

Abstract
Zinc finger proteins (ZNF) play important roles in various physiological processes. Here we report that ZNF300, a novel zinc finger protein, identified specifically in humans, promotes tumour development by modulating the NF-κB pathway. Inflammatory factors were found to induce ZNF300 expression in HeLa cell line, and ZNF300 expression further enhanced NF-κB signalling by activating TRAF2 and physically interacting with IKKβ. Furthermore, ZNF300 overexpression increased ERK1/2 phosphorylation and the expression of c-myc, IL-6, and IL-8 but decreased the expression of p21waf-1 and p27Kip1; whose down-regulation led to the opposite effect. Most importantly, ZNF300 overexpression stimulated cancer cell proliferation in vitro and significantly enhanced tumour development and metastasis in mouse xenograft model, while knocking down ZNF300 led to the opposite effects. We have identified a novel function for ZNF300 in tumour development that may uniquely link inflammation and NF-κB to tumourigenesis in humans but not in mice.
Keywords: zinc finger protein, TRAF2, NF-κB, cancer progression
Introduction
The zinc finger domain is an ancient structural motif containing conserved cysteine and histidine ligands, which mediates protein–protein interactions and binds both DNA and RNA [1-3]. More than 20 classes of structurally distinct modules, termed ZNFs, have been identified, and the classical C2H2-type ZNFs have been proven to be common in complex organisms. In humans, more than 15,000 such domains are predicted to exist in about 1000 different proteins [4, 5]. Many multifunctional zinc finger proteins, such as CTCF, WT1, GLI, ZNF74 and A20, are linked to human development and diseases [6-9]. For instance, mutations in the human WT1 gene are responsible for the development of Wilms’s tumour [10,11]; A20, which is a member of the zinc finger protein family and acts as a key regulator of NF-κB activity, has recently been identified as a novel tumour suppressor gene in Hodgkin’s lymphoma and primary mediastinal B-cell lymphoma [12]. Furthermore, several zinc finger proteins have been shown to be required in morphogenesis and neoplastic transformation [13-15]. However, the mechanism by which these zinc finger proteins (ZFPs) contribute to tumourigenesis is largely unknown.
ZNF300, a novel KRAB/C2H2 gene encoding a 68-kD ZFP, was originally cloned from a human early embryo [16]. ZNF300 is expressed primarily in heart, skeletal muscle and brain, and the KRAB domain of the ZNF300 protein exhibits transcriptional repressor activity [16]. ZNF300 binds to C(t/a)GGGGG(g/c)G sequences that are found in the promoter regions of some genes, such as those encoding IL-2, IL-2Rβ, CD44, p53, tumour necrosis factor-α (TNF-α) and TNF-α receptor associated factor 2 (TRAF2), which play crucial roles in cell proliferation, apoptosis and immune response. Endogenous ZNF300 binds directly to the IL2RB gene promoter and potentially activates its expression [17, 18]. Moreover, ZNF300 is located on chromosome 5q33.1, the deletion of which is a frequent clonal chromosomal abnormality in human myelodysplastic syndrome (MDS, a pre-leucemic disorder) [19]. More recently, ZNF300 was identified as one of the genes most strongly associated with Crohn’s disease, a chronic disorder that causes inflammation of the digestive tract [20]. Thus, the literature suggests that ZNF300 may play an important role in cell proliferation, cell apoptosis, cell differentiation, embryonic development, tumour transformation and immune response.
Evidence from the past decade has shown a close link between inflammation and tumourigenesis. Up to 20% of cancers are linked to chronic inflammation. The tumour microenvironment, which is largely orchestrated by inflammatory cells, is an indispensable participant in the neoplastic process, fostering proliferation, survival and migration [21, 22]. One of the most well-studied factors, NF-κB, functions as a direct link between inflammation and cancer [23]. Activating NF-κB can lead to the production of more cytokines, which, in turn, attract more inflammatory cells into the tumour. ZFPs, such as A20 and KLF4, play important roles in immune regulation and inflammation [24]. Thus, ZFPs are also candidates that may act as links between inflammation and cancers.
We report that ZNF300 promotes tumour progression by modulating NF-κB pathway. We found that exogenous pro-inflammatory factors induced ZNF300 expression, which further induced NF-κB activity, a critical factor mediating inflammation. Overexpression of ZNF300 further induced IL-6 and IL-8, which may exacerbate inflammation and promote tumour metastasis. The induction of IL-6 and IL-8 promoted tumour metastasis in a xenograft mouse model. We therefore identified a unique regulatory pathway in humans that is absent in mice and revealed a novel function for ZNF300 that may bridge inflammation and tumour development.
Materials and methods
Plasmids construction
pEGFP-N1 was obtained from Clontech, Inc. (Mountain view, CA, USA) pGL3(−800/+95) contained the full promoter region of ZNF300. Nested PCR was used to clone the 5’ flanking region of TRAF2. The first primer pair was TRAF2-s (sense)/TRAF2-a (antisense). The primer pairs TRAF2-nest-s/TRAF2-nest-a, TRAF2-nest-a/TRAF2-S1-s and TRAF2-S2-a/TRAF2-nest-s were used to clone pGL3-TRAF2-W, pGL3-TRAF2-S1 and pGL3-TRAF2-S2, respectively. A mutation in the TRAF2 ZNF300 binding site and in the c-Ets-2 binding site was introduced using the overlap extension PCR method, using primer pairs TRAF2-mut-a/TRAF2-mut-s and Ets-2-mut-a/Ets-2-mut-s, respectively.
The full-length ZNF300 was cloned into the pCMV vector (kindly provided by Prof. Daowen Wang, Huazhong University of Science and Technology), pCDNA3.0 and the pIRES-EGFP vector, to create pCMV-ZNF300, pCDNA-ZNF300 and pCMV-ZNF300-IRES-EGFP, respectively. The full-length antisense cDNA of ZNF300 was also cloned into pCMV and pIRES-EGFP to create pCMV-AS-ZNF300 and pCMV-AS-ZNF300-IRES-EGFP, respectively.
pM-ZNF300 and pV-IKKβ were constructed using primer sets PM-ZNF300-s/PM-ZNF300-a and PV-IKKβ-s/PV-IKKβ-a, respectively.
Cell lines, TNF-α and LPS treatment and transient transfection
The human cervical cancer derived HeLa cell line was purchased from CCTCC (Wuhan, China) and cultured in DMEM at 37°C in 5% CO2. HeLa cells were seeded at 2 χ 106/well and treated with TNF-α (10 ng/ml) or LPS (200 ng/ml) (Sigma-Alrich, St. Louis, MO, USA). Protein samples were collected at 24, 48, and 72 hrs after treatment for Western blot analysis. HeLa cells were grown in 6-well plates to 60% confluence and transiently co-transfected with empty vector, pCMV-ZNF300 or pCMV-AS-ZNF300 in combination with a series of ZNF300 or TRAF2 reporter constructs using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY, USA) following the manufacturer’s instructions. A single plate of transfected cells were then used to set up the experimental cultures required for various downstream assays.
