

Conspectus
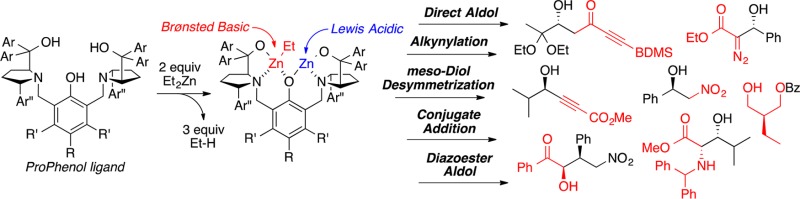
The development of catalytic enantioselective transformations has been the focus of many research groups over the past half century and is of paramount importance to the pharmaceutical and agrochemical industries. Since the award of the Nobel Prize in 2001, the field of enantioselective transition metal catalysis has soared to new heights, with the development of more efficient catalysts and new catalytic transformations at increasing frequency. Furthermore, catalytic reactions that allow higher levels of redox- and step-economy are being developed. Thus, alternatives to asymmetric alkene dihydroxylation and the enantioselective reduction of α,β-unsaturated ketones can invoke more strategic C–C bond forming reactions, such as asymmetric aldol reactions of an aldehyde with α-hydroxyketone donors or enantioselective alkynylation of an aldehyde, respectively. To facilitate catalytic enantioselective addition reactions, including the aforementioned aldol and alkynylation reactions, our lab has developed the ProPhenol ligand.
In this Account, we describe the development and application of the ProPhenol ligand for asymmetric additions of both carbon- and heteroatom-based nucleophiles to various electrophiles. The ProPhenol ligand spontaneously forms chiral dinuclear metal complexes when treated with an alkyl metal reagent, such as Et2Zn or Bu2Mg. The resulting complex contains both a Lewis acidic site to activate an electrophile and a Brønsted basic site to deprotonate a pronucleophile. Initially, our research focused on the use of Zn-ProPhenol complexes to facilitate the direct aldol reaction. Fine tuning of the reaction through ligand modification and the use of additives enabled the direct aldol reaction to proceed in high yields and stereoselectivities with a broad range of donor substrates, including acetophenones, methyl ynones, methyl vinyl ketone, acetone, α-hydroxy carbonyl compounds, and glycine Schiff bases. Additionally, an analogous magnesium ProPhenol complex was used to facilitate enantioselective diazoacetate aldol reactions with aryl, α,β-unsaturated, and aliphatic aldehydes.
The utility of bimetallic ProPhenol catalysts was extended to asymmetric additions with a wide range of substrate combinations. Effective pronucleophiles include oxazolones, 2-furanone, nitroalkanes, pyrroles, 3-hydroxyoxindoles, alkynes, meso-1,3-diols, and dialkyl phosphine oxides. These substrates were found to be effective with a number of electrophiles, including aldehydes, imines, nitroalkenes, acyl silanes, vinyl benzoates, and α,β-unsaturated carbonyls. A truly diverse range of enantioenriched compounds have been prepared using the ProPhenol ligand, and the commercial availability of both ligand enantiomers makes it ideally suited for the synthesis of complex molecules. To date, enantioselective ProPhenol-catalyzed reactions have been used in the synthesis of more than 20 natural products.
Introduction
The efficient and selective synthesis of chiral compounds for which asymmetric catalysis has played a pivotal role is essential to the pharmaceutical and agrochemical industries.1 Studies on how these chiral catalysts induce enantioselectivity have inspired the evolution of new chiral catalysts and the development of subsequent generations of enantioselective reactions.
The ProPhenol ligand (1a) is a salient member of the aza-crown family of ligands,2 whose origin was inspired by the pioneering work of Cram on chiral crown compounds.3 Proline-derived 1a forms dinuclear main group metal catalysts via the direct deprotonation of three hydroxyl groups (Scheme 1). Both gas titration experiments and electrospray mass spectrometry (ESI-MS) have been used to support the formation of the Zn-ProPhenol complex 2 from diethyl zinc and 1a.4 Furthermore, Ding and co-workers were able to characterize a p-nitrophenoxy derivative (3) of compound 2 by X-ray crystallography (Scheme 2).5 In addition, the crystal structure of an analogous dinuclear magnesium ProPhenol complex has also been reported.6 The ProPhenol ligand has been used to ligate a number of different main group metals including lithium, magnesium, zinc, zinc/magnesium (heterobimetallic), cobalt, and bismuth (vide infra).7 These bimetallic complexes contain both a Lewis acidic and Brønsted basic site and are capable of activating both an electrophile and a nucleophile within the chiral pocket created by the two diaryl(oxy)methyl groups (Figure 1).
Scheme 1. Formation of Bimetallic Zn-ProPhenol Complexes.

