

Abstract
Cocaine is a widely abused drug without an FDA-approved medication. It has been recognized as an ideal anti-cocaine medication to accelerate cocaine metabolism producing biologically inactive metabolites via a route similar to the primary cocaine-metabolizing pathway, i.e. human butyrylcholinesterase (BChE)-catalyzed hydrolysis. However, the native human BChE has a low catalytic activity against cocaine. We recently designed and discovered a BChE mutant (A199S/F227A/S287G/A328W/Y332G) with a high catalytic activity (kcat = 5700 min−1, KM = 3.1 μM) specifically for cocaine, and the mutant was proven effective in protecting mice from acute cocaine toxicity of a lethal dose of cocaine (180 mg/kg, LD100). Further characterization in animal models requires establishment of a high-efficiency stable cell line for the BChE mutant production in a relatively larger scale. It has been extremely challenging to develop a high-efficiency stable cell line expressing BChE or its mutant. In the present study, we successfully developed a stable cell line efficiently expressing the BChE mutant by using a lentivirus-based repeated-transduction method. The scale-up protein production enabled us to determine for the first time the in vivo catalytic activity and the biological half-life of this high-activity mutant of human BChE in accelerating cocaine clearance. In particular, it has been demonstrated that the BChE mutant (administrated to mice 1 min prior to cocaine) can quickly metabolize cocaine and completely eliminate cocaine-induced hyperactivity in rodents, implying that the BChE mutant may be developed as a promising therapeutic agent for cocaine abuse treatment.
Keywords: Enzyme, protein drug, protein production, drug abuse, stable cell line
Introduction
Cocaine is one of the most addictive drugs. Despite decades of efforts in developing effective anti-cocaine therapies, there is still no FDA-approved pharmacological treatment specific for cocaine overdose or addiction. Enzyme therapy has been recognized as an ideal approach to anti-cocaine medication for accelerating cocaine metabolism, producing biologically inactive metabolites via a route similar to the primary cocaine-metabolizing pathway, i.e. hydrolysis catalyzed by human butyrylcholinesterase (BChE).[1–3] Unfortunately, the native BChE has a low catalytic activity against naturally-occurring, biologically active (−)-cocaine (kcat = 4.1 min−1, KM = 4.5 μM). We developed unique computational strategies and protocols based on virtual screening of rate-determining transition states of the enzymatic reaction to design enzyme mutants with an improved catalytic activity.[4–9] The computational design was followed by in vitro experiments including site-directed mutagenesis, protein expression, and fast enzyme activity screening. The integrated computational-experimental studies have led to the discovery of a set of high-activity BChE mutants known as cocaine hydrolases.[4–10] One of our designed and discovered mutants, i.e. A199S/S287G/A328W/Y332G, [11–12] has been validated by an independent research group[13–16] and recognized as “a truly cocaine hydrolase with a catalytic efficiency that is 1000 folds greater than wild-type BChE”.[16] Our more recently reported A199S/F227A/S287G/A328W/Y332G mutant (denoted as CocH3 for convenience) demonstrated a ~2000-fold improved catalytic activity (kcat = 5700 min−1, KM = 3.1 μM)[6] against (−)-cocaine compared to the wild-type BChE. In the in vivo potency test, pretreatment of 10 μg CocH3 fully protected the mice from the acute toxicity of 180 mg/kg cocaine (LD100). These high-activity mutants of human BChE have been recognized as promising candidates of therapeutic enzymes for anti-cocaine medication.[1–3, 17] In the initial screening of those BChE mutants, the genes of the mutants were transiently expressed for the in vitro and in vivo examination. Further characterization of this enzyme in animal models requires a stable expression cell line efficiently producing the enzyme in large quantity.
Traditionally, generation of a stable cell line for therapeutic protein production begins with construction of an expression vector. A suitable vector (plasmid or virus) usually carries the gene for target protein and a metabolic selectable marker or an antibiotics selectable marker for the cell line of choice. After transfected with the vector, the cells are then grown under the selection pressure and screened for the expression level of the target protein. The high-productivity clones are selected and amplified for scale-up production. Here the integration position and plasmid copy number affect the productivity of the cells. One of the more recently developed strategies is site-specific integration. Since only 0.1~1% of genome region are actively transcribed, integration of the target gene into specific loci would ensure high and stable productivity.[18–20] Several recombinases, such as Cre and Flp, were utilized for this purpose because of their capability to identify specific sequences and to mediate the integration of the foreign gene for the therapeutic proteins.[18–19] In all the approaches mentioned above, the selection and screening for highly productive single clones is a time-consuming process which may take months.
Currently, Chinese Hamster Ovary (CHO) cells are used for production of about 70% of the therapeutic proteins.[20] They have been proven to be safe and able to provide efficient proper post-translational modification for proteins. However, CHO cells are also difficult to transfect and unstable for the foreign genes. So the commonly used forms of CHO cells are genetically modified, such as dihydrofolate reductase (DHFR)-deficit CHO cell lines DUXB11 and DG44.[19–20] The expression vector will carry both the gene encoding the lacked enzyme (such as DHFR) with an impaired promoter and the gene for expression of the target protein. After the transfection and metabolic selection, the target gene could be stabilized and amplified by addition of increasing dose of an inhibitor for the lacked enzyme (such as DHFR). Recombinase aided site-specific integration is also applied alone or in combination with the gene amplification system to achieve establishment of highly efficient stable cell lines.[18–19]
There have been many efforts in developing high-efficiency stable cell lines to produce BChE in a relatively larger scale. Traditional transfection-selection method has been used to generate stable CHO cell lines that yield about 3 to 5 mg/L pure BChE or mutant.[21] Insect cells, which can produce monoclonal antibody with the yield of 52 mg/L/day in batch culture[22] are generally considered as a highly efficient expression system to produce recombinant proteins. However, in expression of a truncated human BChE mutant, insect cells can only achieve 4 mg/L of production level.[23] It is difficult to generate cell lines that can stably express BChE or its mutant at a high level. Alternatively, transgenic animals/plants strategies[24–25] have succeeded in yielding recombinant BChE and mutants at high production levels. But the produced proteins have other problems. For example, recombinant BChE produced in transgenic goats has a short in vivo half-life.[24] In addition, it requires a long period of time to generate the transgenic animals or plants. It is challenging to develop an effective method for scale-up production of BChE or its mutant.