Dual luciferase assay
Transient transfection was performed as previously reported [17]. To test the transcriptional regulation of ZNF300 at the TRAF2 gene promoter, the expression vector pCDNA-ZNF300 was co-transfected into HeLa cells with pGL3-TRAF2 or a luciferase reporter driven by a composite promoter containing three copies of the wild-type NF-κB responsive element (three times NF-κB-Luc) [34]. pRL-TK expressing Renilla luciferase constitutively was used as an internal control for all dual luciferase assay experiments.
Establishment of stable ZNF300 overexpressing and ZNF300 knockdown cell lines and colony assays
Approximately 5 χ 105/well HeLa cells were seeded. After 24 hrs of incubation at 37°C and 5% CO2, the cells were gently washed with 2 ml of PBS and transfected with pCMV-ZNF300-IRES-EGFP, pCMV-AS-ZNF300-IRES-EGFP or empty vector using Lipofectamine 2000, following the manufacturer’s protocol. At 24 hrs after transfection, standard medium was replaced with medium containing 600 ng/ml G418 for selection. After 1–2 weeks, the colonies that survived the G418 screening were counted and detected using a fluorescence microscope to identify GFP-positive cells. The expression of ZNF300 in selected colonies was examined by Western blot. After ensuring the overexpression of the ZNF300 gene, negative or positive colonies were grown in 6-well plates and used for various assays.
RT-PCR analysis
Total RNA (1 μg, extracted using Trizol reagent according to the manufacturer’s instructions) from HeLa cell lines was reverse-transcribed using oligo (dT) and subjected to PCR with an RNA PCR kit (Takara, Kyoto, Japan). The primers used for PCR amplification (35 cycles) with Taq polymerase are shown in Table S1. The amplified products were separated on a 1.5% agarose gel and photographed.
MTT assay
Cell viability was determined using the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) dye uptake method as reported [35, 36].
ELISA analysis
HeLa cells were seeded at a density of 105/ml in 24-well culture plates and transiently transfected as described earlier. Forty-eight hours after transfection, the supernatant was collected; IL-6 and IL-8 production in the supernatant was measured by ELISA, using a human IL-6 Minikit and IL-8 Minikit (Endogen, Woburn, MA, USA) according to the manufacturer’s instructions.
Western blotting
Western blot analysis was performed as previously reported [17]. Antibodies against phospho-ERK1/2, ERK1/2, c-myc, p21waf-1 and p27Kip1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used to examine ERK/c-myc pathway activity and cell cycle proteins. Antibodies against TRAF2 (Cell Signaling Technology, Inc., Danvers, MA, USA) and phosphorylated IκB kinase (IKK; Invitrogen) were used to analyse NF-κB pathway activity. GAPDH (Santa Cruz Biotechnology) or β-actin antibody was used as a loading control. The ECL Western blot detection kit was purchased from Millipore Ltd. (Watford, UK).
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed as described previously [37]. Briefly, HeLa cells were cross-linked with 1% formaldehyde at 37°C for 15 min. Cells were lysed in lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris-HCl, pH 8.0) and DNA was sheared into fragments of 150–600 bp by sonication. The cell lysate was pre-cleared using 2 μg of sheared salmon sperm DNA (Invitrogen) and 40 μl of protein A-Sepharose (50% slurry in dilution buffer; Sigma-Aldrich) for 6 hrs at 4°C. Supernatants were collected after centrifugation at low speed and subjected to immunoprecipitation with 5 μl of polyclonal anti-ZNF300 antibody overnight at 4°C. In parallel, supernatants were incubated with anti-GFP antibody (Santa Cruz Biotechnology) or without antibody as controls. Then, 40 μl of protein A-Sepharose were added to incubate for 4 hrs. After spinning down at low speed, the pellets were washed and extracted with 1% SDS (v/v), 0.1M NaHCO3, and heated at 65°C overnight to reverse the cross-links. DNA fragments were precipitated with 3 volumes of 100% ethanol and 0.1 volumes of 3M ammonium acetate and resuspended in 25 μl TE buffer. PCR amplification was performed with 35 cycles using the primers TRAF2-chip-S1/TRAF2-chip-A1 for the promoter region and TRAF2chip-dzs/TRAF2chip-dza as the negative control (Table S1).
Protein interaction analysis
Co-immunoprecipitation was performed as previously reported [38]. Briefly, HeLa cells were collected and washed in PBS, then cells were lysed in lysis buffer (50 mM HEPES, pH 7.5, 250 mM NaCl, 0.1% Nonidet P-40, 1 mM EDTA and 1 mM dithiothreitol) containing 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin and 1 μg/ml aprotinin. The cell lysates were pre-cleared with protein G-Sepharose (Amersham Pharmacia Biotech, Piscataway, NJ, USA) for 2 hrs at 4°C and centrifuged at low speed to collect the supernatant. The pre-cleared HeLa extracts were immunoprecipitated with rabbit anti-ZNF300 antibody or rabbit IgG (control; Pierce Biotechnology, Inc., Rockford, IL, USA), and the immunoprecipitates were subjected to Western blot with anti-ZNF300 or anti-IKKβ, respectively.
Immunofluorescence and confocal microscopy were carried out as previously reported [39]. IKKβ was visualized with a mouse anti-IKKβ monoclonal antibody (1:300, sc-8014; Santa Cruz Biotechnology) and a tetramethyl rhodamine isothiocyanate-conjugated goat antimouse secondary antibody (ProteinTech Group, Chicago, IL, USA), whereas ZNF300 was detected with the rabbit anti-ZNF300 antibody and a fluorescein isothiocyanate-conjugated goat anti-rabbit secondary antibody (ProteinTech Group). Cell nuclei were then stained for 10 min. at 37°C using 0.5 mg/ml of DAPI in PBS.
The Matchmaker Mammalian Assay Kit 2 (Clontech, Inc.) was employed for the mammalian two-hybrid assay, except the SEAP reporter gene of pG5SEAP was replaced with the Firefly luciferase reporter gene. The expression vector pM-ZNF300, which expresses a fusion protein of the DNA binding domain of GAL4 (GAL4-DBD) with ZNF300, was co-transfected with the pV-IKKβ vector, which expresses a fusion protein of the transactivation domain of the herpes simplex virus VP16 protein (VP16AD) with IKKβ, as well as the Firefly luciferase reporter construct pG5LUC (containing five consensus GAL4 binding sites and the minimal promoter of the adenovirus E1B gene driving the luciferase gene) and an internal control vector, pRL-TK, into HeLa cells. Dual luciferase assays were performed as described earlier. Fold induction was calculated by dividing the values obtained with the individual samples by the values obtained with the pM(GAL4-DBD) and pV(VP16AD) constructs. Mean values were calculated from at least three independent experiments with triplicate measurements.
Murine xenograft model experiments
Forty nude mice (4 weeks old, 20–25 g, the Center for Disease Control of Hubei Province, China) were randomly divided into four groups (n = 10) and subsequently injected subcutaneously with 200 μl of saline (Control) or with HeLa cells (∼1.5 χ 106 cells) stably transfected with a GFP construct, the ZNF300-overexpression construct or the ZNF300-knockdown construct, and maintained in an aseptic facility. Tumour growth was monitored every 3 days by measuring tumour size using a caliper, and the size of the tumour was determined by the following formula [40]: volume = (length χ width2) χ 0.4. Approximately 30 days later, the mice were sacrificed. Tumours and other tissues were harvested and fixed in 10% formalin, embedded in paraffin, sectioned at 4 μm and stained with haematoxylin and eosin or immunohistochemically stained with anti-ZNF300 antibody. Mouse sera were collected for ELISA assay.