Scheme 2. X-ray Crystal Structure of Zn-ProPhenol Complex 3.
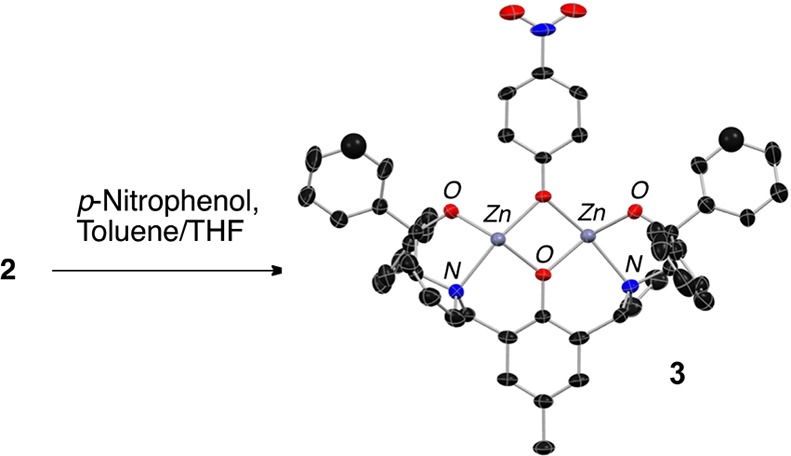
THF omitted for clarity.
Figure 1.
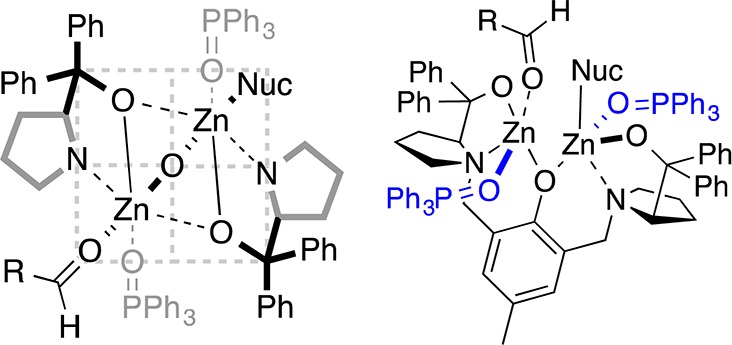
Quadrant view of the zinc-prophenol chiral pocket and additive coordination.
The concise and modular route used to prepare the ProPhenol ligand has enabled tailoring of the chiral pocket for specific reactions by introducing additional substituents on the pyrrolidine rings and the aryl ring (vide infra). A facile procedure has been developed for the recovery of the ProPhenol ligand 1a on multigram scale, whereby the bis-sodium salt is filtered off and 1a is recovered in 95% yield.8
In general, dual catalysis has proven a powerful approach to asymmetric catalysis.9 However, ProPhenol complexes have the added benefit of dictating the orientation of both electrophile and nucleophile by binding the two substrates within the same chiral pocket. Formation of this type of ternary complex followed by product dissociation bears a striking resemblance to the catalytic mechanism used by a number of enzymes.
Enantioselective Direct Aldol Reaction
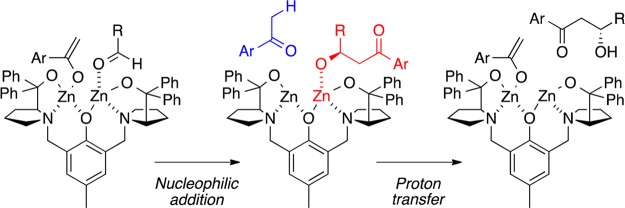
At the time that we began to explore the direct aldol reaction, a potentially powerful and efficient method for the enantioselective formation of carbon–carbon bonds by avoiding the use of chiral auxiliaries and the need for prior formation of an enolate or enol silane, this topic was just starting to become a major endeavor.10 The ProPhenol ligand (1a), which was initially developed for the direct aldol reaction, was based on the hypothesis that an aza-semicrown ligand would bind metal ions tightly enough to provide a good template for the design of a chiral pocket but not too tightly to prevent catalytic turnover. Furthermore, the increased basicity of zinc bound in this motif was envisioned to provide, after C–C bond formation, a Zn-alkoxide (shown in red) that could serve as a base for subsequent enolate formation and facilitate catalyst turnover while also avoiding unwanted epimerization and product elimination (Scheme 3).
Scheme 3. Catalyst Turnover Mechanism.
The first report of the ProPhenol ligand by Trost and Ito in 2000 showed that just 5 mol % of the ProPhenol ligand and 10 mol % Et2Zn could be used to facilitate the asymmetric aldol reaction with a variety of acetophenone derivatives (Scheme 4).4a
Scheme 4. Catalytic Asymmetric Aldol Reaction with Acetophenone Derivatives.

Reaction performed at −20 °C.
96% recovery of excess ketone.
Zn-ProPhenol-catalyzed reactions have proven to be highly tunable, and a variety of additives have been used to boost both yield and enantioselectivity. The benefits of most additives appear to be somewhat substrate specific and, in aldol reactions, have been postulated to reduce aggregation, fill open coordination sites on the catalyst, and mitigate base-mediated side reactions. The direct aldol reaction with methyl vinyl ketone (4, MVK), a donor that has not been reported with any other asymmetric catalyst system, was performed with 5 equiv of iPrOH and provided a variety of highly versatile β-hydroxy enones (Scheme 5).11
Scheme 5. Synthetic Utility of Asymmetric Aldol Reactions with MVK.

These β-hydroxy enones are versatile precursors to the corresponding 1,3-syn or 1,3-anti diols through stereocontrolled reduction of the ketone. In addition, the alkene could be functionalized using either alkene cross metathesis, to give 5, or a diastereoselective [3 + 2] cycloaddition with a nitrile oxide to provide 6. Furthermore, the Zn-ProPhenol catalyzed aldol reaction with methyl vinyl ketone was recently used in the synthesis of the ring A and B subunits of the bryostatins.12
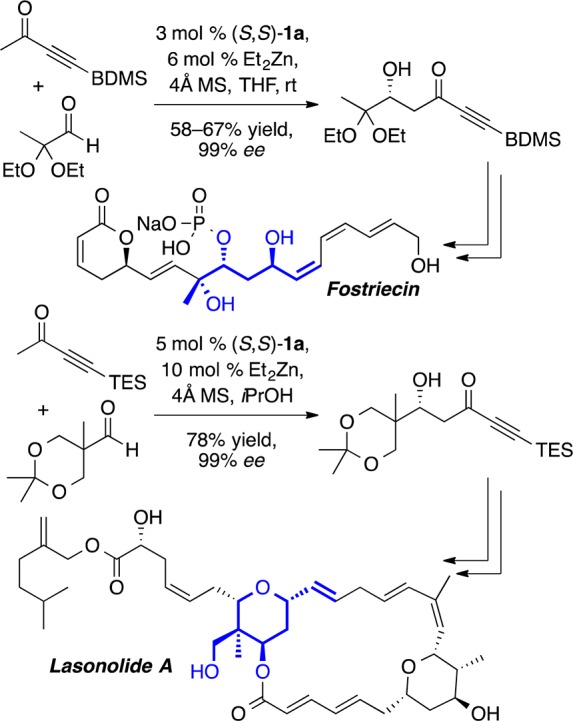
The use of Lewis basic additives in aldol reactions with methyl ynone donors gave rise to a number of surprising results (Scheme 6).13 Astonishingly, it was discovered that the sense of enantioinduction changes over the course of the reaction, with ent-7 being formed as the major product (69% ee) during the first 5 min. This led to the hypothesis that aldol product 7 might be involved in the enantiodetermining step. Indeed, the addition of either chiral or achiral β-hydroxy carbonyl compounds led to improvements in enantioselectivity during the first 45 min of the reaction (Scheme 6). Interestingly, catalyst loadings as low as 2 mol % gave good results (Scheme 6, entry 6). Ultimately, the use of additive-free conditions and higher temperature in the direct aldol reaction was found to provide excellent enantioselectivity and good to moderate yields with silyl, alkyl, and unsubstituted butyn-2-ones (Scheme 7). The latter two results are particularly noteworthy given the propensity of these substrates to undergo unwanted conjugate addition. Furthermore, this reaction was used to great effect in the total synthesis of lasonolide A and fostriecin (Scheme 8).14
Scheme 6. Optimization of the Direct Aldol Reaction with Methyl Ynones.

Reaction performed with 2 mol % catalyst loading.
Scheme 7. Catalytic Enantioselective Aldol Reaction with Ynone Donors.

