function of diseased and injured tissues [20-24]. One of the most interesting characteristics of MSC is their ability to home into sites of damage or inflammation, when they are transplanted via intravenous infusion in patients [25]. This extraordinary ability of MSC has been demonstrated in the bone fracture, cerebral ischaemia and the infarcted heart as well [7, 8, 18, 26–28].
Our previous study showed that intrastromal xenograft transplantation of human umbilical MSC cured cloudy and thin corneas of Lum−/− mice; while transplanted human HSC failed to do so, instead they triggered severe inflammatory and immune responses [29]. This observation prompts us to investigate whether corneal keratocyte dysfunction can be restored by BM cells that egress from BM and home into the diseased cornea. To examine this hypothesis, chimera mice are prepared by transplantation of BM cells isolated from EGFP mice into Kera−/− or Lum−/− mice. Our results show that EGFP+ BM cells appear in naive and/or injured corneas of chimera mice, but do not assume a keratocyte phenotype, whereas a direct transplantation of MSC into diseased corneas is necessary for assuming keratocyte phenotype by transplanted MSC.
Materials and methods
Animal welfare
All animal protocols conformed to the Statement of Association for Research in Vision and Ophthalmology for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Isolation of BM cells
Experimental mice were killed by CO2 asphyxiation. The femurs of 6- to 8-week-old male EGFP mice expressing EGFP by all cell types were excised, and all connective tissues attached to the bones were removed under a stereomicroscope. Following removal of both ends, a 21-gauge needle fitted to a 5 ml syringe was inserted into marrow cavity and marrow was flushed out with 5 ml of DMEM. Bone marrow cells were collected by centrifugation at 250 χ g for 5 min. and resuspended in 500 μl of TAC buffer (170 mM Tris base and 160 mM ammonium chloride) for 3 min. at room temperature to lyse erythroid cells. After adding DMEM, BM cells were collected by centrifuging at 250 χ g for 5 min. and the supernatant was discarded. Finally, the cells were re-suspended with 1 ml PBS. Cell density was determined by hemacytometer, and 10 μl of cell suspension was mixed with an equal volume of 0.4% trypan blue. In another series of experiments, BM cells were isolated from Kera-Cre/ZEG mice in which cells expressing keratocan were EGFP+ due to the synthesis of Cre recombinase driven by the keratocan promoter [30, 31].
Isolation of BM-MSC
Briefly, according to the method of isolation of BM described above, BM cells from 8- to 12-weeks-old wild-type mice were cultured in alpha-MEM supplemented with 10% foetal bovine serum (Invitrogen Corporation, Carlsbad, CA, USA) in an incubator at 37°C and 5% CO2. Next day, non-adherent cells were removed. The medium was changed every 3–4 days. The plastic adherent cells near confluence were harvested by Trypsin/EDTA, subcultured, and used for transplantation after six passages.
Preparation of chimera mice
EGFP-BM cells (1 χ 106 cells) were infused via tail vein injection into Kera−/−and Lum−/− recipient mice that were γ-irradiated with a total of 1200 rads (12 greys) delivered in two doses of 600 rads (6 greys) 3 hrs apart as previously described [32]. The experimental mice were allowed to recover for at least 2 weeks and then subjected to corneal epithelium debridement.
Intrastromal transplantation of BM cells and BM-MSC
Mice were anaesthetized by intraperitoneal injection of ketamine hydrochloride (80 mg/kg) and xylazine (10 mg/kg). The eye was rinsed with PBS and topically anaesthetized with a drop of proparacaine. A small corneal intrastromal tunnel was created with a 32-gauge needle. 2 χ 104 BM cells or 1 χ 104 MSC in 2 μl of PBS were injected into each cornea with a 33-gauge needle (Hamilton Company, Reno, NV, USA). Afterwards, ophthalmic antibiotic ointment was applied to the eye and recipient animals were examined weekly by fluorescence stereomicroscopy [29].
Immunofluorescence staining of cryosections and whole mount immunostaining
For conventional immunostaining, cryosections (10 μm thick) were used as described previously [29]. For whole mount immunostaining, corneas were incubated with primary antibody in 0.3% Triton X-100 in PBS overnight, and then washed with 0.02% Triton X-100 in PBS at room temperature and followed by incubation with fluorescence-conjugated secondary antibody in 0.02% Triton X-100 in PBS for 4–6 hrs. Corneas were subsequently washed with 0.02% Triton X-100 in PBS overnight. Finally, the cornea was mounted on a slide with Mowiol medium (Calbiochem, La Jolla, CA, USA) for confocal laser scanning microscopy or fluorescent microscopy.
Western blot
Individual corneas were cut into small pieces under a stereomicroscope and suspended in 100 μl alkali extraction buffer containing 250 μ/ml Benzonase nuclease (EMD Chemicals, Gibbstown, NJ, USA) and 1χ protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) in 50 mM Tris (NaOH) pH 12 at 4°C overnight. After clarification by centrifugation, the supernatant was neutralized with 10 mM Tris/HCl (pH 6.0) to pH 7–8 and digested in 0.5 μ/ml Keratanase (SEIKAGAKU CORP., Tokyo, Japan) at 37°C overnight. The protein was separated on a 4–20% SDS-PAGE mini gel and then transferred to an immobilon transfer membrane (Millipore Corp., Bedford, MA, USA). The membrane was subjected to Western blot analysis as described previously [29]. Immune reactivity was visualized with fluorescence imaging (secondary fluorescence conjugates) using Odyssey Infrared Imaging System (Li-COR Biosciences, Lincoln, NE, USA).