Statistical analysis
Data are expressed as mean ± S.D. from at least three independent experiments, and statistical significance was analysed using paired or unpaired Student’s t-test or one-way ANOVA followed by a Student–Neuman–Keuls test. P < 0.05 was considered statistically significant.
Results
TNF-α and LPS induce ZNF300 expression in HeLa cells
ZNF300 is expressed in many tissues during early embryogenesis [16]. The findings of high level of ZNF300 expression in embryonic liver [16] and several types of cancer biopsies (data not shown) promoted us to link ZNF300 expression with inflammation. To test whether inflammation could induce ZNF300 expression, we treated HeLa cells with TNF-α (10 ng/ml) or LPS (200 ng/ml) and measured ZNF300 expression. ZNF300 expression was elevated dynamically by either TNF-α or LPS treatment (Fig. 1A). With TESS online prediction program, we identified that there was a binding site for c-Ets-2 in the proximal region of ZNF300 promoter. Luciferase assay showed that the ZNF300 promoter with the wild-type putative c-Ets-2 binding site (WT) was dramatically up-regulated upon TNF-α stimulation (Fig. 1B) or LPS treatment (Fig. 1C). In contrast, when the putative c-Ets-2 binding site was inactivated through PCR-mediated mutagenesis, gene expression from the mutated promoter (Ets2-mut) was significantly reduced under the same condition of TNF-α stimulation (Fig. 1B) or LPS treatment (Fig. 1C), respectively, compared to that from the WT promoter.
Fig 1.
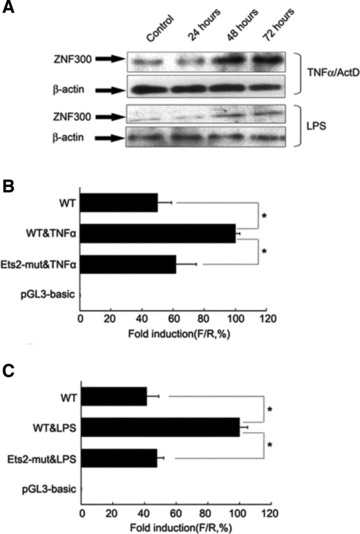
TNF-α/ActD and LPS induced ZNF300 expression in HeLa cells. (A) Time course of TNF-α/ActD or LPS induced up-regulation on ZNF300 expression as revealed by Western blot. β-Action served as loading control. (B, C) The wild-type ZNF300 promoter (WT) or a mutant ZNF300 promoter (Ets2-mut, whose Ets-2 binding site was inactivated) driving luciferase expression was transfected into cells that were treated with TNF-α (B) or LPS (C). Then luciferase activity was assayed after 48 hrs. F/R represents the ratio of Firefly luciferase activity to Renilla luciferase activity. The F/R for WT without TNF-α or LPS treatment was set at 100%, respectively.
ZNF300 overexpression promotes cancer cell growth through NF-κB pathway
Inflammation promotes tumour development, which involves cancer cell proliferation. To investigate the effect of ZNF300 on cell growth, the ZNF300 expression vector pCMV-ZNF300 or antisense ZNF300 expression vector pCMV-AS-ZNF300 was transiently transfected into HeLa cells. Western blots showed that the pCMV-ZNF300 effectively increased ZNF300 expression and that pCMV-AS-ZNF300 significantly knocked down ZNF300expression (Fig. 2A). An MTT assay showed that transient transfection of pCMV-ZNF300 promoted cell growth in a time-dependent (Fig. 2B) and dose-dependent manner (Fig. 2C). In contrast, knocking down ZNF300 expression through pCMV-AS-ZNF300 transfection slowed down cell growth in a time-dependent (Fig. 2B) and dose-dependent manner (Fig. 2C). These results indicate that ZNF300 has a cell growth promoting effect and knocking down ZNF300 expression inhibits cell growth.
Fig 2.
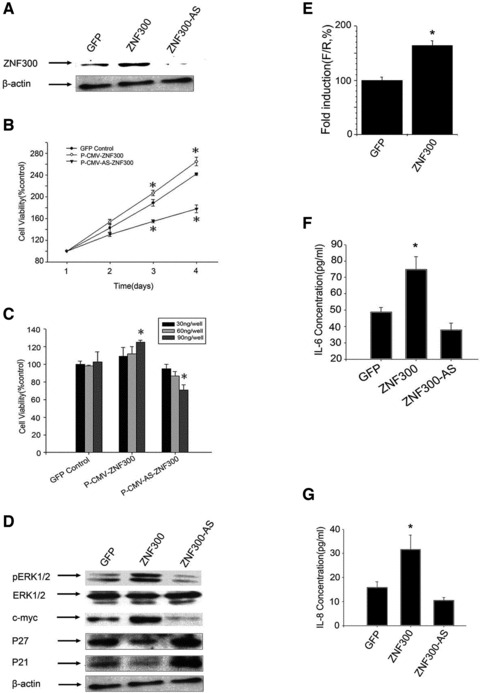
ZNF300 promotes cell proliferation through the NF-κB–dependent pathway. (A) Western blots showed that the pCMV-ZNF300 (ZNF300) effectively increased ZNF300 expression and that pCMV-AS-ZNF300 (ZNF300-AS) significantly knocked down ZNF300expression. (B) Time course of HeLa cell proliferation stimulated by ZNF300 overexpression. *P < 0.05. (C) Dose dependence of the stimulation on HeLa cell proliferation induced by ZNF300 overexpression. Different amounts of the construct pCMV-GFP (GFP), pCMV-ZNF300 or pCMV-AS-ZNF300 were transiently transfected into HeLa cells, and the MTT assay was preformed after 48 hrs; *P < 0.05. (D) ZNF300 expression regulates cell proliferation-related gene expression in HeLa cells. Forty-eight hours after transfecting pCMV-GFP (GFP), pCMV-ZNF300 (ZNF300) or pCMV-AS-ZNF300 (ZNF300-AS) in HeLa cells, cell lysates were prepared for Western blot using the antibodies indicated. (E) Dual luciferase assays of pcDNA-ZNF300 (ZNF300) transiently co-transfected with the three times NF-κB-Luc reporter in HeLa cells for 48 hrs. The ratio F/R for the three times NF-κB-Luc reporter co-transfected with pcDNA3.0 (GFP) was set at 100%; *P < 0.05. (F, G) Construct as indicated was transfected into HeLa cells, respectively, and ELISA assay was performed to measure the levels of secreted IL-6 (F) and IL-8 (G); *P < 0.05.