Here we report the use of a lentivirus-based repeated transduction approach which transduces the prepared lentivirus into CHO cells repeatedly for stable cell line generation. This approach has led us to successfully generate a stable cell line which can efficiently produce CocH3 (the high-activity mutant of human BChE). In addition, a stable human embryonic kidney (HEK) 293F cell line was also developed for comparison. The prepared CocH3 protein was characterized in vivo for its pharmacokinetic property and effectiveness in accelerating cocaine metabolism and eliminating cocaine-induced hyperactivity.
Results and Discussion
Productivity of the stable cell line
We first wanted to explore an efficient method to generate a high-productivity cell line stably expressing a BChE mutant. The BChE mutant cDNA in lentivirus plasmid was constructed into pCSC-SP-PW vector. The lentivirus was packed in 293FT cells by transfection of pCSC-SP-PW-BChE along with three other packaging plasmids and then purified by centrifugation. The CHO-S cells were loaded in 12-well plate and transduced by adding purified lentivirus repeatedly. After each infection, the yield of transduced cells was determined in a 9-day fermentation of 30 ml cell culture. The use of the lentivirus-based repeated transduction method resulted in a stable CHO-S cell line efficiently expressing CocH3 (BChE mutant). The production level is related to the number of times of transduction performed, as seen in Fig. 1. The productivity of the cell pool was increased after each additional round of transduction-recovery cycle until after the 7th time (Fig. 1B). The data suggested that, during the process, more copies of the target gene were integrated into the genome for enhanced target protein expression. After the 7th time, further transduction no longer increased the yield of the 9-day fermentation. In fact, after the 7th transduction, further transduction decreased the yield gradually. The decrease in the yield might be due to the possibility that the hot-spots of chromosomal loci were already saturated by the foreign genes after the 7th transduction and the random insert into other regions of genome might harm the health of the cells. The amount of produced enzyme was proportional to the incubation time during the first a few days when the nutrient was sufficient (Fig. 1A). Then the cells started to die due to lack of nutrients and accumulation of toxins like lactate. The production rate was gradually slowing down until the production level hit the plateau when all the cells died.
Figure 1.

Dependence of the CocH3 production rate on the number of rounds of transduction-recovery cycles. (A) Time course of the CocH3 production in the transduced cells. (B) CocH3 production rate in a 9-day fermentation versus the number of rounds of transduction-recovery cycles.
We also selected single clones from the pool and determined the yields associated with the single clones (Fig. 2). The single clones did not display significantly higher or lower yields in the 9-day fermentation test compared to the pool, suggesting that the cells were transduced rather homogenously such that almost all of the cells had a similar productivity. Based on this observation, in the future work, we may simply skip the step of the selection of single clones so as to save a lot of time during the development of stable cell lines for other BChE mutants.
Figure 2.

CocH3 production rates in a 9-day fermentation using the 7-time transduced cell pool and the single clones selected from the pool.
Since CHO cell is known to be difficult to sustain the expression level of foreign genes, we also evaluated the scalability and stability of the established CocH3-expressing cell line. For the stability test, the cells were seeded at 6 × 105 cells/ml after each passage and cultured for three days; then one eighth of them was passed while the rest were cultured continuously for 6 days to determine the yield of the 9-day fermentation. As seen in Fig. 3, after several times of passages, the cells did not significantly decrease the productivity. In the scalability test, the yield rate did not significantly change by increasing the volume of the cell culture from milliliters to liters. So, the cells were sustainable and scalable for the protein production.
Figure 3.

Stability of the CocH3 production using the stable CHO-S cells obtained from the 7-time transduction. The cells were passed several times. The yield was determined after each passage.
Depicted in Fig. 4 is nondenaturing gel (8%) stained for the BChE activity of the CocH3 materials expressed in the HEK293F cells (with 1 μM poly-L-proline in the medium) and the CHO-S cells. According to the data in Fig. 4A, CocH3 materials expressed in the HEK293 cells (with 1 μM poly-L-proline in the medium) and the CHO-S cells all predominantly existed in tetramer. For CocH3 expressed in the HEK293F cells (with 1 μM poly-L-proline in the medium), there was no band observed for the monomer or dimer. For CocH3 expressed in the CHO-S cells, a very weak (wide) band was noted for the monomer (with a negligible amount), but no dimer was noted. In comparison, weak (wide) bands were noted for both the monomer and dimer in CocH3 expressed in the transiently transfected CHO-S cells (with 1 μM poly-L-proline in the medium).
Figure 4.