RT-PCR
Briefly, total RNA was isolated from BM cells and cornea using a reagent (Trizol Reagent, Invitrogen Life Technologies, Carlsbad, CA). cDNA was synthesized (AMV Reverse Transcriptase; Promega, Madison WI, USA) and subjected to RT-PCR for detection of keratocan and lumican mRNA with the following primer pairs: mKeraRT-F, 5′-CTAACCTGCAGCACCTTCACCTTGAT, and mKeraRT-R, 5′-GTTCTGCACGCAGTGTGGTGGGAC, for keratocan mRNA; primer mLumRT-F, 5-ATAGTGGGGTACCTGGAAACTCGT, and mLumRT-R, 5′-CCAGGATCTTACAGAAGCTCTTCA, for lumican mRNA; primer mGAPDH-F, 5′- AAGGTGGTGAAGCAGGCATCTGAG, and mGAPDH-R, 5′- TCTTACTCCTTGGAGGCCATGTAG, for GAPDH mRNA as control. PCR was performed as follows: 94°C for 5 min., followed by 34 cycles of 94°C for 30 sec., 65°C for 30 sec. and 72°C for 30 sec., and 72°C for 5 min. PCR products were analysed by electrophoresis in 3% agarose gels.
Statistical analysis
Data were presented as mean ± S.D. Two-side paired or non-paired Student’s t-test was used to compare the mean values of two sets of data. ANOVA tests were used to analyse the mean values of multiple group data. Differences were considered significant at P < 0.05.
Results
The characteristics of BM cells
To characterize mouse BM cells, the expression pattern of several HSC and MSC markers was performed by fluorescent immunostaining analysis on smear slide before BM cell transplantation. Figure 1 showed representative images of fluorescence staining for CD34, CD45, CD90 and CD44. Table 1 demonstrated that more than 55% of BM cells were CD34+, a marker of HSC, and 65% CD45+, a marker of leucocytes; meanwhile, only a small number of cells expressed CD44 (2.4%) and CD90 (Thy1, a thymocyte marker) (3.3%), also markers of mesenchymal cell lineage (Fig. 1A). These indicate that cells of haematopoietic lineage are the major cell population used for BM transplantation. To verify the expression of keratocan and lumican by BM cells, immunostaining and RT-PCR were also performed before transplantation. Immunostaining showed a devoid of keratocan expression in BM cells; however, there was an average of three lumican-positive cells in six visual fields. RT-PCR revealed that BM cells lacked keratocan mRNA expression, but displayed a weak lumican band (Fig. 1B) consistent with the observation in which a very small number of lumican positive cells were found by immunofluorescence staining (Fig. 1A and Table 1).
Fig 1.

The characteristics of BM cells. (A) Immunostaining indicated that most BM cells injected expressed CD34 and CD45 (HSC markers), and a few cells were positive to CD90 and CD44; these cells were devoid of the expression of keratocan and lumican. (B) RT-PCR revealed that BM cells lacked keratocan mRNA expression, but displayed a very weak lumican band before transplantation. Scale bar: 50 μm; Blue: DAPI.
Table 1.
The distribution of cell types isolated from mouse bone marrow
| Marker | % of total nucleated cells (mean ± S.D.) |
|---|---|
| CD34 | 55.3 ± 1.0 |
| CD45 | 63.5 ± 1.7 |
| CD44 | 2.4 ± 0.2 |
| CD90 | 3.3 ± 0.3 |
| Lumican | + |
| Keratocan | − |
Note: More than 55% of BM cells injected expressed haematopoietic stem cell markers, CD34 and CD45 (red), and only few BM cells expressed mesenchymal stem cell markers, CD44 and CD90 (red), before the transplantation.
EGFP+ BM cells in the cornea of chimeric mice did not assume keratocyte phenotype for the expression of keratocan and lumican
Following BM cell transplantation into sub-lethally irradiated Kera−/− and Lum−/− mice through tail vein infusion, a small number of EGFP+ cells were present in the corneal limbal area within 1 week (Fig. S1A). EGFP+ cells migrated into the corneal periphery and assumed a dendritic morphology in 2 weeks (Fig. S1B). After 4 weeks, green cells were homogenously distributed in both the periphery and the centre of the cornea and displayed large and dendritic cell morphology (Fig. S1C and D).
To determine the expression of keratocan and lumican by BM-derived cells in chimeric Kera−/− and Lum−/− mice, immunostaining with antibodies directed against keratocan and lumican was performed on tissue isolated from mice 6 and 12 weeks after BM transplantation. The results indicate that no keratocan or lumican is detected in cornea of chimeric Kera−/− and Lum−/− mice, respectively (Fig. S1E and G).
To examine whether transdifferentiation of BM cells may occur in injured cornea of chimeras, a 2 mm central corneal epithelial debridement wound was created using an ALGERBRUSH-II in chimeric Kera−/− and Lum−/− mice 6 weeks after BM cell transplantation and allowed to heal for 6 weeks, a time point the transplanted BM would have stably re-established immune system of the experimental animals that had received sub-lethal irradiation. Immunostaining failed to detect the presence of keratocan or lumican in healed corneas (Fig. S1F and H), suggesting that cells that egressed from BM in response to injury did not transdifferentiate and assume a keratocyte phenotype. Although our data revealed that a large number of BM-derived cells appeared in the mouse corneal stroma, we never found any CD90/Thy-1 (also expressed by MSC) positive BM-derived cells in the cornea before or after corneal epithelial debridement (data not shown); in contrast, these green cells expressed CD45 (a pan-leucocyte marker) (data not shown). Thus, we conclude that BM-MSC rarely egress from BM and home into the injured and un-injured cornea. We therefore examined if a direct introduction of BM cells into the corneal stroma is essential to allow for differentiation into a keratocyte phenotype to heal injured/diseased corneas similar to what we have shown with umbilical MSC [29].