Scheme 8. Asymmetric Aldol Reaction in the Synthesis of Fostriecin and Lasonolide A.
Zn-ProPhenol complexes have also been shown to facilitate acetone aldol reactions in good yield and enantioselectivity (Scheme 9).15 While the standard ProPhenol ligand (1a) provided good results with a variety of aliphatic and aryl aldehydes, the use of 1b, containing a 3,5-dimethyl phenol motif, provided slightly improved yield and enantioselectivity in a number of cases. The two methyl groups in this motif are believed to have a buttressing effect on the prolinol groups to reinforce the chiral pocket. A major challenge in aldol reactions with acetone is the avoidance of unwanted elimination, which is particularly problematic in the reaction of benzaldehyde with acetone; only 12% yield of 8 was obtained when using 10 mol % 1b. However, lowering the catalyst loading to 5 mol % provided a much improved 78% yield and 83% ee of the desired product.
Scheme 9. Catalytic Enantioselective Acetone Aldol Reaction.

The syn-1,2 diol motif is common in a number of bioactive natural products, and in contrast to dihydroxylation, the asymmetric aldol reaction represents a particularly efficient means of constructing this unit.16 Using just 2.5 mol % of the ProPhenol ligand and a slight excess of an α-hydroxy acetophenone donor, we could obtain a range of syn-1,2 diols in high yield and stereoselectivity (Scheme 10).4b This methodology was used to facilitate the direct aldol reaction of 2-hydroxyacetylfuran with an enolizable aldehyde and enable the enantioselective total synthesis of boronolide.17 Interestingly, the absolute configuration of the β-hydroxy group is the opposite of that obtained with acetophenone donors. Presumably, this reversal is due to the bridging of the α-oxygenated enolate between the two zinc atoms, which results in addition to the opposite enantiotopic face of the aldehyde (Figure 2).
Scheme 10. Direct Aldol Reaction with α-Hydroxy Acetophenone Donors.

Reaction performed with (R,R)-1a for 12 h on a 16 mmol scale.
Figure 2.

Proposed bidentate coordination of α-hydroxyketone donors.
Relative to ketone-based donors, ester enolate equivalents have proven much less reactive as aldol donors. Furthermore, ester equivalents are typically not amenable to organocatalytic direct aldol reactions, thus making an efficient synthesis of β-hydroxy esters particularly valuable. The development of a formal α-hydroxyacetate aldol reaction began with the screening of a number of different activated ester equivalents in reactions with the ProPhenol ligand 1a and Et2Zn (Scheme 11).18 The use of an N-acylpyrrole donor was found to be superior to both 2-acylimidazole and N-acylbenzoxazolinone donors. Futhermore, substitution at the 2-position of the pyrrole provided improved diastereoselectivity, and therefore N-(α-hydroxyacetyl)-2-ethylpyrrole (9) was chosen as the activated ester equivalent. Both of the ProPhenol ligands 1a and 1b provided good yield and stereoselectivity in aldol reactions of 9 with aliphatic, aryl, and α,β-unsaturated aldehydes, with ligand 1b leading to slightly improved results in some cases (Scheme 12). The products from these aldol reactions have shown great synthetic utility because the N-acylpyrrole group can be converted to the corresponding ester, amide, or ketone in a single step (Scheme 13). Furthermore, this methodology provided a key step in the total synthesis of laulimalide.19
Scheme 11. Initial Optimization of the α-Hydroxyacetate Aldol Reaction.

Scheme 12. ProPhenol-Catalyzed Hydroxyacetate Equivalent Aldol Reaction.

Reaction performed at −15 °C for 72 h.
Scheme 13. Synthetic Utility of Hydroxyacetate Aldol Products.

Esters, in the form of glycine Schiff bases (i.e., 10), also could be used directly as donors in ProPhenol-catalyzed aldol reactions (Scheme 14).20 Optimization by providing additional steric constraint in the chiral pocket led to higher stereoselectivity. The ProPhenol ligand [(S,S),(S,S)]-1c proved best. High yield and stereoselectivity were obtained with a range of aliphatic aldehydes (Scheme 15). Furthermore, catalyst control was found to override the influence of α-chiral aldehydes, and while a small matched/mismatched effect was observed, high diastereoselectivity was obtained regardless of the configuration of the α-stereocenter of the acceptor. Both the methyl and t-butyl esters of the glycinate Schiff base proved effective, in contrast to other chiral catalysts for this transformation.21
Scheme 14. Ligand Screening with Glycinate Schiff Base Donors.

Yield of the isolated pure 1,2-syn diastereomer.
Scheme 15. Asymmetric Aldol Reaction with Glycinate Schiff Base Donors.

A versatile alternative to the aforementioned α-hydroxy and α-amino ester enolate equivalents is the diazo ester aldol reaction.22 The unique reactivity of the resulting β-hydroxy-α-diazo esters can be used to prepare a wide range of 1,2-diols and α-hydroxy-β-amino esters in a stereoselective manner (vide infra). For this process, the use of di-n-butylmagnesium, in place of diethylzinc, provided the desired product 12 in a modest 27% ee (Scheme 16).23 Guided by the notion that open coordination sites on zinc were occupied by multiple equivalents of ethyl diazoacetate, the use of a bidentate coordinating agent, cis-1,2-cyclopentanediol 16 (Scheme 16), combined with a higher reaction concentration (1.0 M) and slow addition of ethyl diazoacetate led to further improvements, providing 12 in 95% yield and 95% ee. Scheme 17 illustrates the scope of the reaction. Further, the resulting enantioenriched β-hydroxy-α-diazo esters serve as versatile synthetic intermediates with a wealth of possible reactivity as depicted in Scheme 18, whereby oxidation of the diazo group with dimethyldioxirane (DMDO) is followed by nucleophilic addition to the resulting ketone. This reaction sequence enables the rapid and stereoselective preparation of vicinal diols of considerable molecular complexity. For example, a three step sequence was used to convert a β-hydroxy-α-diazo ester into compound 17, a fragment present in a number of polyketide antibiotics including azithromycin.23 Interestingly, the ProPhenol ligand was used to facilitate the diastereoselective methylation of a β-hydroxy-α-keto ester en route to 17. In the absence of 1a, a complex mixture of products was obtained from this methyl addition.
Scheme 16. Optimization of the Mg-ProPhenol Diazo Ester Aldol Reaction.

Scheme 17. Mg-ProPhenol-Catalyzed Aldol Reaction with Ethyl Diazoacetate.

Reaction performed with (R,R)-1a.
Reaction conducted at 0.5 M.
Aldehyde (2 equiv) added.
Scheme 18. Synthetic Versatility of Diazo Ester Aldol Products.

β-Amino acid derivatives can also be prepared from diazo ester aldol adducts using trichloroacetonitrile and sodium hydride to convert the alcohol to the corresponding trichloroacetamide (Scheme 19).24 This C–O to C–N transfer occurs with retention of absolute configuration.
Scheme 19. Conversion of Diazo Ester Aldol Adducts into β-Amino Acid Derivatives25.