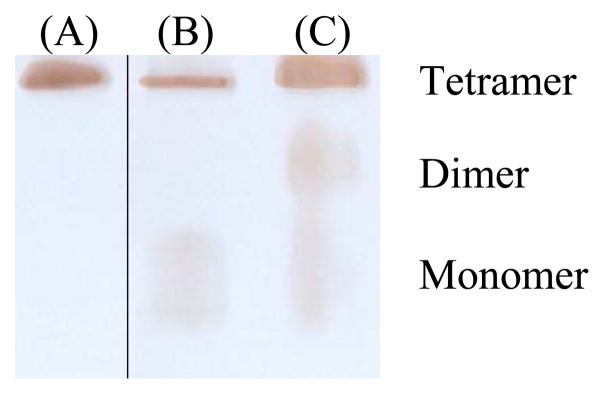
Nondenaturing gel (8%) stained for the BChE activity of the CocH3 materials expressed in (A) the stable HEK293F cells (with 1 μM poly-L-proline in the medium), (B) the stable CHO-S cells, and (C) the transiently transfected CHO-S cells (with 1 μM poly-L-proline in the medium). The three lanes came from the same gel where lane A was not adjacent to lane B. The gel was run with the constant current of 8 mA at 4°C overnight. The gel was stained for the BChE activity with butyrylthiocholine iodide as substrate at room temperature for 1 to 4 hr until the protein bands with the enzymatic activity were clearly identified.
Concerning the catalytic activity of the BChE mutant, according to our previously reported in vitro assay,[6] we determined that kcat = 5700 min−1 and KM = 3.1 μM for CocH3 expressed in HEK293F cells. The same in vitro assay (the sensitive radiometric assay) was employed to determine the enzymatic activity of CocH3 expressed in the stable CHO-S cells, showing no significant change in the catalytic activity (data not shown).
Pharmacokinetic profiles of CocH3
The CocH3 protein material expressed in the stable CHO-S cells was tested for its pharmacokinetic (PK) profile in rats. Rats (n=5) were administered intravenously (i.v., via tail vein) with 0.15 mg/kg of the purified enzyme. The blood was sampled at 2, 15, and 30 min, and 1, 2, 3, 5, 8, 12, 24, and 48 h after the enzyme injection. For comparison, the protein material expressed in the stable HEK293F cells was also tested in rats. Depicted in Fig. 5 are the time courses of the active enzyme concentrations after the i.v. injection of the enzyme materials. The measured time-dependent concentrations of the active enzyme were fitted to a well-known double exponential equation ([E]t = Ae−k1t + Be−k2t) which accounts for both the enzyme distribution process (the fast phase, associated with k1) and elimination process (the slow phase, associated with k2). The half-life associated with the enzyme elimination rate constant k2 is called the biological half-life (the usually referred in vivo half-life).
Figure 5.

Time-dependent concentrations of the active enzyme (CocH3) materials expressed in the CHO-S and HEK293F cells after the 5 mg/kg cocaine injection (i.v.). Rats (n=5) were injected with 0.15 mg/kg CocH3 expressed in the CHO-S cells. For comparison, the same dose of CocH3 expressed in the HEK293 cells was injected to rats. The enzyme concentrations were determined by using a sensitive radiometric assay.
The CHO cells-expressed CocH3 displayed a biological half-life of 7.3 hours, which is significantly longer than that (2.8 hours) of the same enzyme expressed in the HEK293F cells, in rats. The native human BChE (purified from human serum) has a half-life of 7~12 days in humans and 24 hours in rats.[26] Recombinant BChE has a much shorter biological half-life.[27] It should be noted that multiple factors could affect the biological half-life of a protein, including oligomerization and glycosylation. As the nondenaturing gel staining (Fig. 4) revealed that the CocH3 proteins expressed in the HEK293F and CHO-S cells were predominantly tetramer, there was no significant difference in oligomerization between the two protein forms. Thus, the difference in the biological half-life is mainly due to the difference in post-translational modification, particularly the glycosylation. The CHO cells-expressed CocH3 may have glycosylation closer to the native enzyme compared to that for the HEK293F cells-expressed CocH3.
Generally speaking, it is desirable to have a long in vivo half-life for the potential therapeutic protein in cocaine addiction treatment. The longer the enzyme can stay in the body, the longer it can protect the subjects, and the lower dosing and/or lower frequency the therapeutic protein would be needed for administration to the patients. It also lowered the potential risk of unexpected adverse effects, and increased the chances of full protection of patients against cocaine effects. Based on the data depicted in Fig. 5, future CocH3 production should use the stable CHO-S cell line.
Cocaine clearance accelerated by CocH3
In order to examine the in vivo potency of CocH3 for metabolizing cocaine, we characterized the pharmacokinetic profiles of cocaine clearance with and without the presence of CocH3 in rats by using a chromatographic assay. The data are depicted in Fig. 6. The rats (n=4) were injected with saline or 0.1 mg/kg CocH3, followed by i.v. injection of 5 mg/kg cocaine. The blood was sampled at 2, 5, 10, 15, 30, and 60 min after the cocaine injection. The blood samples were analyzed for the concentrations of cocaine and benzoic acid by using a High-Performance Liquid Chromatographic (HPLC) method. CocH3 can hydrolyze cocaine to produce benzoic acid and ecgonine methyl ester, and greatly accelerate the clearance of cocaine from the body. It has been known that the endogenous BChE in rats is very inefficient in metabolizing cocaine and, for this reason, cocaine was mainly metabolized by carboxylesterase in the blood to produce benzoylecgonine and methanol in rats.[28] The control curves in Fig. 6 reflect the overall effects of the all cocaine elimination pathways. As seen in Fig. 6, in the control rats, the average concentration of cocaine at the first time point (2 min) was 7.4 μM, while the average concentration of benzoic acid (metabolite) was 0.5 μM. In the presence of CocH3, the average concentration of cocaine at ~2 min in the blood sample was below the detectable level (Fig. 6A), while the average concentration of benzoic acid at the first time point (2 min) was 12.2 μM (Fig. 6B). Most of the cocaine was hydrolyzed by CocH3 between the i.v. cocaine injection and the first blood sampling at 2 min after the injection. The CocH3-caused dramatic changes in both the cocaine and benzoic acid concentrations clearly indicated that cocaine was metabolized rapidly to benzoic acid in the presence of CocH3.