The environment of corneal stroma induced the synthesis of extracellular matrix by intrastromally transplanted BM cells
Our previous studies have demonstrated that umbilical MSC can differentiate and assume a keratocyte phenotype when they are transplanted into the corneal stroma [29]. We thereby directly injected BM cells into the cornea of experimental mice to examine if the stromal tissue environment might provide a niche essential and sufficient for the differentiation of BM cells into keratocyte phenotypes. BM cells isolated from EGFP mice were directly transplanted into the corneal stroma of Kera−/− or Lum−/− mice. After intrastromal transplantation, green BM-derived cells uniformly distributed in the corneal stroma and never migrated into the corneal epithelium and anterior chamber (Fig. 2A). One week after transplantation, most of BM-derived cells assumed dendritic cell morphology (Fig. 2B). To determine whether BM cells can differentiate to assume a keratocyte phenotype, keratocan and lumican expression was examined by immunostaining and Western blot analysis in the tissue from Kera−/− and Lum−/− mice. Immunostaining results showed that keratocan and lumican were detected around green cells in the corneal stroma of Kera−/− and Lum−/− mice, respectively (Fig. 2C and D). Western blot with anti-keratocan and lumican antibodies identified a single 45 kD core protein presented after the treatment with keratanase and/or endo-β-galactosidase in Kera−/− and Lum−/− corneas 1 week after transplantation, respectively (Fig. 2E and F); without enzyme treatment, Western blots showed smear immune reactivity (data not shown). These results indicate that some BM cells transplanted can differentiate into keratocytes and synthesize corneal stroma-unique KS-keratocan and KS-lumican.
Fig 2.

Green BM-derived cells synthesized keratocan and lumican in the corneal stroma after intrastromal transplantation. (A) Cryosection prepared from eyes after 1 week of corneal intrastromal transplantation with BM cells, green BM cells uniformly distributed in corneal stroma and never migrated into corneal epithelium and anterior chamber. (B) One week after the transplantation, most of BM-derived cells changed their round cell shape to be dendritic under a fluorescent stereomicroscopy (20χ). (C, D) Immunostaining on cryosections revealed that corneal stroma of Kera−/− and Lum−/− mice respectively, expressed keratocan and lumican (red) surrounding green BM-derived cells. (E, F) Following the treatment with keratanase and/or endo-β-galactosidase, a single 45 kD protein was respectively detected in Kera−/− or Lum−/− mouse cornea 1 week after corneal intrastromal transplantation of BM by Western blot with anti-keratocan and lumican antibodies. Scale bar: 50 μm; Blue: DAPI.
To further elucidate that BM can give rise to cells with a keratocyte phenotype, BM cells were isolated from a double transgenic mouse line, Kera-Cre/ZEG mice in which cells expressing keratocan become EGFP+ due to the excision of LacZ gene from the dual reporter ZEG gene by Cre-recombinase driven by the keratocan promoter [30, 31], and intrastromally transplanted into Kera−/− mice. Before transplantation, a smear of BM cells from a Kera-Cre/ZEG mouse was examined and were negative for EGFP expression. BM cells from Kera-Cre/ZEG mice were intrastromally transplanted into Kera−/− mouse. Interestingly, EGFP+ cells appeared in the corneal stroma 72 hrs after transplantation (Fig. 3A and B), before assuming a dendritic morphology. The observation suggests that expression of keratocyte unique gene, keratocan, precede morphological change that might require the coordinated expression of many more genes. Under confocal microscopy, the EGFP+ cells displayed a flat and dendritic cell morphology (Fig. 3B); immunostaining further demonstrated that these green cells expressed keratocan (Fig. 3D). These results suggest that the Cre gene was turned on in BM-cells derived from Kera-Cre/ZEG mouse in the corneal stroma; implying certain BM cell types are capable of assuming keratocyte phenotype. In contrast, when BM cells from Kera-Cre/ZEG mice were transplanted into sub-conjunctival stroma of Kera−/− mouse concurrent with corneal intrastromal transplantation, EGFP+ cells were found in cornea but not in conjunctiva from 72 hrs to 2 weeks after transplantation (Fig. 3E and F). This indicates that corneal stromal environment provides the niche for differentiation of BM cells into the keratocyte phenotype.
Fig 3.