Starting material had an ee of 97%.
For experimental details see ref (25).
Catalytic Enantioselective Mannich and Henry Reactions
The Mannich reaction is a highly efficient method for the preparation of β-amino carbonyl compounds.26 The Zn-ProPhenol catalyst (1a/Et2Zn) was found to facilitate the direct Mannich reaction with a range of α-hydroxy acetophenones (Scheme 20).27 An o-methoxyaryl imine improved the stereocontrol compared with a p-methoxy analog, presumably due to coordination of the ortho methoxy group to zinc. Improved diastereoselectivity was obtained with the ProPhenol ligands 1f and 1g, which contain bulky 4-biphenyl and 2-naphthyl groups, respectively. Substrates containing more labile N-protecting groups on the imine, such as diphenylphosphinoyl (DPP) and Boc, were also found to be effective in the ProPhenol-catalyzed Mannich reaction and enabled the use of enolizable aliphatic imines, which are typically challenging substrates.28 Interestingly, the bulky DPP group led to selective formation of the anti-1,2-amino alcohol (Scheme 21), whereas the Boc-imine resulted in the preferential formation of the corresponding syn-diastereomer (Scheme 22). In both cases, the absolute configuration at the α-position is the same and is likely attributed to bidentate coordination of the α-hydroxy ketone enolate with the dinuclear zinc catalyst.
Scheme 20. ProPhenol-Catalyzed Direct Mannich Reaction.

Scheme 21. anti-Selective Mannich Reaction with DPP-Imines.

Scheme 22. syn-Selective Direct Mannich Reaction with Boc-Imines.

Wang and co-workers have recently reported an elegant application of the Zn-ProPhenol catalyzed Mannich reaction, using 5H-oxazolones as donors to form two adjacent stereocenters, one of which is tetrasubstituted (Scheme 23).29 In this case, a 2-thienyl variant of the ProPhenol ligand, (S,S)-1h, plus the use of an additive, diethyl phosphoramidate (18), to block open coordination sites on zinc was found to provide superior diastereoselectivity, relative to 1a, and the Mannich product 19 was obtained in 89% yield, 97:3 dr, and 95% ee. Furthermore, the preformed Zn-complex derived from 1h/Et2Zn/18 was found to be relatively air stable, and identical results were obtained when the reaction was performed open to air with the preformed complex.
Scheme 23. ProPhenol-Catalyzed Mannich Reaction with 5H-Oxazol-4-ones.

The Zn-ProPhenol catalyst was also found to be effective with nitronate-based nucleophiles in enantioselective nitro-Mannich (or aza-Henry) reactions (Scheme 24).30 While a number of effective catalyst systems have been reported for the enantioselective aza-Henry reaction with aryl- and aliphatic imines,31 the results in Scheme 24 constitute a rare example of asymmetric addition to α,β-unsaturated imines. Given the facile conversion of β-nitro amines into 1,2-diamines and the synthetic utility of enantioenriched allylic amines, these results represent a valuable extension of the aza-Henry reaction. Analogous reactivity was also exploited in a ProPhenol-catalyzed Henry reaction with aryl aldehydes (Scheme 25).32 Subsequent reduction of the nitro groups in 20 and 21 led to the total synthesis of (−)-denopamine and (−)-arbutamine, respectively.
Scheme 24. Zn-ProPhenol-Catalyzed Aza-Henry Reaction.

Reaction performed with 30 mol % ProPhenol, 60 mol % Et2Zn.
Reaction performed at room temperature.
Scheme 25. ProPhenol-Catalyzed Enantioselective Henry Reaction.

Enantioselective Conjugate Additions
The Zn-ProPhenol catalyst was found to facilitate the asymmetric conjugate addition of α-hydroxy ketones to nitroalkenes (Scheme 26) to form the adducts 22.33,34 Interestingly, generating the catalyst with an equimolar mixture of Et2Zn and Bu2Mg led to a synergistic effect, whereby higher diastereoselectivity was obtained while also maintaining high ee. The presence of alkyl, alkynyl, and electron-rich aryl substituents on the nitroalkene typically provided higher diastereoselectivities relative to β-nitrostyrene. Further, 2-hydroxyacetylfuran also served as an excellent donor. The γ-nitro ketones produced from these reactions are particularly useful intermediates in the stereoselective synthesis of pyrrolidines.34
Scheme 26. ProPhenol-Catalyzed Nitroalkene Conjugate Addition.

Oxazolones were found to be effective pronucleophiles in ProPhenol-catalyzed asymmetric conjugate additions with nitroalkenes using the bulky 2-naphthyl variant 1g to provide 23 in excellent yield and stereoselectivity (Scheme 27).35 The aryl group of the oxazolone was found to have a significant impact on diastereoselectivity, with 3-methylbenzene providing the highest stereoselectivity. The products obtained from this reaction can be readily converted to the corresponding α-hydroxy carboxamide using NaOH.35
Scheme 27. Zn-ProPhenol Catalyzed Nitro-Michael Reaction with Oxazolones.

An analogous transformation with 2(5H)-furanone was developed, harnessing vinylogous nucleophilicity to create enantioenriched butenolides (Scheme 28). The reaction concentration at 0.5 M proved significant in obtaining high diastereoselectivity.
Scheme 28. Enantioselective γ-Alkylation of 2-Furanone.

Absolute configuration determined by X-ray crystallography.
Nitroalkenes also serve as efficient acceptors in the ProPhenol-catalyzed Friedel–Crafts alkylation of pyrroles.36 Using 10 mol % catalyst loading and 3 equiv of pyrrole, we obtained high yield and enantioselectivity with both aryl and alkyl substituted nitroalkenes (Scheme 29). Additionally, Wang and co-workers have reported the ProPhenol-catalyzed Friedel–Crafts alkylation of indoles with aldimines.37
Scheme 29. Asymmetric Friedel–Crafts Alkylation of Pyrroles.

Yield based on recovered starting material.
The ProPhenol ligand has also been used to facilitate the enantioselective alkylation of hydroxyoxindoles 24 to form spirocyclic γ-lactones (Scheme 30) in a 2:1 mixture of acetonitrile and toluene at 40 °C.38 Interestingly, the catalyst is involved in both the conjugate addition and the transesterification to form the spirocyclic oxindole product; in the absence of 1a, the open-chained Michael adduct did not cyclize to form a γ-lactone.
Scheme 30. Asymmetric Hydroxyoxindole Annulation.

Absolute configuration determined by X-ray crystallography.
Wang and co-workers have used the ProPhenol ligand to facilitate the asymmetric phospha-Michael reaction with a wide range of substrates (Scheme 31).39 Initial work outlined the use of the standard ProPhenol ligand 1a to facilitate the addition of diethylphosphite to a variety of enones.40 This reactivity was extended to included the addition of less reactive dialkyl phosphine oxides to α,β-unsaturated N-acylpyrroles and ketones.
Scheme 31. ProPhenol-Catalyzed Asymmetric Phospha-Michael Additions.