Figure 6.

Cocaine clearance accelerated by CocH3. Benzoic acid (B) is the product of BChE- or CocH3-catalyzed hydrolysis of cocaine (A). Saline or 0.1 mg/kg CocH3 was injected i.v. in rats (n=4) 1 min before the i.v. injection of 5 mg/kg cocaine.
It should be mentioned that the total plasma concentration of cocaine and benzoic acid (12.2 μM) in the presence of CocH3 (when the benzoic acid concentration was higher) was higher than that (7.4 μM) in the absence of CocH3 (when the cocaine concentration was higher). This observation might be associated with the potentially different distribution volumes of cocaine and benzoic acid in the body. As well known, cocaine is an amine drug which can readily cross cell membranes under physiological condition, while benzoic acid primarily exists in the benzoate ion under physiological condition. So, benzoic acid is expected to have a relatively smaller distribution volume compared to cocaine.
Effects of CocH3 on the hyperactivity induced by cocaine
Development of stable cell lines for the scale-up protein production enabled us to characterize the potency of the enzyme in elimination of the physiological effect of cocaine. In this study, mice (n=6) were injected with cocaine alone, saline alone, or the BChE mutant (CocH3) 1 min before 25 or 90 mg/kg of i.p. cocaine. The mice were returned to the cages and recorded for their locomotor activity for the first 10 min after the cocaine injection. The first cocaine dose used for our locomotor activity tests in mice was 25 mg/kg (i.p.). As seen in Fig. 7, without pretreatment of the enzyme, 25 mg/kg cocaine (i.p.) induced rather strong hyperactivity in mice. With pretreatment of 1.5 mg/kg CocH3 (i.v.), the mice had a slight hyperactivity between 2 and 5 min and then returned back to the baseline level of activity. With pretreatment of 2 mg/kg CocH3 (i.v.), 25 mg/kg cocaine (i.p.) did not induce any significant hyperactivity in mice, suggesting that the minimum dose of CocH3 required to completely block the hyperactivity induced by 25 mg/kg cocaine (i.p.) was 2 mg/kg.
Figure 7.

Effects of the exogenous enzyme (CocH3) on cocaine-induced hyperactivity of mice. Saline or enzyme was injected i.v. through tail veins of mice 1 min before i.p. injection of saline or cocaine. Five dose conditions were tested using five groups of mice, and each group had six mice (n=6). Group a were treated with i.v. saline and i.p. saline; Group b were treated with i.v. saline and i.p. 25 mg/kg cocaine; Group c were treated with i.v. 1.5 mg/kg CocH3 and i.p. 25 mg/kg cocaine; Group d were treated with i.v. 2 mg/kg CocH3 and i.p. 25 mg/kg cocaine; Group e were treated with i.v. 5 mg/kg and i.p. 90 mg/kg cocaine.
We further increased the dose of cocaine to 90 mg/kg (LD50 for i.p. cocaine), and noted that 90 mg/kg cocaine (i.p.) did not induce any significant hyperactivity (or any sign of toxicity in our further observation after the hyperactivity tests) in the mice pretreated with 5 mg/kg CocH3 as seen in Fig. 7. The observation suggests that the physiological effect of such a lethal dose of cocaine can be blocked completely by pretreatment of 5 mg/kg CocH3.
The concept of a possible enzyme therapy for cocaine abuse treatment is based on a hypothesis that the effects of cocaine are dependent on how intensely and how quickly cocaine gets to the brain, and that the therapeutic enzyme can alter the pharmacokinetics of cocaine in a favorable manner by rapidly metabolizing cocaine. In this way, a therapeutic enzyme could reduce cocaine’s entry into the brain to an amount (threshold) that is too low to produce detectable physiological effects. When the cocaine concentration in brain dose not reach the “threshold” value because of the presence of an efficient enzyme in plasma, one may consider that the enzyme has effectively blocked cocaine from reaching brain. Further, the threshold concentration of cocaine in brain is related to the degree of the DAT occupancy by cocaine. Volkow et al. demonstrated that, for humans, “at least 47% of dopamine transporter has to be blocked for subjects to perceive cocaine’s effects”.[29] The threshold concentration of cocaine in brain required to produce physiological effects was estimated to be 0.22±0.07 μM in light of a recently reported cocaine pharmacokinetic modeling.[30] The data in Fig. 7 indicate that, in the presence of the exogenous cocaine hydrolase (5 mg/kg), cocaine can be degraded so rapidly that even a lethal dose of cocaine (90 mg/kg, i.p.) did not produce a brain cocaine concentration being greater than the threshold (0.22±0.07 μM) required to induce detectable physiological effects; the animals did not show any detectable behavioral abnormality. The data indicate that it is possible to completely eliminate cocaine’s physiological effects by administration of a highly efficient cocaine hydrolase, providing a proof of the principle for the desirable enzyme therapy for cocaine abuse treatment.
According to previous studies,[31] one-time use of cocaine could increase dopamine transporter (the primary target protein for cocaine) expression on the cell surface for months so that it takes a long time for the brain’s communication system returning to normal. Repeated administration of cocaine would reinforce the effects on the expression levels of transporters/receptors and continuously change the circuits in the brain. Hence, for effective treatment of cocaine addiction, it is essentially important to completely protect the brain from the cocaine effect so that the brain can gradually recover to function normally. It would be interesting to further test the cocaine hydrolase in a well-established animal addiction model including cocaine self-administration in the near future.