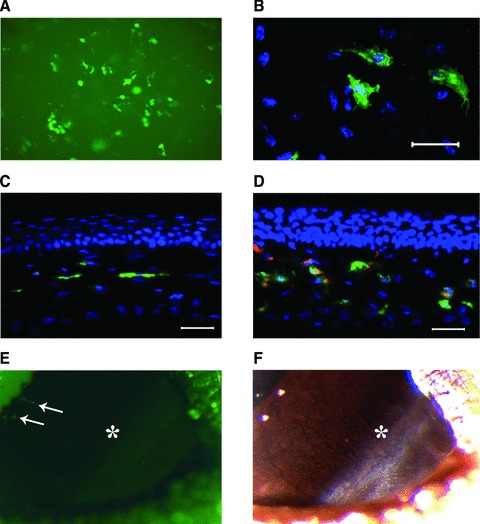
Kera-Cre transgene of BM-derived cells was turned-on in the stroma of Kera−/− mouse after intrastromal transplantation. (A) Green cells presented in the cornea of Kera−/− mouse 1 week after corneal intrastromal transplantation of BM cells isolated from Kera-Cre/ZEG mouse (20χ, fluorescent stereomicroscopy image). (B) Confocal microscopy displayed that green cells display flat and dendritic cell morphology. (C, D) Fluorescent microscopy images of cryosection revealed that green cells of (A) and (B) were located in corneal stroma (C: control) and green cells expressed keratocan (red). (E) Following transplantation of BM cells from Kera-Cre/ZEG mice into the corneal stroma and sub-conjunctiva of Kera−/− mouse at same time, EGFP+ cells (white arrows) presented in the cornea other than conjunctiva 1 week after transplantation. (*: limbal region; 1.6χ, fluorescent stereomicroscope image). (F) The same region with image (E) was taken by a stereomicroscope at same time (*: limbal region, 1.6χ). Scale bar: 50 μm; Green: EGFP; Red: Keratocan; Blue: DAPI.
Transplanted BM cells were rejected by recipient
To elucidate whether intrastromal transplantation of allogeneic BM cells can be used to heal corneal diseases, a crucial requirement is that BM cells can survive and function in an allogeneic cornea. To address this question, excised Kera−/− mouse cornea that had been treated with EGFP+ BM cell transplantation were stained with DAPI and scanned by a ZEISS Observer.Z1 fluorescent microscope with ApoTome Mode and the number of transplanted cells was determined by counting green cells in serial Z-stack images. The results revealed that the number of transplanted green cells rapidly decreased and almost all green cells vanished within 4 weeks after transplantation (Fig. 4).
Fig 4.

The numbers of BM-derived cells decreased with time extension after the transplantation. (A) One week after the injection, a large number of BM-derived cells were distributed in corneal stroma and displayed a variety of cell morphology. (B) Two weeks after the transplantation, green cells displayed dendritic cell shape in corneal stroma; however, the numbers of BM-derived cells obviously decreased. (C) Four weeks after intrastromal injection, only several green cells continuously stayed in the cornea. (D) The number of BM-derived cells was determined by counting green cells in serial Z-stack images and the results revealed that the number of transplanted green cells rapidly decreased and almost all green cells were exhausted 4 weeks after the transplantation. P < 0.001; scale bar: 50 μm; Green: EGFP; Blue: DAPI.
The decrease of transplanted cells might be due to host immune rejection, because the recipient mice were not lethally irradiated prior to intrastromal transplantation and host immune system remained intact. To further elucidate that the disappearance of transplanted BM cells were caused by host inflammation and immune rejection response, immunostaining was performed at different time points after transplantation of BM cells into Kera−/− mouse cornea. Results of immunostaining with anti-CD45 antibody, a pan-leucocyte marker, showed that a large number of recipient CD45+ leucocytes invaded the corneal stroma, which surrounded and engulfed green cells 1 week after the transplantation (Fig. 5A). It appeared that differentiation of BM cells to assuming a keratocyte phenotype might occur concurrently with the invasion of inflammatory cells. However, the reason for such coincidence remains unknown. Afterwards, many transplanted BM cells were phagocytosized by host CD45+ cells; 4 weeks after the transplantation, numerous fragments of green cell debris remained in the corneal stroma. Figure 5B summarizes the leucocyte density at different time points; the results indicate that there are more CD45+ cells at every time point after transplantation compared to uninjured Kera−/− mouse corneas.
Fig 5.
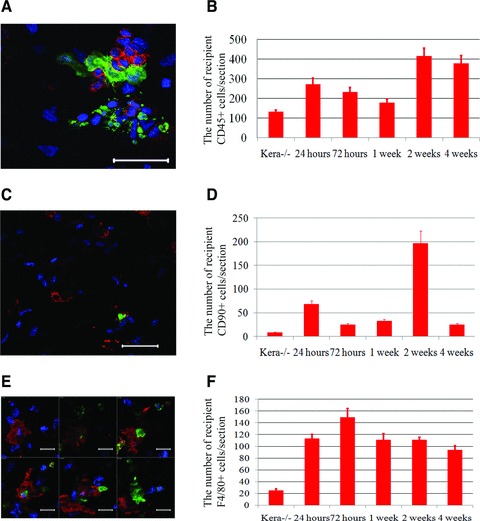
BM cell transplantation induced the infiltration of immune and inflammatory cells into corneal stroma. (A) Confocal image revealed that CD45+ leucocytes (red) invaded the corneal stroma of Kera−/− mouse to surround and engulf green cells in one week after the transplantation. (B) A large number of recipient leucocytes (CD45+) continuously invaded corneal stroma up to 4 weeks of transplantation. (C and D) The numbers of T cells (red CD90/Thy-1+ shown in C) significantly increased and peaked at 2 weeks after the transplantation and then decreased to a lower level in 4 weeks of transplantation. (E) A serial confocal images verified that green cells were phagocytosed by recipient macrophages (red) in the corneal stroma of Kera−/− mouse. (F) There was always a large number of recipient macrophage infiltrations in the corneal stroma after the transplantation in comparison to normal cornea. P < 0.001; scale bar: 20 μm; Green: EGFP; Red: F4/80; Blue: DAPI.