Me2Zn was used in place of Et2Zn.
Reaction run at 40 °C.
Absolute configuration determined by X-ray crystallography.
This reaction inspired the design of a new ProPhenol ligand, 1h, containing four 2-thienyl groups in place of the typical phenyl rings (see Scheme 23). The smaller heteroaromatic groups are postulated to create a larger chiral pocket and serve as an internal Lewis base, which occupies the open coordination sites of each metal, thereby increasing the ee and enhancing the scope to β,β-disubstituted acceptors, thus generating a tetrasubstituted stereocenter.
ProPhenol-Catalyzed Asymmetric Alkyne Addition
Chiral propargylic alcohols serve as both robust and versatile synthetic intermediates, providing a wealth of potential reactivity with a high degree of orthogonality. Furthermore, propargylic alcohols are present in numerous natural products and therapeutic agents.41 The success of the Zn-ProPhenol catalyst with stabilized nucleophiles prompted the examination of this system to facilitate the addition of zinc acetylides to aldehydes (Scheme 32). Initial results revealed that, in contrast to previous ProPhenol-catalyzed methodologies, asymmetric alkynylation required 2–3 equiv of dialkynylzinc for efficient catalyst turnover and enantioinduction.42 Nonetheless, good yield and enantioselectivity was obtained in the addition of aryl, alkyl, and silyl acetylenes to aryl and α,β-unsaturated aldehydes with 10 mol % of (S,S)-1a (Scheme 33).43 Extension to the addition of diynes to aldehydes required the addition of 20 mol % triphenylphosphine oxide (TPPO) for higher enantioselectivity as illustrated by the formation of 27 and 28.(44) The efficiency of the catalytic diyne addition to form 28, an intermediate for the synthesis of the antitumor agent strongylodial, contrasts with the use of an ephedrine mediated reaction, which required 4 equiv of the “catalyst” to give the adduct in 68% yield and 80% ee compared with the ProPhenol method, which utilized only 20 mol % catalyst to give the desired adduct in 90% yield and 87% ee.45 Combining the use of TPPO with higher reaction concentrations enabled use of just 1.2 equiv of alkyne more generally, while maintaining good enantioselectivity and yield. For example, these conditions allowed use of an equimolar amount of a precious alkyne with fumaraldehyde dimethyl acetal to provide the desired product 30, an intermediate in the total synthesis of aspergillide B, in 82% yield and 19:1 dr.46 The alkynol 29 served as a substrate in a second ProPhenol catalyzed alkynylation leading ultimately to the bioactive natural product spirolaxine methyl ether.51 The asymmetric addition of methyl propiolate using nearly stoichiometric amounts of aldehyde and alkyne typically led to exemplary results, as seen in the yield and ee of 25 and 26. This strategy was recently applied to the total synthesis of asteriscunolide D to convert methyl propiolate into the chiral butenolide 31 (Scheme 34).47
Scheme 32. Proposed Mechanism for ProPhenol-Catalyzed Alkyne Addition.

Scheme 33. ProPhenol-Catalyzed Asymmetric Alkyne Addition.

Scheme 34. Alkyne Addition in the Enantioselective Synthesis of Asteriscunolide D.

Enolizable aldehydes, most notably acetaldehyde, are particularly challenging substrates for enantioselective alkyne addition due to self-aldol side reactions. The prevalence of chiral 1-methyl propargylic alcohols (i.e., 32) in natural products prompted attempts to form these products using the Zn-ProPhenol catalyst.48 Minimizing the concentration of acetaldehyde by its slow addition over 30 min provided high yield and enantioselectivity with a variety of alkynes (Scheme 35). This methodology was applied to the synthesis of minquartynoic acid,48 tetrahydropyrenophorol, and the proposed structure of trocheliophorolide B.49
Scheme 35. ProPhenol-Catalyzed Alkynylation of Acetaldehyde.

Reaction performed at −20 °C.
Reaction performed at 0 °C.
Marek and co-workers have recently reported an elegant one pot application of the Zn-ProPhenol asymmetric alkynylation in tandem with a Brook-type rearrangement and ene–allene cyclization (Scheme 36) ultimately to provide a 1,3-diol 33 via oxysilanes 34.50 Overall, this sequence forms three new bonds and two new stereocenters in a highly stereoselective manner.
Scheme 36. Asymmetric Alkynylation/Brook Rearrangement/Ene–Allene Cyclization.

Catalytic Desymmetrization of meso-1,3-Diols and Epoxides
The asymmetric desymmetrization of meso-diols provides rapid access to a number of versatile chiral building blocks.51 The Zn-ProPhenol catalyst has been successfully used to differentiate the two enantiotopic groups of 2-alkyl- and 2-aryl-1,3-propanediols to form the corresponding monobenzoates in high yield and enantioselectivity (Scheme 37).52 The bulky 4-biphenyl ProPhenol ligand 1f gave significantly higher enantioselectivity than the standard ligand 1a, with products such as 35 often being formed in >90% ee. This transformation was found to be both practical and scalable as the chiral benzoate 36 was prepared on a 18 g scale and used in the total synthesis of (−)-18-epi-peloruside A.53
Scheme 37. ProPhenol-Catalyzed Desymmetrization of meso-1,3-Diols.

Reaction performed on a 96.5 mmol scale.
Reaction performed with 5 mol % (S,S)-1f at −15 °C.
In related research, Ding and co-workers used a Mg-ProPhenol catalyst to facilitate the enantioselective ring opening of meso-epoxides with anilines (Scheme 38).54 This reaction was used to prepare a variety of 1,2-amino alcohols in high yield and enantioselectivity, once again highlighting the versatile nature of the ProPhenol ligand.
Scheme 38. ProPhenol-Catalyzed Desymmetrization of meso-Epoxides.