Conclusion
In the present study, we have successfully developed a stable cell line efficiently expressing a promising cocaine hydrolase, CocH3 (the A199S/F227A/S287G/A328W/Y332G mutant of human BChE, with a yield of 1.4 pg/cell/day), by using a lentivirus-based repeated-transduction method. This is the first report to use lentivirus vector to express human BChE or its mutant. The scale-up protein production enabled us to characterize the cocaine hydrolase in rodents concerning the biological half-life and potency in accelerating cocaine clearance. In particular, it has been demonstrated that the exogenous enzyme (cocaine hydrolase) can rapidly metabolize cocaine and completely eliminate cocaine-induced hyperactivity in rodents. The administration of CocH3 can completely eliminate the physiological effects of cocaine, providing a proof of the principle for a desirable enzyme therapy using CocH3 for cocaine abuse treatment.
Materials and Methods
Preparation of lentivirus encoding the gene of the BChE mutant
The BChE mutant (CocH3) cDNA in lentivirus plasmid was constructed into pCSC-SP-PW vector at ApaI and XhoI sites. The A199S/F227A/S287G/A328W/Y332G mutations were generated on cDNA of full-length human BChE (access number: P06276 in the Swiss Protein Database) on pRC/CMV-BChE by using site-directed mutagenesis. The CocH3 gene was amplified by PCR with pRC/CMV–CocH3 as a template. The forward primer is gagggcccaaggtgcacggcccacgt (ApaI), and backward primer is ccgctcgagttagagacccacacaactttct (XhoI). Then the CocH3 cDNA was ligated with pCSC-SP-PW fragment that was double digested by ApaI and XhoI. The sequence of construct was confirmed by DNA sequencing. To package the lentivirus particles carrying the gene of CocH3, HEK293FT cells were cultured in DMEM-10% Fetal Bovine Serum (FBS) (Life Technologies, Grand Island, NY) with 250 ng/ml G418 (Life Technologies, Grand Island, NY). Cells were transfected at approximately 70% confluence by lentivirus plasmid encoding CocH3 (CocH3/pCSC-SP-PW) along with three other packaging plasmids, pMDL-pg.RRE, pRSV.rev, and pVSVG, at a mass ratio of 10:6.5:2.5:3.5. Transfection was achieved by lipofection. In brief, for a 10 cm dish of cells, total DNAs about 22.5 μg were mixed first, and then diluted in 1.5 ml of Opti-MEM® I Reduced Serum Medium (Invitrogen) without serum. 60 μl of Lipofetamine™ 2000 (Invitrogen) was then mixed with 1.5 ml of Opti-MEM® I Reduced Serum Medium and incubated at room temperature for 5 min. The diluted DNAs and Lipofetamine™ 2000 were mixed and incubated at room temperature for 20 min before added dropwise onto the cell culture. The cells were cultured at 3% CO2 at 37°C. Culture medium was changed 12 to 16 h after transfection. The medium was collected three times at a 24 h-interval beginning 24 h after the post-transfection change of medium. The medium was filtered through a 0.45-μm cellulose acetate filter and spun in Beckman SW28 rotor at 19,400 rpm for 2 h at 4°C to pellet the virus particles. Lentivirus was then suspended in Hank’s balanced salt solution and aliquoted to be stored at −80°C. The physical concentration of lentivirus was determined by using QuickTiter™ lentivirus rapid quantitation kit (Cell Biolabs, San Diego, CA).
Generation of stable cell line by lentivirus infection
Scale-up preparation of enzyme was first achieved by infecting CHO-S cells with lentivirus followed by resuspending attached CHO-S cells in protein free suspension culture in Gibco® FreeStyle™ CHO expression medium (Life Technologies, Grand Island, NY) with 8 mM L-glutamine. The day before infection, the cells were loaded at 1 × 105 cells/ml in 12-well plate and cultured steadily in freestyle CHO expression medium with 8 mM L-glutamine and 1% FBS. Cells began to attach to the plate soon in the presence of FBS after the change of culture condition. Lentivirus was then added to infect the cells for one day. The cross linker polybrene (Santa Cruz Biotechnology, Santa Cruz, CA) was also added to cell culture to increase the infection efficiency by neutralizing the charge repulsion between the virus and cell culture. The infection was stopped by change of medium. The infected cells were allowed to recover from the infection for one to two days (or more days, depending on the status of the cells), then were suspended by 0.05% trypsin-EDTA and split into two halves. One half was cultured in a 6-well plate, and then transferred to a 10 cm plate in 1% FBS freestyle CHO expression medium. Then the cells were changed back to suspension culture, and the culture volume increased from 6-well plate to 125-ml shake flask for yield determination. The other half was seeded in a 12-well plate for the next round of infection. After each infection, the yield was determined for a 9-day fermentation of 30 ml cell culture. The pool with the highest yield was selected and amplified for scale-up production of the enzyme. Culture medium was changed every two to three days, and collected to store at 4°C. The scalability and stability of the production was also evaluated during the process. The stable cell line in HEK293F was also generated by using the same method. For the difference, the HEK293F cells were cultured in Gibco® FreeStyle™ 293 medium (Life technologies, Grand Island, NY), with addition of 1 μM poly-L-proline (Sigma Aldrich, St. Louis, MO) to facilitate the formation of the CocH3 tetramer.
To compare the productivity of the pool with that of single clones, the cells from the pool were seeded at 2–10 cells/well in 96-well plates in free-style CHO expression medium with 1% FBS to select the single clones. The cells were cultured for another 14 to 21 days without changing medium and shaking until single clones clearly appeared. Single clone cell lines were chosen to culture in 48-well plates, then 12-well plates and 6-well plates in 1% FBS-freestyle CHO expression medium. High-expression single clones were selected by determining the enzyme activity in the medium. Selected cell-line cells were then changed to suspension culture and the culture volume was changed from 6-well plate to 125-ml flask. The yields of single clones were determined for a 9-day fermentation of 30 ml cell culture.