T cells and macrophages are known to play pivotal roles in graft rejection. To detect the presence of T cells and macrophages, anti-CD90 (Thy-1) and anti-F4/80 antibodies were used to detect the presence of pan-T cells and macrophages after transplantation. The results revealed that the number of pan-T cells and macrophages were significantly increased at different time points after transplantation (Fig. 5C–F). This is concurrent with the time period of graft rejection.
MSC isolated from BM assume keratocyte phenotypes and suppress host immune and inflammatory responses
We previously demonstrated that the xenograft transplantation of human umbilical HSC into mouse corneas triggered immense immune rejection response. In contrast, transplantation of umbilical MSC survived and healed cloudy and thin corneas of Lum−/− mice [29]. Thus, the synthesis of KS-keratocan and KS-lumican following direct BM cell transplantation is likely derived from cells of mesenchymal stem cell lineage rather than haematopoietic stem cell lineage. We therefore hypothesize that transplantation of BM-MSC may be used as treatment regimens for congenital corneal disease caused by genetic mutation. To examine the hypothesis, MSC were isolated from BM of wild type mice as described in Materials and methods. These plastic adherent cells were CD90+, CD44+, CD34−, CD45− and keratocan negative prior to transplantation (Fig. 6A).
Fig 6.

BM-MSC assumed the phenotypes of keratocytes after corneal intrastromal transplantation. (A) The plastic adherent cells isolated from bone marrow were positive to CD44 and CD90/Thy-1, and negative to CD34 and CD45, and also did not express keratocan and lumican before the transplantation. (B) The images of in vivo fluorescent stereomicroscopy showed that, DiI-labelled BM-MSC (red) displayed a round-like cell shape at the first week after the transplantation; later, the cells extended cell body to be dendritic cells (magnification, 20χ). (C) Confocal microscopy revealed that BM-MSC-derived cells displayed dendritic cell characters and the density of dye labelling became lower with the time points. Scale bar: 50 μm; Red: antibody; Blue: DAPI.
For tracing the morphological changes of BM-derived MSC in the corneal stroma, MSC were ex vivo labelled with DiI (red fluorescence) prior to transplantation and examined by fluorescent stereomicroscopy. Following the transplantation of DiI-labelled MSC, most of MSC initially stayed near the site of injection and then gradually migrated to the corneal periphery. Morphologically, MSC began to display dendritic morphology within 1–2 weeks after transplantation. Four and six weeks later, transplanted MSC were homogeneously distributed in the entire cornea as revealed by fluorescent stereomicroscopy (Fig. 6B). The morphological change was better illustrated by confocal microscopy with the whole mount Kera−/− mouse cornea (Fig. 6C) and also the intensity of dye decreased, suggesting that BM-MSC might undergo cell division after transplantation, same as what had been shown in the transplantation of umbilical MSC into the mouse cornea [29].
To verify that BM-MSC could indeed assume a keratocyte phenotype, the synthesis of KS-keratocan was determined by immunostaining with anti-human keratocan antibody in Kera−/− mouse cornea. Results displayed that BM-MSC expressed keratocan in 3 days after transplantation as shown in Figure 7A. Whether transplanted cells can be used for cell therapy, a critical criterion is that the cells escape from the host immune rejection. To address this point, we did immunostaining on cryosections at different time-points following BM-MSC transplantation. Compared to BM-transplanted Kera−/− mouse cornea, there is very few inflammatory cell infiltration 1 week after BM-MSC transplantation (Fig. 7B).
Fig 7.

Transplanted BM-MSC synthesized keratocan in Kera−/− mouse cornea without an obvious infiltration of immune and inflammatory cells. (A) Immunostaining with anti-keratocan antibody displayed that keratocan (green) distributed surrounding transplanted BM-MSC (red) in the corneal stroma of Kera−/− mouse. (B) Compared to BM-transplanted Kera−/− mouse cornea, there was an obvious increase in inflammatory cell infiltration after BM cell transplantation, but very few inflammatory cell infiltration after BM-MSC transplantation at same stage. Scale bar: 50 μm; Green: Keratocan; Red: BM-MSC; Blue: DAPI.
Discussion
In this study, we have demonstrated that EGFP+ BM-derived cells of chimeric mice generated by BM transplantation via tail vein infusion can egress and migrate into injured and un-injured cornea, where they display dendritic cell morphology but failed to synthesize corneal-specific keratan sulfate proteoglycans, that is KS-lumican and KS-keratocan. In contrast, some BM cells that are directly transplanted into the corneal stroma assume a keratocyte phenotype, as evidenced by dendritic cell morphology, synthesis of corneal stromal-specific proteins, and expression of Kera-Cre transgene following BM cell transplantation. Although these results demonstrate a potential for cell therapy with direct transplantation of BM cells, subsequent inflammatory and immune response eventually leading to the elimination of implanted cells preclude the possibility of a direct allogeneic BM cell transplantation as a treatment regimen for corneal disease. It should be noted, however, that autologous transplantation of BM cells would ameliorate corneal diseases because this type of autograft would likely not trigger immune rejection.