Conclusion
Since the initial report of the ProPhenol ligand in 2000, a wealth of catalytic enantioselective transformations have been developed with dinuclear metal complexes derived from this semi-aza crown motif. The use of ProPhenol ligands in the direct aldol, Mannich, and Henry reactions has provided an atom economic alternative to the use of chiral auxiliaries and the preformation of enolates or silyl enol ethers and a complement to organocatalytic methods. Furthermore, asymmetric conjugate additions to nitro alkenes and α,β-unsaturated carbonyl compounds enable the mild and selective preparation of tertiary carbon stereocenters. The Zn-ProPhenol-catalyzed alkynylation of aldehydes provides access to chiral propargylic alcohols with immense synthetic utility. Consequently, this transformation has been used in the synthesis of more than a dozen complex natural products. Lastly, the desymmetrization of meso-1,3-diols provides an efficient method for the large scale preparation of chiral 1,3-oxygenated building blocks, a motif ubiquitous in polyketide natural products. The continued pursuit of complex target structures and new synthetic methodology is likely to result in many more applications for this privileged ligand scaffold.
Biographies
Mark Bartlett was born in New Zealand and completed a bachelor’s degree at Stanford University in 2007 under the tutelage of Prof. Barry Trost. He subsequently worked for two years as a medicinal chemist at Roche Palo Alto before returning to New Zealand, where he obtained a Ph.D. from Victoria University of Wellington in 2013. Mark is currently a postdoctoral researcher at UC Berkeley working under the guidance of Prof. John Hartwig.
Barry Trost, born in Philadelphia, Pennsylvania, obtained a B.A. degree from the University of Pennsylvania in 1962 and Ph.D. degree just three years later at the Massachusetts Institute of Technology (1965). He directly moved to the University of Wisconsin where he was promoted to Professor of Chemistry in 1969 and subsequently became the Vilas Research Professor in 1982. He joined the faculty at Stanford as Professor of Chemistry in 1987 and became Tamaki Professor of Humanities and Sciences in 1990. In addition, he has been a Visiting Professor in Germany (Universities of Marburg, Hamburg, Munich, and Heidelberg), Denmark (University of Copenhagen), France (Universities of Paris VI and Paris-Sud), Italy (University of Pisa), and Spain (University of Barcelona). He received honorary degrees from the Université Claude-Bernard (Lyon I), France (1994), and the Technion, Haifa, Israel (1997). In recognition of his many contributions, Professor Trost has received a large number of awards, a few among which are the ACS Award in Pure Chemistry (1977), the Dr. Paul Janssen Prize (1990), the ASSU Graduate Teaching Award (1991), Bing Teaching Award (1993), the ACS Roger Adams Award (1995), the Presidential Green Chemistry Challenge Award (1998), the Belgian Organic Synthesis Symposium Elsevier Award (2000), the ACS Nobel Laureate Signature Award for Graduate Education in Chemistry (2002), the ACS Cope Award (2004), the Nagoya Medal (2008), the Ryoji Noyori Prize (2013), and the German Chemical Society’s August-Wilhelm-von-Hofmann Denkmuenze (2014). Professor Trost has been elected a fellow of the American Academy of Sciences (1992) and a member of the U.S. National Academy of Sciences (1990).
Supporting Information Available
Detailed procedure for the recovery of the ProPhenol ligand. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† M.J.B.: Department of Chemistry, University of California, Berkeley, CA 94720-1460.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
We thank the NSF (Grant CHE-1145236) for support of our efforts in new reaction discovery and NIH (Grant GM-033049) in developing new strategies for total synthesis of biologically active targets.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Synthesis, Design, and Molecular Function”.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Busacca C. A.; Fandrick D. R.; Song J. J.; Senanayake C. H. The Growing Impact of Catalysis in the Pharmaceutical Industry. Adv. Synth. Catal. 2011, 353, 1825–1864. [Google Scholar]
- For selected examples of aza-crown ligands, see:; a Zhang W.; Loebach J. L.; Wilson S. R.; Jacobsen E. N. Enantioselective Epoxidation of Unfunctionalized Olefins Catalyzed by Salen Manganese Complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar]; b Sawamura M.; Nagata H.; Sakamoto H.; Ito Y. Chiral Phosphine Ligands Modified by Crown Ethers: An Application to Palladium-Catalyzed Asymmetric Allylation of β-Diketones. J. Am. Chem. Soc. 1992, 114, 2586–2592. [Google Scholar]
- a Cram D. J. The Design of Molecular Hosts, Guests, and Their Complexes (Nobel Lecture). Angew. Chem., Int. Ed. Engl. 1988, 27, 1009–1020. [PubMed] [Google Scholar]; b Kyba E. B.; Koga K.; Sousa L. R.; Siegel M. G.; Cram D. J. Chiral Recognition in Molecular Complexing. J. Am. Chem. Soc. 1973, 95, 2692–2693. [Google Scholar]
- a Trost B. M.; Ito H. A Direct Catalytic Enantioselective Aldol Reaction via a Novel Catalyst Design. J. Am. Chem. Soc. 2000, 122, 12003–12004. [Google Scholar]; b Trost B. M.; Ito H.; Silcoff E. R. Asymmetric Aldol Reaction via a Dinuclear Zinc Catalyst: α-Hydroxyketones as Donors. J. Am. Chem. Soc. 2001, 123, 3367–3368. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Wang Z.; Ding K. Copolymerization of Cyclohexene Oxide with CO2 by Using Intramolecular Dinuclear Zinc Catalysts. Chem.—Eur. J. 2005, 11, 3668–3678. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Wang Z.; Ding K. Intramolecularly Dinuclear Magnesium Complex Catalyzed Copolymerization of Cyclohexene Oxide with CO2 under Ambient CO2 Pressure: Kinetics and Mechanism. Macromolecules 2006, 39, 128–137. [Google Scholar]
- a Co-ProPhenol:Netto C. G. C. M.; Toma H. E. CO2 Complexation and Activation by a Trost-Bis(ProPhenol)Cobalt Complex. Eur. J. Inorg. Chem. 2013, 5826–5830. [Google Scholar]; b Bi-ProPhenol:Li Z.; Plancq B.; Ollevier T. Bismuth Triflate-Catalyzed Asymmetric Allylation of Aromatic Aldehydes. Chem.–Eur. J. 2012, 18, 3144–3147. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Rohbogner C. Unpublished results. See Supporting Information.