Enzyme purification
Scale-up purification of the enzyme in the medium was achieved by a two-step purification using ion exchange chromatography followed by affinity chromatography. In brief, the crude medium was diluted with the same volume of 20 mM Tris-HCl, pH 7.4. Equilibrated QFF anion exchanger was added to diluted medium in 1% of its volume and incubated at 4°C with occasional stirring for 1 h. More than 95% enzyme activity was found to bind to the resin after the incubation. The suspension was then packed in a column and the medium was allowed to flow through rapidly with the aid of suction of (50–100 ml /min). The QFF resin was repacked again in a washing buffer after the entire medium was excluded. After washing the column with 20 mM Tris-HCl, pH 7.0, overnight at 4°C, the enzyme was eluded by 20 mM Tris-HCl, pH 7.0, plus 0.3 M NaCl. The eluate was desalted to 20 mM Tris-HCl, pH 7.0, by Millipore centrifugal filter device. The desalted eluate was applied to a hydroxyapatite column (Clarkson Chem. Co., Williamsport, PA) (2.5 × 22 cm), which was packed with fibrous cellulose powder CF11 at a ratio of 1:1. The column was washed by 20 mM Tris-HCl, pH 7.0, and then the enzyme was eluted by 10 mM sodium phosphate buffer, pH 7.0, containing 0.3 M NaCl. The purified enzyme was dialyzed against phosphate-buffered saline and stored at 4 °C or −80 °C. The purified enzyme had a catalytic activity of 34 U/mg against (−)cocaine.
Nondenaturing gel electrophoresis
Activity-stained nondenaturing polyacrylamide gel was ultilized to estimate the relative amount of tetramers, dimers, and monomers. 4% polyacrylamide stacking gel and 8 % separating gel were prepared in a Biorad gel apparatus. Electrophoresis was at 8 mA constant current for 6 h at 4 °C. The gel was stained for BChE activity against 2 mM butyrylthiocholine iodide by using the method described by Karnovsky and Roots.[32]
Subjects for in vivo studies
Sprague-Darley (male or female) rats (200–250 g) and male CD-1 mice (27–30 g) were ordered from Harlan (Harlan, Indianapolis, IN). The rats were housed initially in 2 to 4 rats per cage. The mice were housed in groups of 2 to 5 mice per page. All the animals were allowed ad libitum access to food and water and were maintained on a 12-hour light and dark cycle with lights on at 8 AM in a room kept at a temperature of 21 to 22°C. Each animal was only used once. Experiments were performed in same colony room in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. The experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky.
Determination of in vivo half-life in rats
Rats were injected intravenously (i.v., via tail vein) with 0.15 mg/kg of the purified enzyme expressed in the stable CHO-S or HEK293F cells. The saphenous veins were punctured with a needle. About 50 to 75 μl of blood was collected into a capillary tube at various time points after the enzyme injection. The blood samples were centrifuged at 5000 g for 15 min. The serum was tested for the enzyme activity using a radiometric method as described in our previous reports.[6–7, 10] The data for elimination of the enzyme from the circulation were fitted to a double-exponential equation which was described by Kronman.[33]
Characterization of cocaine clearance accelerated by CocH3
General anesthetic isoflurane was utilized with nose cone during the course of study. Four rats were injected with saline through tail vein 1 min before i.v. injection of 5 mg/kg cocaine, and other four rats were injected with 0.1 mg/kg of CocH3 followed by i.v. injection of the same dose of cocaine. About 50 to 75 μl of blood from saphenous veins was collected into capillary tubes and immediately diluted in 100 μl of 250 μM paraoxon at 2, 5, 10, 15, 30, and 60 min after the i.v. injection of cocaine. Paraoxon is an irreversible BChE inhibitor that can stop the enzymatic hydrolysis of cocaine between sampling and analysis. The diluted blood samples were stored at −70°C and assayed by using a High-Performance Liquid Chromatographic (HPLC) method.
Benzoic acid is the product of cocaine hydrolysis by the enzyme. The standard cocaine and benzoic acid were purchased through Sigma Aldrich (Sigma Aldrich, St. Louis, MO). Cocaine and benzoic acid concentrations in the blood samples were assayed by the following method. The frozen whole blood samples were thawed on ice for 3 hours. Then 150 μl of mobile phase (74% acetonitrile and 0.26% TFA) was mixed with each sample. Then 50 μl of 7% HClO4 was added to break the blood cell membrane. The mixture was vortexed for 1 min and then centrifuged at 25,000 g for 15 min, and the supernatant was transferred to an autosampler vial of which 200 μl was injected into the chromatographic system.
Chromatography was performed using a Waters 1525 binary HPLC pump (Waters Corporation, Milford, MA), a Waters 2487 dual λ absorbance detector, a Waters 2475 multi λ fluorescence detector, and a Waters 717 plus autosampler. The mobile phase is 74% acetonitrile and 0.26 % TFA. The flow rate was 1 ml/min. The eluent was monitored at 230 nm for absorbance of benzoic acid and at 465 nm for fluoresce of cocaine when exciting at 383 nm. The cocaine peaks appeared at 10.5 min, and the benzoic acid peaks occurred at 14.5 min. The quantification of cocaine and benzoic acid was performed by comparing the corresponding HPLC peak areas with those of authentic standards.
Locomotor activity
The mice were acclimated to live individually in the cages for at least 1 hour. Mice were injected with cocaine alone, saline alone, or the BChE mutant (CocH3) 1 min before 25 or 90 mg/kg of i.p. cocaine. The CocH3 doses were varied from 1.5 to 5 mg/kg (i.v.). The mice were returned to cages after the i.p. injection of cocaine, and recorded for their locomotor activities for 10 min. The distances travelled within the time were evaluated.