There is evidence showing that BM cells can migrate into tissues under normal conditions, such in skin, brain, liver and cornea [17, 33-36]. These circulating cells can home into damaged tissues and are involved in tissue defence and inflammatory response [17, 37, 38]. In addition, BM-derived cells not only contribute to inflammatory response, but also give rise to tissue-specific cell phenotypes and participate in tissue regeneration and restoration of function. For example, Eglitis and Mezey show that BM-derived cells express the astroglial marker, glial fibrillary acidic protein in brain, and Theise et al. find that BM cells could transdifferentiate and replenish a large number of hepatic parenchymal cells in humans [33, 34]. Although cornea is devoid of blood vessels, studies have demonstrated the presence of lymphoid and myeloid cell types, for example, circulating BM-derived CD45+ leucocytes belonging to haematopoietic cell lineage in corneal stroma besides keratocytes. The observation shows that these cells display a dendritic morphology and about half of them co-expressed F4/80 (a macrophages marker) [35, 36]. However, the normal cornea does not contain T cells, as evidenced by the absence of CD90+ cells in the stroma.
BM contains two major stem cell populations, that is HSC and MSC. The latter is multi-potent and capable of differentiating into various progenitor cells of connective tissues and these fibroblast-like cells are defined by multiple markers (CD90+, CD44+, CD73+, CD105+, CD45−, CD34−, CD14−) and only represent a minor cell population in BM, representing about 0.001–0.01% of the nucleated cells [4, 39-47]. Our previous study demonstrates that umbilical MSC transplanted into the corneal stroma of Kera−/− and Lum−/− mice can assume a keratocyte phenotype [29]. In this study, although BM cells show dendritic cell morphology after transplantation via tail vein infusion, they never express keratocan and lumican in both un-injured and injured corneas of chimeric mice. In contrast, after direct transplantation of BM cells into the corneal stroma, some BM-derived cells display the characteristics of keratocytes, namely dendritic cell morphology and the synthesis of KS-keratocan and KS-lumican. Taken together, our results suggest that BM-derived MSC do not normally egress and home into the corneal stroma under normal circumstances or upon injury. As mentioned earlier, there is circumstantial evidence that BM-derived cells may repair the injured tissue; however, our observation implies that the capacity of spontaneous repair via egression of BM-MSC may be minimal in respect to the injured cornea. Pluripotency of MSC has attracted great interest for stem cell therapy. However, whether transplanted cells can be used for cell therapy, a critical issue is that the cells must escape from the host immune rejection and survive in the tissue. Our previous study demonstrates that human umbilical MSC transplanted into the corneal stroma of Kera−/− and Lum−/− mice can survive for more than 3 months with little or no noticeable graft rejection; whereas transplanted HSC trigger severe host inflammatory and immune rejection responses [29]. When un-fractionated BM cells are transplanted, those cells that differentiate and assume a keratocyte phenotype are consumed due to host immune rejection triggered by the presence of haematopoietic cells present in the allogeneic transplantation. In contrast, transplanted allogeneic MSC survive in the cornea up to 6 weeks without triggering noticeable inflammation and host immune rejection response. Coincidentally, our recent data from another experiment transplanting human umbilical MSC into an alkali-burned mouse cornea show that MSC transplantation can significantly suppress the infiltration of inflammatory cells (our unpublished observation). Although several in vitro studies have demonstrated that cellular and molecular mechanisms by which MSC suppress the host immune and inflammatory response benefited from the expression of TNF-α stimulated genes (TSG6 and TSG14) [48, 49], iNOS (inducible nitric oxide synthetase) [50], IDO (Indoleamine-pyrrole 2,3-dioxygenase) [51] by transplanted MSC. Our in vivo study has showed that MSC expressed TSG14 (PTX3) in alkali-burned mouse cornea 72 hrs after the transplantation. Therefore, this may explain why very few inflammatory cells are presented in the corneas of Kera−/− mice after MSC transplantation as seen in this study (Fig. 7). Based on our finding, these long-term survived BM-MSC might continuously secrete keratocan to meet the requirement of corneal stroma; thus, transplantation of BM-MSC could be an effective treatment regimens for corneal diseases. Moreover, it should also be noted that autologous BM cell transplantation may be a potential alternative treatment regimen for severely traumatized corneas, for example chemical burns, which frequently lead to limbal stem cell deficiency because of persistent inflammation.
In conclusion, we demonstrate that BM-derived haematopoietic cells can home into naive un-injured and injured corneas via circulation, but these cells cannot give rise to keratocytes using chimeric mice via tail vein transplantation of EGFP+ bone marrow cells. However, direct-introduction of BM cells into the corneal stroma is rejected soon after transplantation albeit some BM-derived cells assume a keratocyte phenotype, for example synthesis KS-keratocan and KS-lumican. In comparison, BM-derived MSC can survive in the corneal stroma up to 6 weeks and assume a keratocyte phenotype after intrastromal transplantation. This study suggests that allogeneic corneal intrastromal transplantation of BM-MSC may be an effective treatment regimen for the treatment of corneal diseases involving dysfunction of keratocytes, which is similar to that of umbilical mesenchymal stem cell transplantation into lumican null mice [29]. It should be noted that autograft of BM cells may also be feasible to treat traumatized corneas, for example chemical burns.
Acknowledgments
This study is in part supported by grants NIH/NEI EY 011845, EY013755, Research to Prevent Blindness Inc. and Ohio Lions Eye Research Foundation. Authors thank Dr Mindy Call for her assistance in proof reading the manuscript.