- a Shibasaki M.; Kanai M.; Matsunaga S.; Kumagai N. Recent Progress in Asymmetric Bifunctional Catalysis Using Multimetallic Systems. Acc. Chem. Res. 2009, 42, 1117–1127. [DOI] [PubMed] [Google Scholar]; b Paull D. H.; Abraham C. J.; Scerba M. T.; Alden-Danforth E.; Lectka T. Bifunctional Asymmetric Catalysis: Cooperative Lewis Acid/Base Systems. Acc. Chem. Res. 2008, 41, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews, see:; a Trost B. M.; Brindle C. S. The Direct Catalytic Asymmetric Aldol Reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Notz W.; Tanaka F.; Barbas C. F. III Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels–Alder Reactions. Acc. Chem. Res. 2004, 37, 580–591. [DOI] [PubMed] [Google Scholar]; c Palomo C.; Oiarbide M.; García J. M. Current Progress in the Asymmetric Aldol Addition Reaction. Chem. Soc. Rev. 2004, 33, 65–75. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Shin S.; Sclafani J. A. Direct Asymmetric Zn–Aldol Reaction of Methyl Vinyl Ketone and Its Synthetic Applications. J. Am. Chem. Soc. 2005, 127, 8602–8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Yang H.; Brindle C. S.; Dong G. Atom-Economic and Stereoselective Syntheses of the Ring A and B Subunits of the Bryostatins. Chem.–Eur. J. 2011, 17, 9777–9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Fettes A.; Shireman B. T. Direct Catalytic Asymmetric Aldol Additions of Methyl Ynones. Spontaneous Reversal in the Sense of Enantioinduction. J. Am. Chem. Soc. 2004, 126, 2660–2661. [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Stivala C. E.; Hull K. L.; Huang A.; Fandrick D. R. A Concise Synthesis of (−)-Lasonolide A. J. Am. Chem. Soc. 2014, 136, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost B. M.; Frederiksen M. U.; Papillon J. P. N.; Harrington P. E.; Shin S.; Shireman B. T. Dinuclear Asymmetric Zn Aldol Additions: Formal Asymmetric Synthesis of Fostriecin. J. Am. Chem. Soc. 2005, 127, 3666–3667. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Silcoff E. R.; Ito H. Direct Asymmetric Aldol Reactions of Acetone Using Bimetallic Zinc Catalysts. Org. Lett. 2001, 3, 2497–2500. [DOI] [PubMed] [Google Scholar]
- For a representative example, see:Kumagai N.; Matsunaga S.; Kinoshita T.; Harada S.; Okada S.; Sakamoto S.; Yamaguchi K.; Shibasaki M. Direct Catalytic Asymmetric Aldol Reaction of Hydroxyketones: Asymmetric Zn Catalysis with a Et2Zn/Linked-BINOL Complex. J. Am. Chem. Soc. 2003, 125, 2169–2178. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Yeh V. S. C. Stereocontrolled Synthesis of (+)-Boronolide. Org. Lett. 2002, 4, 3513–3516. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Michaelis D. J.; Truica M. I. Dinuclear Zinc–ProPhenol-Catalyzed Enantioselective α-Hydroxyacetate Aldol Reaction with Activated Ester Equivalents. Org. Lett. 2013, 15, 4516–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Amans D.; Seganish W. M.; Chung C. K. Evaluating Transition-Metal-Catalyzed Transformations for the Synthesis of Laulimalide. J. Am. Chem. Soc. 2009, 131, 17087–17089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Miege F. Development of ProPhenol Ligands for the Diastereo- and Enantioselective Synthesis of β-Hydroxy-α-amino Esters. J. Am. Chem. Soc. 2014, 136, 3016–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ooi T.; Kameda M.; Taniguchi M.; Maruoka K. Development of Highly Diastereo- and Enantioselective Direct Asymmetric Aldol Reaction of a Glycinate Schiff Base with Aldehydes Catalyzed by Chiral Quaternary Ammonium Salts. J. Am. Chem. Soc. 2004, 126, 9685–9694. [DOI] [PubMed] [Google Scholar]; b Mettath S.; Srikanth G. S. C.; Dangerfield B. S.; Castle S. L. Asymmetric Synthesis of β-Hydroxy Amino Acids via Aldol Reactions Catalyzed by Chiral Ammonium Salts. J. Org. Chem. 2004, 69, 6489–6492. [DOI] [PubMed] [Google Scholar]
- For representative examples of the diazoester aldol reaction, see:; a Yao W.; Wang J. Direct Catalytic Asymmetric Aldol-Type Reaction of Aldehydes with Ethyl Diazoacetate. Org. Lett. 2003, 5, 1527–1530. [DOI] [PubMed] [Google Scholar]; b Arai S.; Hasegawa K.; Nishida A. One-Pot Synthesis of α-Diazo-β-hydroxyesters under Phase-Transfer Catalysis and Application to the Catalytic Asymmetric Aldol Reaction. Tetrahedron Lett. 2004, 45, 1023–1026. [Google Scholar]
- a Trost B. M.; Malhotra S.; Fried B. A. Magnesium-Catalyzed Asymmetric Direct Aldol Addition of Ethyl Diazoacetate to Aromatic, Aliphatic and α,β-Unsaturated Aldehydes. J. Am. Chem. Soc. 2009, 131, 1674–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost B. M.; Malhotra S.; Koschker P.; Ellerbrock P. Development of the Enantioselective Addition of Ethyl Diazoacetate to Aldehydes: Asymmetric Synthesis of 1,2-Diols. J. Am. Chem. Soc. 2012, 134, 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Malhotra S.; Ellerbrock P. Exploring the Unique Reactivity of Diazoesters: An Efficient Approach to Chiral β-Amino Acids. Org. Lett. 2013, 15, 440–443. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Shi W.; Zhang J.; Zhang B.; Liu B.; Liu Y.; Fan B.; Xiao F.; Xu F.; Wang J. RhII-Catalyzed Reaction of α-Diazocarbonyl Compounds Bearing β-Trichloroacetamino Substituent: C–H Insertion versus 1,2-H Shift. Chem.—Asian J. 2010, 5, 1112–1119. [DOI] [PubMed] [Google Scholar]
- For leading references, see:Arrayás R. G.; Carretero J. C. Catalytic Asymmetric Direct Mannich Reaction: A Powerful tool for the Synthesis of α,β-Diamino Acids. Chem. Soc. Rev. 2009, 38, 1940–1948Also see reference (10b). [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Terrell L. R. A Direct Catalytic Asymmetric Mannich-type Reaction to syn-Amino Alcohols. J. Am. Chem. Soc. 2003, 125, 338–339. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Jaratjaroonphong J.; Reutrakul V. A Direct Catalytic Asymmetric Mannich-type Reaction via a Dinuclear Zinc Catalyst: Synthesis of Either anti- or syn-α-Hydroxy-β-Amino Ketones. J. Am. Chem. Soc. 2006, 128, 2778–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Wang L.; Yang D.; Zhang Y.; Wang R. Highly Diastereo- and Enantioselective Synthesis of α-Alkyl Norstatine Derivatives: Catalytic Asymmetric Mannich Reactions of 5H-Oxazol-4-ones. Angew. Chem. 2012, 124, 7641–7645. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Lupton D. W. Dinuclear Zinc-Catalyzed Enantioselective Aza-Henry Reaction. Org. Lett. 2007, 9, 2023–2026. [DOI] [PubMed] [Google Scholar]