Acknowledgments
This work was supported by the NIH (grants R01DA035552, R01 DA032910, R01 DA013930, and R01 DA025100 to Zhan).
Abbreviations used
- BChE
butyrylcholinesterase
- FDA
U.S. Food and Drug Administration
- CocH
cocaine hydrolase
- CocH3
the A199S/F227A/S287G/A328W/Y332G mutant of human BChE
- CHO
Chinese Hamster Ovary
- DHFR
dihydrofolate reductase
- HEK
human embryonic kidney
- HPLC
High-Performance Liquid Chromatographic
- FBS
Fetal Bovine Serum
- IACUC
Institutional Animal Care and Use Committee
- PK
pharmacokinetics
- NIH
National Institutes of Health
Footnotes
AUTHOR CONTRIBUTION
Liu Xue prepared the lentivirus encoding the gene of the enzyme for development of stable cell lines, infected CHO-S cells with the lentivirus, performed HPLC analysis and the in vivo studies, and drafted the paper. Shurong Hou performed in the in vivo studies together with Liu Xue. Min Tong prepared the lentiviral vector and infected cells with the lentivirus in early stage of the work. Lei Fang ran the activity-stained nondenaturing polyacrylamide gel and participated in the locomotor activity experiments. Xiabin Chen participated in the HPLC analysis. Zhenyu Jin performed cell culture and protein purification. Hsin-Hsiung Tai supervised Min Tong for the work related to lentivirus and stable cell lines. Fang Zheng and Chang-Guo Zhan designed the project, with Chang-Guo Zhan finalizing the paper.
References
- 1.Zheng F, Zhan CG. Recent progress in protein drug design and discovery with a focus on novel approaches to the development of anti-cocaine medications. Future Med Chem. 2009;1:515–528. doi: 10.4155/fmc.09.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng F, Zhan CG. Enzyme-therapy approaches for the treatment of drug overdose and addiction. Future Med Chem. 2011;3:9–13. doi: 10.4155/fmc.10.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng F, Zhan CG. Are pharmacokinetic approaches feasible for treatment of cocaine addiction and overdose? Future Med Chem. 2012;4:125–128. doi: 10.4155/fmc.11.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan Y, Gao D, Yang W, Cho H, Yang G, Tai HH, Zhan CG. Computational redesign of human butyrylcholinesterase for anticocaine medication. Proc Natl Acad Sci USA. 2005;102:16656–16661. doi: 10.1073/pnas.0507332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang W, Xue L, Fang L, Chen X, Zhan CG. Characterization of a high-activity mutant of human butyrylcholinesterase against (−)-cocaine. Chem Biol Interact. 2010;187:148–152. doi: 10.1016/j.cbi.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng F, Yang W, Ko MC, Liu J, Cho H, Gao D, Tong M, Tai HH, Woods JH, Zhan CG. Most efficient cocaine hydrolase designed by virtual screening of transition states. J Am Chem Soc. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Y, Gao D, Yang W, Cho H, Zhan CG. Free Energy Perturbation (FEP) Simulation on the Transition States of Cocaine Hydrolysis Catalyzed by Human Butyrylcholinesterase and Its Mutants. J Am Chem Soc. 2007;129:13537–13543. doi: 10.1021/ja073724k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan Y, Gao D, Zhan CG. Modeling the Catalysis of Anti-Cocaine Catalytic Antibody: Competing Reaction Pathways and Free Energy Barriers. J Am Chem Soc. 2008;130:5140–5149. doi: 10.1021/ja077972s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W, Pan Y, Zheng F, Cho H, Tai HH, Zhan CG. Free-Energy Perturbation Simulation on Transition States and Redesign of Butyrylcholinesterase. Biophys J. 2009;96:1931–1938. doi: 10.1016/j.bpj.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue L, Ko MC, Tong M, Yang W, Hou S, Fang L, Liu J, Zheng F, Woods JH, Tai HH, Zhan CG. Design, preparation, and characterization of high-activity mutants of human butyrylcholinesterase specific for detoxification of cocaine. Mol Pharmacol. 2011;79:290–297. doi: 10.1124/mol.110.068494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan Y, Gao D, Yang W, Cho H, Yang G, Tai HH, Zhan CG. Computational redesign of human butyrylcholinesterase for anticocaine medication. Proc Natl Acad Sci USA. 2005;102:16656–16661. doi: 10.1073/pnas.0507332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W, Xue L, Fang L, Chen X, Zhan CG. Characterization of a high-activity mutant of human butyrylcholinesterase against (−)-cocaine. Chem Biol Interact. 2010;187:148–152. doi: 10.1016/j.cbi.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll ME, Zlebnik NE, Anker JJ, Kosten TR, Orson FM, Shen X, Kinsey B, Parks RJ, Gao Y, Brimijoin S. Combined cocaine hydrolase gene transfer and anti-cocaine vaccine synergistically block cocaineinduced locomotion. PLoS One. 2012;7:e43536. doi: 10.1371/journal.pone.0043536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brimijoin S, Orson F, Kosten TR, Kinsey B, Shen XY, White SJ, Gao Y. Anti-cocaine antibody and butyrylcholinesterase-derived cocaine hydrolase exert cooperative effects on cocaine pharmacokinetics and cocaine-induced locomotor activity in mice. Chem Biol Interact. 2013;203:212–216. doi: 10.1016/j.cbi.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y, Geng L, Orson F, Kinsey B, Kosten TR, Shen X, Brimijoin S. Effects of anti-cocaine vaccine and viral gene transfer of cocaine hydrolase in mice on cocaine toxicity including motor strength and liver damage. Chem Biol Interact. 