Conflict of interest
The authors indicate no potential conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Green BM-derived cells displayed dendritic cells morphology in corneas of chimeras after tail vein transplantation of EGFP+ BM cells, but failed to synthesize corneal unique keratan sulfate proteoglycans, that is KS-lumican and KS-keratocan.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Griffiths IR, Burns N, Crawford AR. Early vascular changes in the spinal grey matter following impact injury. Acta Neuropathol. 1978;41:33–9. doi: 10.1007/BF00689554. [DOI] [PubMed] [Google Scholar]
- 2.Gudewicz PW, Frewin MB, Heinel LA, et al. Priming of human monocyte superoxide production and arachidonic acid metabolism by adherence to collagen- and basement membrane-coated surfaces. J Leukoc Biol. 1994;55:423–9. doi: 10.1002/jlb.55.4.423. [DOI] [PubMed] [Google Scholar]
- 3.Lafrenie RM, Wahl LM, Epstein JS, et al. HIV-1-Tat modulates the function of monocytes and alters their interactions with microvessel endothelial cells. A mechanism of HIV pathogenesis. J Immunol. 1996;156:1638–45. [PubMed] [Google Scholar]
- 4.Orlic D, Kajstura J, Chimenti S, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–5. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 5.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95:9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- 6.Sutherland FWH, Perry TE, Yu Y, et al. From stem cells to viable autologous semilunar heart valve. Circulation. 2005;111:2783–91. doi: 10.1161/CIRCULATIONAHA.104.498378. [DOI] [PubMed] [Google Scholar]
- 7.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–8. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 8.Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–12. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- 9.Noel D, Djouad F, Jorgense C. Regenerative medicine through mesenchymal stem cells for bone and cartilage repair. Curr Opin Investig Drugs. 2002;3:1000–4. [PubMed] [Google Scholar]
- 10.Awad HA, Butler DL, Boivin GP, et al. Autologous mesenchymal stem cell-mediated repair of tendon. Tissue Eng. 1999;5:267–77. doi: 10.1089/ten.1999.5.267. [DOI] [PubMed] [Google Scholar]
- 11.Kataoka K, Medina RJ, Kageyama T, et al. Participation of adult mouse bone marrow cells in reconstitution of skin. Am J Pathol. 2003;163:1227–31. doi: 10.1016/S0002-9440(10)63482-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakagawa H, Akita S, Fukui M, et al. Human mesenchymal stem cells successfully improve skin-substitute wound healing. Br J Dermatol. 2005;153:29–36. doi: 10.1111/j.1365-2133.2005.06554.x. [DOI] [PubMed] [Google Scholar]
- 13.Szmitko PE, Fedak PWM, Weisel RD, et al. Endothelial progenitor cells new hope for a broken heart. Circulation. 2003;107:3093–100. doi: 10.1161/01.CIR.0000074242.66719.4A. [DOI] [PubMed] [Google Scholar]
- 14.Grant MB, May WS, Caballero S, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–12. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 15.Murayama T, Teppera OM, Silvera M, et al. Determination of bone marrow-derived endothelial progenitor cell significance in angiogenic growth factor-induced neovascularization in vivo. Exp Hematol. 2002;30:967–72. doi: 10.1016/s0301-472x(02)00867-6. [DOI] [PubMed] [Google Scholar]
- 16.Orlic D, Kajstura J, Chimenti S, et al. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci USA. 2001;98:10344–9. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fathke C, Wilson L, Hutter J, et al. Contribution of bone marrow-derived cells to skin: collagen deposition and wound repair. Stem Cells. 2004;22:812–22. doi: 10.1634/stemcells.22-5-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nygren JM, Jovinge S, Breitbach M, et al. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat Med. 2004;10:494–501. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 19.Balsam LB, Wagers AJ, Christensen JL, et al. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004;428:668–73. doi: 10.1038/nature02460. [DOI] [PubMed] [Google Scholar]
- 20.Popp FC, Eggenhofer E, Renner P, et al. Mesenchymal stem cells can affect solid organ allograft survival. Transplantation. 2009;87:S57–62. doi: 10.1097/TP.0b013e3181a288aa. [DOI] [PubMed] [Google Scholar]
- 21.Dai LJ, Li HY, Guan LX, et al. The therapeutic potential of bone marrow-derived mesenchymal stem cells on hepatic cirrhosis. Stem Cell Res. 2009;2:16–25. doi: 10.1016/j.scr.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 22.Chhabra P, Brayman KL. The use of stem cells in kidney disease. Curr Opin Organ Transplant. 2009;14:72–8. doi: 10.1097/MOT.0b013e328320d2f5. [DOI] [PubMed] [Google Scholar]
- 23.Kitada M, Dezawa M. Induction system of neural and muscle lineage cells from bone marrow stromal cells; a new strategy for tissue reconstruction in degenerative diseases. Histol Histopathol. 2009;24:631–42. doi: 10.14670/HH-24.631. [DOI] [PubMed] [Google Scholar]
- 24.Lasala GP, Minguell JJ. Bone marrow-derived stem/progenitor cells: their use in clinical studies for the treatment of myocardial infarction. Heart Lung Circ. 2009;18:171–80. doi: 10.1016/j.hlc.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Horwitz EM, Prockop DJ, Fitzpatrick LA, et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–13. doi: 10.1038/6529. [DOI] [PubMed] [Google Scholar]