Western blot analysis was used to evaluate the expression ofERK, c-myc, p21waf-1 and p27Kip1, which are associated with the cell cycle. We found that the overexpression of ZNF300 increased the phosphorylation of ERK and the expression of c-myc but decreased the expression of p21waf-1 and p27Kip1 (Fig. 2D). In addition, knocking down ZNF300 expression resulted in decreased ERK phosphorylation and c-myc expression and increased p21waf-1 and p27Kip1 expression (Fig. 2D). The expression of p21waf-1 and p27Kip1, which are cyclin-dependent kinase inhibitors, was previously reported to be suppressed by the ERK/c-myc pathway [25], all of which belong to the NF-κB signalling pathway. These results suggest that ZNF300 plays a role in the NF-κB signalling pathway.
Then we examined whether ZNF300 broadly regulated the NF-κB pathway. To test this, an NF-κB–dependent promoter reporter (three times NF-κB-Luc), in which the Firefly luciferase gene is driven by a promoter containing three copies of the NF-κB binding site, was transiently co-transfected into HeLa cells with pcDNA-ZNF300 or pcDNA3.0, respectively. Forty-eight hours after transfection, cells were harvested for dual luciferase assay. The data indicated that overexpression of ZNF300 in HeLa cells significantly promoted the activity of the NF-κB–dependent promoter (Fig. 2E).
IL-6 and IL-8 are downstream target genes of NF-κB, and higher secreted levels of these cytokines stimulate cancer cell growth. To test whether ZNF300 regulated IL-6 or IL-8 secretion in the NF-κB pathway, we examined the effect of ZNF300 on IL-6 and IL-8 secretion. Overexpression of ZNF300 promoted the secretion of IL-6 and IL-8, whereas knockdown of ZNF300 slightly decreased the secretion of IL-6 and IL-8 (Fig. 2F and G). Together, all these results indicate that ZNF300 overexpression promotes cancer cell growth through NF-κB pathway.
ZNF300 regulates transcription of TRAF2
We next explored how ZNF300 regulate the NF-κB pathway. Our previous study had indicated the presence of putative ZNF300 binding sites (C(t/a)GGGGG(g/c)G) in the promoter regions of genes such as IL2RB, CD44 and TRAF2 [17, 18]. Here, we focused on the putative ZNF300 binding site in the proximity of the promoter of the gene-encoding TRAF2 (Fig. 3A), which is a member of the TRAF protein family that plays important roles in TNF-α-induced NF-κB activation [26].
Fig 3.
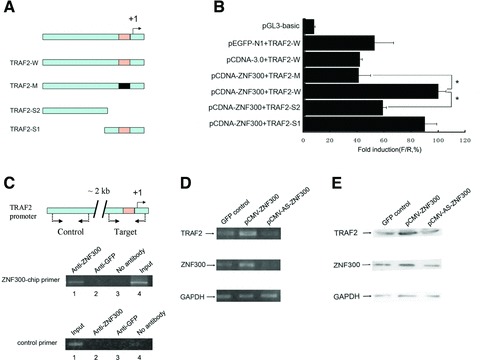
ZNF300 promotes TRAF2 gene expression. (A) Schematic diagram of the human TRAF2 gene promoters used to drive firefly luciferase gene expression in transient transfection experiments. The wild-type promoter and the truncated promoters with or without ZNF300 binding sites deleted are depicted as TRAF2-W, TRAF2-S1 and TRAF2-S2, respectively, whereas the promoter with a mutated ZNF300 binding site is depicted as TRAF2-M. The transcription start site was designated as ‘+1’ and the putative ZNF300 binding site is boxed. (B) Dual luciferase assay showing that ZNF300 binding site is required for ZNF300 promotion on TRAF2 promoter activity; *P < 0.05. Note that overexpression of ZNF300, but not of GFP, increased promoter activity of TRAF2-W. (C) Binding of ZNF300 to the promoter of TRAF2 was analysed by ChIP assays using the anti-ZNF300 antibodies. Anti-ZNF300 antibody was used in immunoprecipitation for endogenous ZNF300 binding on HeLa cell chromatin and anti-GFP was used as a control. PCR analysis of the input (Input), of the precipitates in the absence of antibody (No antibody), and of the immunoprecipitated DNA with anti-ZNF300 (Anti-ZNF300) or anti-GFP (Anti-GFP) was performed. Top: Schematic presentation showing the relative position of ZNF300-chip primers amplifying the region encompassing the putative ZNF300 binding site (Target) or control primers amplifying the region about 2kb upstream of the putative ZNF300 binding site (Control); middle: PCR with ZNF300-chip primers; bottom: PCR with control primers. The transcription start site was designated as ‘+1’ and the putative ZNF300 binding site is boxed. (D, E) ZNF300 expression regulates TRAF2 expression in vivo. Constructs overexpressing GFP, ZNF300 or ZNF300 antisense RNA (AS-ZNF300) were transiently transfected into HeLa cells. Forty-eight hours after transfection, RT-PCR (D) and Western blot (E) were carried out to detect the expression of ZNF300 and TRAF2. GAPDH served as a loading control.
To define the essential elements of the TRAF2 promoter, a putative promoter construct (Fig. 3A) encompassing −1565 to +344 (+1 corresponds to the transcription start site, TSS), −460 to +344 or −1565 to −154 bp of TRAF2 was individually cloned into pGL3-basic (Promega) to generate the construct TRAF2-W, TRAF2-S1 (deletion of the 5’ end of the cloned promoter but containing the putative ZNF300 binding site) or TRAF2-S2 (deletion of the 3’ end and lacking the ZNF300 binding site), respectively. The mutant type of TRAF2 promoter was constructed and named as TRAF2-M (harbouring a inactive mutant ZNF300 binding site). These constructs were co-transfected with pCDNA-ZNF300 or pCDNA3.0 into HeLa cells for dual luciferase. Quantitative analysis of luciferase activity (Fig. 3B) showed that overexpression of ZNF300, but not of GFP, increased promoter activity of TRAF2-W; and mutation inactivating the putative ZNF300 binding site within the promoter (TRAF2-M) markedly reduced the ZNF300-induced promoter activity (Fig. 3B). In case of deletion assays on the promoter, the promoter activity of TRAF2-S1 with deletions up to −460 bp from the 5’ end (−1565 bp) reduced slightly whereas the activity of the TRAF2-S2 promoter, in which the putative ZNF300 binding site was deleted, decreased significantly (Fig. 3B). These findings demonstrate that the putative ZNF300 binding site (TTGGGGG, −19 to −27) within the human TRAF2 promoter plays a critical role in the promoter activity.