2013;203:208–211. doi: 10.1016/j.cbi.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brimijoin S, Gao Y, Anker JJ, Gliddon LA, Lafleur D, Shah R, Zhao Q, Singh M, Carroll ME. A cocaine hydrolase engineered from human butyrylcholinesterase selectively blocks cocaine toxicity and reinstatement of drug seeking in rats. Neuropsychopharmacology. 2008;33:2715–2725. doi: 10.1038/sj.npp.1301666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng F, Zhan CG. Structure-and-mechanism-based design and discovery of therapeutics for cocaine overdose and addiction. Org Biomol Chem. 2008;6:836–843. doi: 10.1039/b716268e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Liu ZG, Sun ZW, Huang Y, Yu WY. Generation of stable cell lines by site-specific integration of transgenes into engineered Chinese hamster ovary strains using an FLP-FRT system. J Biotech. 2010;147:122–129. doi: 10.1016/j.jbiotec.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotech. 2012;93:917–930. doi: 10.1007/s00253-011-3758-5. [DOI] [PubMed] [Google Scholar]
- 20.Li F, Vijayasankaran N, Shen AY, Kiss R, Amanullah A. Cell culture processes for monoclonal antibody production. mAbs. 2010;2:466–479. doi: 10.4161/mabs.2.5.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nachon F, Nicolet Y, Viguie N, Masson P, Fontecilla-Camps JC, Lockridge O. Engineering of a monomeric and low-glycosylated form of human butyrylcholinesterase: expression, purification, characterization and crystallization. European J Biochem. 2002;269:630–637. doi: 10.1046/j.0014-2956.2001.02692.x. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Hu H, Yang J, Wang F, Kaisermayer C, Zhou P. High yield of human monoclonal antibody produced by stably transfected Drosophila schneider 2 cells in perfusion culture using wave bioreactor. Mol Biotech. 2012;52:170–179. doi: 10.1007/s12033-011-9484-5. [DOI] [PubMed] [Google Scholar]
- 23.Brazzolotto X, Wandhammer M, Ronco C, Trovaslet M, Jean L, Lockridge O, Renard PY, Nachon F. Human butyrylcholinesterase produced in insect cells: huprine-based affinity purification and crystal structure. FEBS J. 2012;279:2905–2916. doi: 10.1111/j.1742-4658.2012.08672.x. [DOI] [PubMed] [Google Scholar]
- 24.Huang YJ, Huang Y, Baldassarre H, Wang B, Lazaris A, Leduc M, Bilodeau AS, Bellemare A, Cote M, Herskovits P, Touati M, Turcotte C, Valeanu L, Lemee N, Wilgus H, Begin I, Bhatia B, Rao K, Neveu N, Brochu E, Pierson J, Hockley DK, Cerasoli DM, Lenz DE, Karatzas CN, Langermann S. Recombinant human butyrylcholinesterase from milk of transgenic animals to protect against organophosphate poisoning. Proc Natl Acad Sci USA. 2007;104:13603–13608. doi: 10.1073/pnas.0702756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larrimore KE, Barcus M, Kannan L, Gao Y, Zhan CG, Brimijoin S, Mor T. Plants as a source of butyrylcholinesterase variants designed for enhanced cocaine hydrolase activity. Chem Biol Interact. 2013;203:217–220. doi: 10.1016/j.cbi.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun H, Shen ML, Pang YP, Lockridge O, Brimijoin S. Cocaine metabolism accelerated by a re-engineered human butyrylcholinesterase. J Pharmacol Exp Therap. 2002;302:710–716. doi: 10.1124/jpet.302.2.710. [DOI] [PubMed] [Google Scholar]
- 27.Duysen EG, Bartels CF, Lockridge O. Wild-type and A328W mutant human butyrylcholinesterase tetramers expressed in Chinese hamster ovary cells have a 16-hour half-life in the circulation and protect mice from cocaine toxicity. J Pharmacol Exp Therap. 2002;302:751–758. doi: 10.1124/jpet.102.033746. [DOI] [PubMed] [Google Scholar]
- 28.Sun H, Shen ML, Pang YP, Lockridge O, Brimijoin S. Cocaine Metabolism Accelerated by a Re-Engineered Human Butyrylcholinesterase. J Pharmacol Exp Therap. 2002;302:710–716. doi: 10.1124/jpet.302.2.710. [DOI] [PubMed] [Google Scholar]
- 29.Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, Hitzemann R, Shea CE. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature. 1997;386:827–830. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- 30.Zheng F, Zhan CG. Modeling of pharmacokinetics of cocaine in human reveals the feasibility for development of enzyme therapies for drugs of abuse. PLoS Comput Bio. 2012;8:e1002610. doi: 10.1371/journal.pcbi.1002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt KC, Reith ME. Regulation of the dopamine transporter: aspects relevant to psychostimulant drugs of abuse. Ann New York Acad Sci. 2010;1187:316–340. doi: 10.1111/j.1749-6632.2009.05148.x. [DOI] [PubMed] [Google Scholar]
- 32.Karnovsky MJ, Roots L. A “Direct-Coloring” Thiocholine Method For Cholinesterases. J Histochem Cytochem. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- 33.Kronman C, Chitlaru T, Elhanany E, Velan B, Shafferman A. Hierarchy of post-translational modifications involved in the circulatory longevity of glycoproteins. Demonstration of concerted contributions of glycan sialylation and subunit assembly to the pharmacokinetic behavior of bovine acetylcholinesterase. J Biol Chem. 2000;275:29488–29502. doi: 10.1074/jbc.M004298200. [DOI] [PubMed] [Google Scholar]