- 26.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–53. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 27.Kawada H, Fujita J, Kinjo K, et al. Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood. 2004;104:3581–7. doi: 10.1182/blood-2004-04-1488. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez M, Simon V, Herrera G, et al. Detection of stromal cells in peripheral blood progenitor cell collections from breast cancer patients. Bone Marrow Transplant. 1997;20:265–71. doi: 10.1038/sj.bmt.1700890. [DOI] [PubMed] [Google Scholar]
- 29.Liu H, Zhang J, Liu CY, et al. Cell therapy of congenital corneal diseases with umbilical mesenchymal stem cells: lumican null mice. PloS One. 2010;5:e10707. doi: 10.1371/journal.pone.0010707. . doi: 10.1371/journal.pone.0010707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weng DY, Zhang Y, Hayashi Y, et al. Promiscuous recombination of LoxP alleles during gametogenesis in cornea Cre driver mice. Molec Vision. 2008;14:562–71. [PMC free article] [PubMed] [Google Scholar]
- 31.Kao WW. Corneal morphogenesis during development and diseases. Eye Contact Lens. 2010;36:265–8. doi: 10.1097/ICL.0b013e3181ef0e00. [DOI] [PubMed] [Google Scholar]
- 32.Carlson EC, Lin M, Liu C-Y, et al. Keratocan and lumican regulate neutrophil infiltration and corneal clarity in lipopolysacharide-induced keratitis by direct interaction with CXCL1. J Biol Chem. 2007;282:35502–9. doi: 10.1074/jbc.M705823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eglitis M, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc Natl Acad Sci USA. 1997;94:4080–5. doi: 10.1073/pnas.94.8.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Theise ND, Nimmakayalu M, Gardner R, et al. Liver from bone marrow in humans. Hepatology. 2000;32:11–6. doi: 10.1053/jhep.2000.9124. [DOI] [PubMed] [Google Scholar]
- 35.Brissette-Storkus CS, Reynolds SM, Lepisto AJ, et al. Identification of a novel macrophage population in the normal mouse corneal stroma. Invest Ophthalmol Vis Sci. 2002;43:2264–71. [PMC free article] [PubMed] [Google Scholar]
- 36.Sosnová M, Bradl M, Forrestera JV. CD34+ corneal stromal cells are bone marrow–derived and express hematopoietic stem cell markers. Stem Cells. 2005;23:507–15. doi: 10.1634/stemcells.2004-0291. [DOI] [PubMed] [Google Scholar]
- 37.Deng W, Han Q, Liao L, et al. Engrafted bone marrow-derived flk-(11) mesenchymal stem cells regenerate skin tissue. Tissue Eng. 2005;11:110–9. doi: 10.1089/ten.2005.11.110. [DOI] [PubMed] [Google Scholar]
- 38.Brittan M, Braun KM, Reynolds LE, et al. Bone marrow cells engraft within the epidermis and proliferate in vivo with no evidence of cell fusion. J Pathol. 2005;205:1–13. doi: 10.1002/path.1682. [DOI] [PubMed] [Google Scholar]
- 39.Vaquero J, Zurita M. Bone marrow stromal cells for spinal cord repair: a challenge for contemporary neurobiology. Histol Histopathol. 2009;24:107–16. doi: 10.14670/HH-24.107. [DOI] [PubMed] [Google Scholar]
- 40.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 41.Azizi SA, Stokes D, Augelli BJ, et al. Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats-similarities to astrocyte grafts. Proc Natl Acad Sci U S A. 1998;95:3908–13. doi: 10.1073/pnas.95.7.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pereira RF, Halford KW, O’Hara MD, et al. Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. Proc Natl Acad Sci USA. 1995;92:4857–61. doi: 10.1073/pnas.92.11.4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackson KA, Majka SM, Wang H, et al. Regeneration of ischaemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest. 2001;107:1395–402. doi: 10.1172/JCI12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mangi AA, Noiseux N, Kong D, et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med. 2003;9:1195–201. doi: 10.1038/nm912. [DOI] [PubMed] [Google Scholar]
- 45.Sato Y, Araki H, Kato J, et al. Human mesenchymal stem cells xenografted directly to rat liver are differentiated into human hepatocytes without fusion. Blood. 2005;106:756–63. doi: 10.1182/blood-2005-02-0572. [DOI] [PubMed] [Google Scholar]
- 46.Horwitz EM, Blanc KL, Dominici M, et al. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy. 2005;7:393–5. doi: 10.1080/14653240500319234. [DOI] [PubMed] [Google Scholar]
- 47.Dominici M, Blanc KL, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2005;7:393–5. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 48.Litvack ML, Palaniyar N. Review: soluble innate immune pattern-recognition proteins for clearing dying cells and cellular components: implications on exacerbating or resolving inflammation. Innate Immun. 2010;16:191–200. doi: 10.1177/1753425910369271. [DOI] [PubMed] [Google Scholar]
- 49.Milner CM, Higman VA, Day AJ. TSG6: a pluripotent inflammatory mediator? Biochem Soc Trans. 2006;34:446–50. doi: 10.1042/BST0340446. [DOI] [PubMed] [Google Scholar]
- 50.Ren G, Zhang L, Zhao X, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–50. doi: 10.1016/j.stem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 51.Ren G, Su J, Zhang L, et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunsuppression. Stem Cells. 2009;27:1954–62. doi: 10.1002/stem.118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Green BM-derived cells displayed dendritic cells morphology in corneas of chimeras after tail vein transplantation of EGFP+ BM cells, but failed to synthesize corneal unique keratan sulfate proteoglycans, that is KS-lumican and KS-keratocan.