To demonstrate if ZNF300 binds with the putative ZNF300 binding site of the TRAF2 promoter, we conducted EMSA analysis with HeLa nuclear extract on oligos encompassing the region of the putative ZNF300 binding site (WT probe). The data demonstrated that the HeLa nuclear extract caused the labelled WT probe to shift in gels which could be competed by unlabelled WT probe but not unlabelled mutant probe that was known to lack ZNF300 binding ability (Fig. S1). Anti-ZNF300 antibody reduced the density of the shifted band of labelled WT probe/HeLa nuclear extract (Fig. S1), suggesting the shift of the WT probe caused by the HeLa nuclear extract was due to the endogenous ZNF300 in the nuclear extract. To investigate whether the putative ZNF300 binding site in the proximal promoter of TRAF2 could interact with ZNF300 in vivo, ChIP analysis was performed in HeLa cells. As shown in Figure 3C, a 334-bp fragment was detected when TRAF2-chip-sense/TRAF2-chip-antisense primers (−318 to +15) were used to amplify the immunoprecipitates pulled down with anti-ZNF300 antibody (lane 1, Fig. 3C, middle panel) or the input (lane 4, Fig. 3C, middle panel), but not with anti-GFP antibody (lane 2, Fig. 3C, middle panel) or when no antibody was added (lane 3, Fig. 3C, middle panel). PCR amplification with control primers TRAF2-chip-dzs/TRAF2-chip-dza (Fig. 3C, bottom panel) failed to generate a product from the anti-ZNF300 immunoprecipitates (lane 2, Fig. 3C, bottom panel), but the primer pair amplified the input DNA extensively (lane 1, Fig. 3C, bottom panel). These results indicate that the transcription factor ZNF300 bound to the putative ZNF300 binding site, C(t/a)GGGGG(g/c)G, in the TRAF2 promoter region in vivo.
Having demonstrated that ZNF300 binds to the putative ZNF300 binding site in the proximity of the TRAF2 promoter, we examined whether ZNF300 regulates TRAF2 gene expression. RT-PCR and Western blot analysis showed that overexpression of ZNF300 in HeLa cells enhanced the expression of TRAF2, whereas knocking down ZNF300 inhibited TRAF2 expression (Fig. 3D and E). These results indicate that ZNF300 promotes the expression of TRAF2, which could further activate the NF-κB pathway.
ZNF300 protein interacts with IKKβ protein
As discussed earlier, overexpression of ZNF300 affects the NF-κB signalling pathway by regulating the transcription of TRAF2. The IKK complex is at the convergent point of the NF-κB signalling pathway. We therefore examined whether ZNF300 could affect NF-κB activity by interacting with the IKK complex and modifying its activity. We investigated whether endogenous IKKβ and ZNF300 were co-localized within the cell using immunofluorescent and confocal microscopic analysis of these proteins in HeLa cells. As shown in Figure 4A, ZNF300 was distributed in both the cytoplasm and the nucleus (panel a, Fig. 4A), whereas IKKβ was exclusively located in the cytoplasm (panel b, Fig. 4A). DAPI staining was used to visualize the nuclear region (panel d, Fig. 4A). Together, these results indicate that IKKβ and ZNF300 are co-localized within similar regions of the cytoplasm (panel c).
Fig 4.
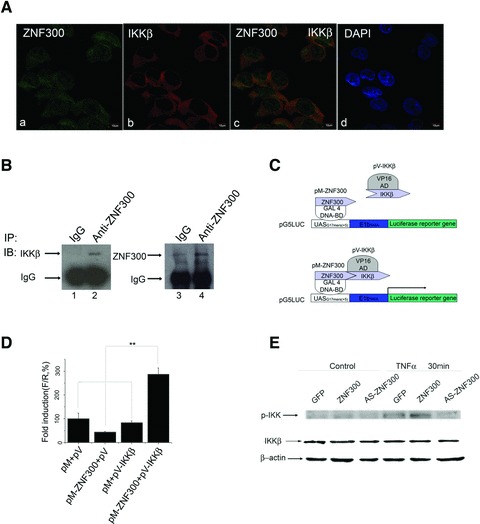
ZNF300 interacts with IKKβ. (A) Cofocal microscopic assays were used to investigate the co-localization of endogenous ZNF300 with IKKβ in HeLa cells. DAPI was used to stain nuclei (d). (B) HeLa cell lysates were immunoprecipitated (IP) with IgG or anti-ZNF300 antibody, and Western blot was performed with anti-IKKβ or anti-ZNF300 antibody. (C) Schematic representation of the mammalian two-hybrid system used in this study. (D) Interaction of ZNF300 with IKKβ. Plasmids pM or pM-ZNF300 was co-transfected with either pV or pV-IKKβ, respectively, together with pG5LUC. pRL-TK was used as an internal control. Forty-eight hours after transfection, a dual luciferase assay was performed; *P < 0.05. (E) Western blot analysis showing overexpression of ZNF300 significantly increased the phosphorylation of IKK 30 min. after TNF-α treatment whereas total IKK was constant.
To further investigate whether endogenous ZNF300 interacted with IKKβ in HeLa cells, co-immunoprecipitation assays were employed. Endogenous IKKβ was detected by anti-IKKβ antibody (Fig. 4B, lane 2) in anti-ZNF300 immunoprecipitates containing ZNF300 (Fig. 4B, lane 4), but not in control IgG immunoprecipitates (Fig. 4B, lane 1) that lacked ZNF300 (Fig. 4B, lane 3), confirming a potential association of IKKβ and ZNF300 in vivo.
To examine if the in vivo interaction of ZNF300 with IKKβ was functional, we employed a mammalian two-hybrid assay. pM-ZNF300, which encodes a fusion protein of GAL DNA binding domain (Gal4-DBD) and ZNF300, was co-transfected into HeLa cells with the pV-IKKβ vector, which expresses a fused protein of the VP16 transactivation domain (VP16AD) and IKKβ (Fig. 4C). As shown in Figure 4D, pM-ZNF300 and pV-IKKβ together promoted transcription, whereas GAL4-DBD alone (pM) did not significantly affect transcription in the presence of either VP16AD alone (pV) or pV-IKKβ. Similarly, transcription was not significantly altered when PM-ZNF300 was co-transfected with VP16AD alone (pV). These results indicate that ZNF300 functionally interacts with IKKβ in vivo.
The phosphorylation of the IKK complex is crucial for its inactivation of the IκB proteins and thus for the control of NF-κB activity. To show that the interaction between ZNF300 and IKKβ was able to activate the NF-κB signalling pathway, Western blots were performed to examine whether ZNF300 overexpression enhanced the phosphorylation of IKK. After performing transient transfection as described earlier, HeLa cells were treated with TNF-α, and the cell lysates were collected for Western blot analysis using an anti-phospho-IKK antibody. As shown in Figure 4E, with TNF-α treatment, ZNF300 overexpression enhanced the phosphorylation of IKK, while the level of unphosphorylated IKKβ was constant. These results indicate that ZNF300 overexpression could activate the kinase activity of IKKβ and further enhance the NF-κB activity.
ZNF300 overexpression promotes colony formation and cell viability of transfected HeLa cells
To confirm that ZNF300 overexpression promotes cancer cell growth through the activation of the NF-κB–dependent pathway, we established HeLa cell lines stably transfected with pCMV-ZNF300-IRES-EGFP to overexpress ZNF300, with pCMV-AS-ZNF300-IRES-EGFP to knock down ZNF300 or with the empty vector as a control, in preparation for the construction of murine xenograft models. After 2 weeks of selection in G418, colonies were counted and detected using a fluorescent microscope to identify GFP-positive cells. Compared to the transfection with empty vector, there was increased colony formation after transfection with ZNF300 and significantly decreased colony formation in the AS-ZNF300–transfected group (Fig. 5A). Western blot analysis revealed that stably transfected HeLa cells expressed ZNF300 at different levels (Fig. 5B). We selected three clones stably overexpressing ZNF300 at three different levels, which we named ZNF300(+)C1, C2 and C3. In addition, we isolated one clone from the antisense-ZNF300 group (AS-ZNF300) and one clone from the empty vector-transfected (GFP control) group (Fig. 5B). MTT assays showed that the cell viability of ZNF300-overexpressing clones ZNF300(+)C1, C2 and C3 increased in dose-dependent manner consistent with the expression level of ZNF300(all higher than control group; Fig. 5C). In contrast, the AS-ZNF300 clone had the opposite effect on cell viability (Fig. 5C) over time. These results suggest that overexpression of ZNF300 promotes cancer cell growth in vitro.
Fig 5.
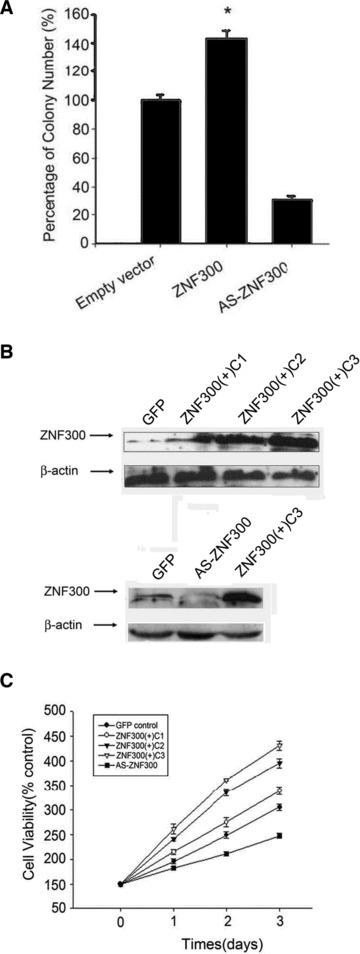
ZNF300 expression promotes colony formation and cell viability of the transfected HeLa cells. (A) ZNF300 expression promotes colony formation. HeLa cells were transfected with pCMV-ZNF300-IRES-EGFP or pCMV-AS-ZNF300-IRES-EGFP to overexpress ZNF300 or antisense ZNF300 RNA, respectively, and subjected to G418 selection. HeLa cells transfected with empty vector served as a control. After 2 weeks of selection in G418, colonies were counted, and the viability of empty vector transfected cells was set at 100%. *P < 0.05. (B) Colonies were picked and subjected to Western blot analysis with anti-ZNF300 antibody. The clones from the empty vector transfection (GFP) served as a control. (C) ZNF300 promotes cell viability of the stably transfected clones. Stably transfected cells were cultured under the same conditions, and cell viability was measured daily by MTT assay.
ZNF300 overexpression enhances tumour formation and metastasis in murine xenograft model
To test if ZNF300 promotes tumourigenesis in vivo, we selected the ZNF300(+)C3 and AS-ZNF300 clones described earlier for xenograft model construction and the GFP clone as a control, which were named as ZNF300, AS-ZNF300 and GFP group, respectively. Approximately 1.5 χ 106 cells of each clone were injected subcutaneously into male BALB/c-nu mice. The ZNF300 group showed a significant difference in the tumour formation rate compared with the GFP control and AS-ZNF300 groups. ZNF300 mice developed tumours earlier (day 7 versus day 10 in GFP control and day 13 in AS-ZNF300), with higher penetrance (70% of mice in the ZNF300 group developed tumours versus 30% in the control group and 0% in AS-ZNF300 group on day 13). On day 20, 100% of the mice in the ZNF300 group developed tumours, compared to 70% mice in the control group and 50% in the AS-ZNF300 group (Fig. 6A). A significant enhancement of tumour size was observed in mice injected with the stable ZNF300-overexpressing cell line compared to the GFP and AS-ZNF300 groups. In addition, tumour volume in the AS-ZNF300 group was slightly smaller than in the GFP control group (Fig. 6B).
Fig 6.
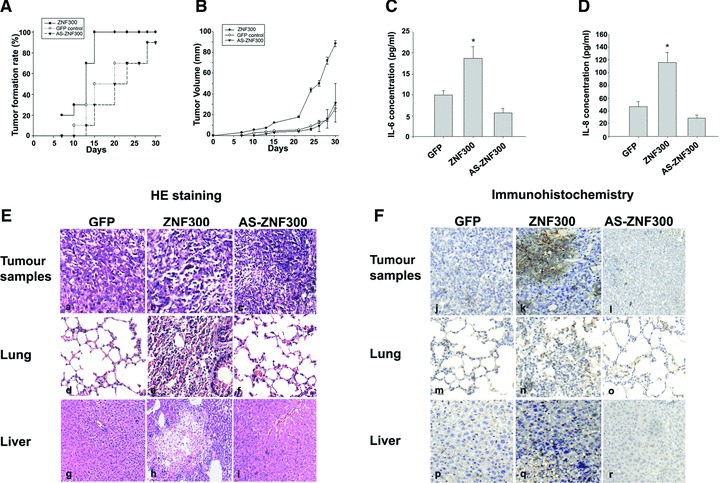
ZNF300 overexpression enhances tumour formation and metastasis in a murine xenograft model. (A) Tumour formation rates in the xenograft models. Nude mice were subcutaneously injected with HeLa cells stably transfected with GFP, ZNF300 or AS-ZNF300 constructs and monitored for tumour formation at different time points. (B) Tumour volume in the xenograft models was measured with a caliper. (C, D) ZNF300-overexpressing HeLa cells secrete elevated levels of IL-6 and IL-8 in the xenograft model. Sera were colleted to measure IL-6 (C) and IL-8 (D) by quantitative ELISA. *P < 0.05. (E, F) Anatomic analysis of ZNF300 promotion of tumour formation and metastasis. Nude mice were sacrificed to obtain tumour tissues at the injected location as well as from lung and liver. (E) HE staining of sectioned tissues. (F) Immunohistochemical staining with anti-ZNF300 antibody.
All of the animals were sacrificed 30 days after cancer cell injection, at which point blood sera were collected for IL-6 and IL-8 concentration determination by enzyme-linked immunosorbent assay (ELISA; Fig. 6C and D). Both IL-6 and IL-8 are transcriptionally regulated targets of NF-κB, and higher secreted levels of these cytokines stimulate cancer cell growth and contribute to metastasis. Elevation of their concentrations in serum might be utilized as a marker for monitoring cancer progression. The ELISA showed two- to threefold higher serum concentrations of IL-6 and IL-8 in ZNF300 mice compared to GFP controls. AS-ZNF300 had an opposite effect (Fig. 6C and D). These results confirm that ZNF300 overexpression can activate the NF-κB-dependent pathway, resulting in increased IL-6 and IL-8 secretion, which contributes to tumour growth and metastasis in murine xenograft models.
In addition, the mouse tumour, lung, liver and other tissue samples were collected for HE staining (Fig. 6E) and immunohistochemical analysis. Immunohistochemical analysis showed higher expression of the ZNF300 protein in tumour cells in the ZNF300 group than in the GFP control or AS-ZNF300 group (Fig. 6F, panels j, k and l). Interestingly, five of ten animals in the ZNF300 group showed metastasis of cancer cells into liver and lung, while no metastasis was observed in the GFP control or AS-ZNF300 animals (Fig. 6E, panels d–i). Immunohistochemical analysis showed that in cancer cells that had metastasized to liver and lung tissues, many cells had a higher expression of ZNF300 protein (Fig. 6F). These results indicate that the increased tumour size and metastasis by stably transfected HeLa cells were due to the overexpression of ZNF300.
Discussion
Our previous studies have revealed several unique features of the ZNF300 gene [16], suggesting that it plays a crucial role in cell proliferation, apoptosis and immune response [17]. In this study, we examined the mechanism by which ZNF300 exerts its function. We showed that TNF-α and LPS stimulation, both of which are major players in inflammation and NF-κB activators, up-regulated ZNF300 (Fig. 1). Furthermore, ZNF300 positively affected NF-κB signalling by up-regulating TRAF2 expression and binding IKK. We also showed that overexpression of ZNF300 in HeLa cells in the present of TNF-α treatment led to increased phosphorylation on IKK, although the enhancement on phosphorylated IKK from overexpression of ZNF300 alone was weak or barely detectable (Fig. 4E). These results suggest that ZNF300 plays important roles in promoting IKK phosphorylation, although it may need other cofactors to help exert such a role. Nevertheless, we have therefore identified a unique signalling regulator in the human NF-κB– dependent pathway (Fig. S2) and revealed several novel aspects of tumourigenesis and inflammation in humans.
Our findings highlight how human and mouse cells may behave differently, despite possessing conserved signalling pathways such as the NF-κB pathway. The NF-κB pathway is an important convergent point for various signalling pathways and participates in many physiological functions [27]. In this study, we identified ZNF300 as a novel regulatory factor promoting NF-κB pathway activity in human cells. This unique mechanism is not found in mouse cells because no ZNF300 gene or homologue has been identified in mouse cells. Such differences affect aspects of biological function such as the promotion of tumour colony formation in vitro and tumour formation in vivo by overexpression of ZNF300. Although the significance of such differences in tumour development remains unclear, we speculate that it may partially contribute to the inconsistent results found in many mouse models and clinical trials. It is known that many chemotherapeutic drugs that work well in mouse models have failed in clinical trials [28]. One can speculate that drug development based on mouse models may prove fruitless in humans due to these unique regulatory pathways that are only found in human and not in mouse cells. Our findings emphasize how cautious we should be when we evaluate the data from mouse studies. The extensive use of mouse models to approximate human cancer pathogenesis has yielded a wealth of insight into the mechanistic details of tumour progression in humans and mice. Nonetheless, the great evolutionary distance separating humans and mice has led to substantial differences in the biology of these two mammalian species [29]. Therefore, it is important to study the differences between mice and humans. These differences may reveal new targets or provide auxiliary targets to enhance existing tumour therapies.
Our study has shown that ZNF300 may bridge inflammation and tumourigenesis. Inflammation tightly correlates with tumourigenesis [30], and NF-κB is a critical factor mediating inflammation [31]. In this study, we found that proinflammatory factors such as TNF-α and LPS up-regulated ZNF300. Furthermore, overexpression of ZNF300 promoted colony formation in vitro and tumour formation in vivo, suggesting that ZNF300 may be a putative oncogene. This evidence may directly link proinflammatory factors to a putative oncogene. Our findings also reveal that overexpression of ZNF300 can further enhance NF-κB signalling by up-regulating TRAF2 and interacting with IKKβ. These pathways may each combine to exacerbate inflammation.
Our findings suggest that ZNF300 may be a novel oncogene in humans. Many multifunctional zinc finger proteins, such as CTCF, WT1, GLI, ZNF74 and A20, have been linked to human development and disease, usually acting as oncogenic or tumour suppressor proteins [5–8, 32]. Previous studies have shown that ZNF300 is associated with MDS [33] and Crohn’s disease [20]. Our data demonstrate that ZNF300 may promote proliferation by suppressing p21 and p27 and activating NF-κB and ERK. This mechanism may not be the only one by which ZNF300 contributes to human disease. As a zinc finger protein, ZNF300 potentially regulates more target genes. Studies on global target genes or interacting partners of ZNF300 may reveal a more complete picture of the ZNF300 regulatory network and add new knowledge on human embryo and tumour development.
Taken together, our findings suggest that ZNF300 acts as a potential oncogenic gene, is a downstream target gene of c-Ets-2 and is a potent activator of the NF-κB–dependent pathway, as depicted in Figure S2, ZNF300 is likely to be a novel molecular factor important in human embryogenesis, cancer progression and malignant transformation. This study can also provide insight into the mechanism of tumourigenesis caused by species-specific genes.
Acknowledgments
This work was supported by the China Postdoctoral Science Foundation (Nos. 2005038548) and Development 863-Program of China (2006AA02A306). We thank Z. Huang (Wuhan University) for helpful comments on the manuscript. TW, XGW, JHX designed the research study, performed the experiments and analysed the data; XPW and HLQ performed the experiments; HY analysed the data and wrote the paper; WXL designed the research study and wrote the paper. All authors approved the final version of the paper.
Conflict of interest
The authors confirm that there is no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
ZNF300 bind to the putative binding site (TTGGGGG, −19 to −27) in the TRAF2 promoter. EMSA analysis was preformed with HeLa nuclear extract (lanes 2, 3, 4, 5, 6, 7). Labelled WT probe (putative ZNF300 binding site) probe was added to all reactions (lanes 1, 2, 3, 4, 5, 6, 7). Unlabelled wild-type WT probe (lane 3) or unlabelled mutant probe (lane 4) was added during preincubation prior to probe addition. Anti-ZNF300 antibody (lane 6) or anti-GFP antibody (lane 7) was incubated with nuclear extracts before adding to the reaction.
ZNF300 acts as a potential oncogenic gene through regulating NF-κB-dependent pathway. ZNF300 acts as a potential oncogenic gene and is a downstream target gene of c-Ets-2. ZNF300 can enhance NF-κB signalling by up-regulating TRAF2 and interacting with IKKβ. ZNF300 also suppresses p21 and p27 and activates ERK, but the mechanisms need to be identified.
Gene specific primers and probes
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Miller J, McLachlan AD, Klug A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. Embo J. 1985;4:1609–14. doi: 10.1002/j.1460-2075.1985.tb03825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee MS, Gippert GP, Soman KV, et al. Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science. 1989;245:635–7. doi: 10.1126/science.2503871. [DOI] [PubMed] [Google Scholar]
- 3.Gamsjaeger R, Liew CK, Loughlin FE, et al. Sticky fingers: zinc-fingers as protein-recognition motifs. Trends Biochem Sci. 2007;32:63–70. doi: 10.1016/j.tibs.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Rubin GM, Yandell MD, Wortman JR, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–15. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. 2001;11:39–46. doi: 10.1016/s0959-440x(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 6.Barbaux S, Niaudet P, Gubler MC, et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. 1997;17:467–70. doi: 10.1038/ng1297-467. [DOI] [PubMed] [Google Scholar]
- 7.Ladomery M, Dellaire G. Multifunctional zinc finger proteins in development and disease. Ann Hum Genet. 2002;66:331–42. doi: 10.1017/S0003480002001215. [DOI] [PubMed] [Google Scholar]
- 8.De La Rosa-Velazquez IA, Rincon-Arano H, Benitez-Bribiesca L, et al. Epigenetic regulation of the human retinoblastoma tumour suppressor gene promoter by CTCF. Cancer Res. 2007;67:2577–85. doi: 10.1158/0008-5472.CAN-06-2024. [DOI] [PubMed] [Google Scholar]
- 9.Song XT, Evel-Kabler K, Shen L, et al. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14:258–65. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumour locus. Cell. 1990;60:509–20. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 11.Zhuang Z, Merino MJ, Vortmeyer AO, et al. Identical genetic changes in different histologic components of Wilms’ tumours. J Natl Cancer Inst. 1997;89:1148–52. doi: 10.1093/jnci/89.15.1148. [DOI] [PubMed] [Google Scholar]
- 12.Schmitz R, Hansmann ML, Bohle V, et al. TNFAIP3 (A20) is a tumour suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981–9. doi: 10.1084/jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebelein B, Fernandez-Zapico M, Imoto M, et al. KRAB-independent suppression of neoplastic cell growth by the novel zinc finger transcription factor KS1. J Clin Invest. 1998;102:1911–9. doi: 10.1172/JCI1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuramoto K, Uesaka T, Kimura A, et al. ZK7, a novel zinc finger gene, is induced by vascular endothelial growth factor and inhibits apoptotic death in hematopoietic cells. Cancer Res. 2000;60:425–30. [PubMed] [Google Scholar]
- 15.Qi CF, Martensson A, Mattioli M, et al. CTCF functions as a critical regulator of cell-cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc Natl Acad Sci USA. 2003;100:633–8. doi: 10.1073/pnas.0237127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gou D, Wang J, Gao L, et al. Identification and functional analysis of a novel human KRAB/C2H2 zinc finger gene ZNF300. Biochim Biophys Acta. 2004;1676:203–9. doi: 10.1016/j.bbaexp.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Qiu H, Xue L, Gao L, et al. Identification of the DNA binding element of the human ZNF300 protein. Cell Mol Biol Lett. 2008;13:391–403. doi: 10.2478/s11658-008-0005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue L, Qiu H, Ma J, et al. ZNF300, a recently identified human transcription factor, activates the human IL-2Rbeta promoter through the overlapping ZNF300/EGR1 binding site. Cell Mol Biol Lett. 2010;15:530–40. doi: 10.2478/s11658-010-0025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao H, Zhu C, Zhao Z, et al. KRAB-containing zinc finger gene ZNF268 encodes multiple alternatively spliced isoforms that contain transcription regulatory domains. Int J Mol Med. 2006;18:457–63. [PubMed] [Google Scholar]
- 20.Ferguson LR, Philpott M, Dryland P. Nutrigenomics in the whole-genome scanning era: Crohn’s disease as example. Cell Mol Life Sci. 2007;64:3105–18. doi: 10.1007/s00018-007-7303-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 23.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 24.Wertz IE, O’Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 25.Gartel AL, Shchors K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp Cell Res. 2003;283:17–21. doi: 10.1016/s0014-4827(02)00020-4. [DOI] [PubMed] [Google Scholar]
- 26.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 27.Tergaonkar V. NFkappaB pathway: a good signaling paradigm and therapeutic target. Int J Biochem Cell Biol. 2006;38:1647–53. doi: 10.1016/j.biocel.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 28.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 29.Rangarajan A, Weinberg RA. Opinion: comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer. 2003;3:952–9. doi: 10.1038/nrc1235. [DOI] [PubMed] [Google Scholar]
- 30.Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–25. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 32.Krig SR, Miller JK, Frietze S, et al. ZNF217, a candidate breast cancer oncogene amplified at 20q13, regulates expression of the ErbB3 receptor tyrosine kinase in breast cancer cells. Oncogene. 2010;29:5500–10. doi: 10.1038/onc.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu TX, Becker MW, Jelinek J, et al. Chromosome 5q deletion and epigenetic suppression of the gene encoding alpha-catenin (CTNNA1) in myeloid cell transformation. Nat Med. 2007;13:78–83. doi: 10.1038/nm1512. [DOI] [PubMed] [Google Scholar]
- 34.DiDonato JA, Hayakawa M, Rothwarf DM, et al. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 35.Freimoser FM, Jakob CA, Aebi M, et al. The MTT [3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide] assay is a fast and reliable method for colorimetric determination of fungal cell densities. Appl Environ Microbiol. 1999;65:3727–9. doi: 10.1128/aem.65.8.3727-3729.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 37.Guo MX, Wang D, Shao HJ, et al. Transcription of human zinc finger ZNF268 gene requires an intragenic promoter element. J Biol Chem. 2006;281:24623–36. doi: 10.1074/jbc.M602753200. [DOI] [PubMed] [Google Scholar]
- 38.Agata Y, Matsuda E, Shimizu A. Two novel Kruppel-associated box-containing zinc-finger proteins, KRAZ1 and KRAZ2, repress transcription through functional interaction with the corepressor KAP-1 (TIF1beta/KRIP-1) J Biol Chem. 1999;274:16412–22. doi: 10.1074/jbc.274.23.16412. [DOI] [PubMed] [Google Scholar]
- 39.Hagemeier C, Bannister AJ, Cook A, et al. The activation domain of transcription factor PU.1 binds the retinoblastoma (RB) protein and the transcription factor TFIID in vitro: RB shows sequence similarity to TFIID and TFIIB. Proc Natl Acad Sci USA. 1993;90:1580–4. doi: 10.1073/pnas.90.4.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kyriazis AP, Kyriazis AA, Scarpelli DG, et al. Human pancreatic adenocarcinoma line Capan-1 in tissue culture and the nude mouse: morphologic, biologic, and biochemical characteristics. Am J Pathol. 1982;106:250–60. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ZNF300 bind to the putative binding site (TTGGGGG, −19 to −27) in the TRAF2 promoter. EMSA analysis was preformed with HeLa nuclear extract (lanes 2, 3, 4, 5, 6, 7). Labelled WT probe (putative ZNF300 binding site) probe was added to all reactions (lanes 1, 2, 3, 4, 5, 6, 7). Unlabelled wild-type WT probe (lane 3) or unlabelled mutant probe (lane 4) was added during preincubation prior to probe addition. Anti-ZNF300 antibody (lane 6) or anti-GFP antibody (lane 7) was incubated with nuclear extracts before adding to the reaction.
ZNF300 acts as a potential oncogenic gene through regulating NF-κB-dependent pathway. ZNF300 acts as a potential oncogenic gene and is a downstream target gene of c-Ets-2. ZNF300 can enhance NF-κB signalling by up-regulating TRAF2 and interacting with IKKβ. ZNF300 also suppresses p21 and p27 and activates ERK, but the mechanisms need to be identified.
Gene specific primers and probes