- For a recent review on the asymmetric Henry reaction, see:Boruwa J.; Gogoi N.; Saikia P. P.; Barua N. C. Catalytic Asymmetric Henry Reaction. Tetrahedron: Asymmetry 2006, 17, 3315–3326. [Google Scholar]
- Trost B. M.; Yeh V. S. C.; Ito H.; Bremeyer N. Effect of Ligand Structure on the Zinc-Catalyzed Henry Reaction. Asymmetric Syntheses of (−)-Denopamine and (−)-Arbutamine. Org. Lett. 2002, 4, 2621–2623. [DOI] [PubMed] [Google Scholar]
- For a recent review, see:Christoffers J.; Koripelly G.; Rosiak A.; Rössle M. Recent Advances in Metal-Catalyzed Asymmetric Conjugate Additions. Synthesis 2007, 1279–1300. [Google Scholar]
- Trost B. M.; Hisaindee S. A Heterodinuclear Asymmetric Catalyst for Conjugate Additions of α-Hydroxyketones to β-substituted Nitroalkenes. Org. Lett. 2006, 8, 6003–6005. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Hirano K. Highly Stereoselective Synthesis of α-Alkyl-α-Hydroxycarboxylic Acid Derivatives Catalyzed by a Dinuclear Zinc Complex. Angew. Chem. 2012, 124, 6586–6589. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Müller C. Asymmetric Friedel-Crafts Alkylation of Pyrroles with Nitroalkenes Using a Dinuclear Zinc Catalyst. J. Am. Chem. Soc. 2008, 130, 2438–2439. [DOI] [PubMed] [Google Scholar]
- Wang B.-L.; Li N.-K.; Zhang J.-X.; Liu G.-G.; Liu T.; Shen Q.; Wang X.-W. Dinuclear Zinc Catalyzed Asymmetric Friedel-Crafts Amidoalkylation of Indoles with Aryl Aldimines. Org. Biomol. Chem. 2011, 9, 2614–2617. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Hirano K. Dinuclear Zinc Catalyzed Asymmetric Spiroannulation Reaction: An Umpolung Strategy for Formation of α-Alkylated-α-Hydroxyindoles. Org. Lett. 2012, 14, 2446–2449. [DOI] [PubMed] [Google Scholar]
- For leading references, see:Zhao D.; Wang R. Recent Developments in Metal Catalyzed Asymmetric Addition of Phosphorus Nucleophiles. Chem. Soc. Rev. 2012, 41, 2095–2108. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Mao L.; Wang L.; Yang D.; Wang R. Catalytic Asymmetric Construction of Tetrasubstituted Carbon Stereocenters by Conjugate Addition of Dialkyl Phosphine Oxides to β,β-Disubstituted α,β-Unsaturated Carbonyl Compounds. Chem. Commun. 2012, 48, 889–891and references cited therein. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Trost B. M.; Weiss A. H. The Enantioselective Addition of Alkyne Nucleophiles to Carbonyl Groups. Adv. Synth. Catal. 2009, 351, 963–983. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Turlington M.; Pu L. Asymmetric Alkyne Addition to Aldehydes Catalyzed by BINOL and Its Derivatives. Synlett 2012, 23, 649–684. [Google Scholar]; c Frantz D. E.; Fässler R.; Tomooka C. S.; Carreira E. M. The Discovery of Novel Reactivity in the Development of C–C Bond-Forming Reactions: In Situ Generation of Zinc Acetylides with ZnII/R3N. Acc. Chem. Res. 2000, 33, 373–381. [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Weiss A. H.; von Wangelin A. J. Dinuclear Zn-Catalyzed Asymmetric Alkynylation of Unsaturated Aldehydes. J. Am. Chem. Soc. 2006, 128, 8–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost B. M.; Bartlett M. J.; Weiss A. H.; von Wangelin A. J.; Chan V. S. Development of Zn-ProPhenol-Catalyzed Asymmetric Alkyne Addition: Synthesis of Chiral Propargylic Alcohols. Chem.–Eur. J. 2012, 18, 16498–16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Weiss A. H. An Alkyne Strategy for the Asymmetric Synthesis of Natural Products: Application to (+)-Spirolaxine Methyl Ether. Angew. Chem., Int. Ed. 2007, 46, 7664–7666. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Chan V. S.; Yamamoto D. Enantioselective ProPhenol-Catalyzed Addition of 1,3-Diynes to Aldehydes to Generate Synthetically Versatile Building Blocks and Diyne Natural Products. J. Am. Chem. Soc. 2010, 132, 5186–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reber S.; Knöpfel T. F.; Carreira E. M. Enantioselective Total Synthesis of (R)-Strongylodiols A and B. Tetrahedron 2003, 59, 6813–6817. [Google Scholar]
- Trost B. M.; Bartlett M. J. Transition-Metal-Catalyzed Synthesis of Aspergillide B: An Alkyne Addition Strategy. Org. Lett. 2012, 14, 1322–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Burns A. C.; Bartlett M. J.; Tautz T.; Weiss A. H. Thionium Ion Initiated Medium-Sized Ring Formation: The Total Synthesis of Asteriscunolide D. J. Am. Chem. Soc. 2012, 134, 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Quintard A. Asymmetric Catalytic Alkynylation of Acetaldehyde: Application to the Synthesis of (+)-Tetrahydropyrenophorol. Angew. Chem., Int. Ed. 2012, 51, 6704–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Quintard A. Asymmetric Catalytic Synthesis of the Proposed Structure of Trocheliophorolide B. Org. Lett. 2012, 14, 4698–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov P.; Mathew J.; Nijs A.; Katan E.; Karni M.; Bolm C.; Apeloig Y.; Marek I. One-Pot Zinc-Promoted Asymmetric Alkynylation/Brook-Type Rearrangement/Ene-Allene Cyclization: Highly Selective Formation of Three New Bonds and Two Stereocenters in Acyclic Systems. Angew. Chem. 2013, 125, 13962–13966. [DOI] [PubMed] [Google Scholar]
- For representative examples, see:; a Lewis C. A.; Sculimbrene B. R.; Xu Y.; Miller S. J. Desymmetrization of Glycerol Derivatives with Peptide-Based Acylation Catalysts. Org. Lett. 2005, 7, 3021–3023. [DOI] [PubMed] [Google Scholar]; b Honjo T.; Nakao M.; Sano S.; Shiro M.; Yamaguchi K.; Sei Y.; Nagao Y. Nonenzymatic Enantioselective Monoacetylation of Prochiral 2-Protected Amino-2-alkyl-1,3-propanediols Utilizing a Chiral Sulfonamide-Zn Complex Catalyst. Org. Lett. 2007, 9, 509–512. [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Mino T. Desymmetrization of Meso 1,3- and 1,4-Diols with a Dinuclear Zinc Asymmetric Catalyst. J. Am. Chem. Soc. 2003, 125, 2410–2411. [DOI] [PubMed] [Google Scholar]; b Trost B. M.; Malhotra S.; Mino T.; Rajapaksa N. S. Dinuclear Zinc-Catalyzed Asymmetric Desymmetrization of Acyclic 2-Substituted-1,3-Propanediols: A Powerful Entry in Chiral Building Blocks. Chem.–Eur. J. 2008, 14, 7648–7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Michaelis D. M.; Malhotra S. Total Synthesis of (−)-18-epi-Peloruside A: An Alkyne Linchpin Strategy. Org. Lett. 2013, 15, 5274–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H.; Wang Z.; You T.; Ding K. Enantioselective Ring Opening of meso-Epoxides with Aromatic Amines Catalyzed by Dinuclear Magnesium Complexes. Chin. J. Chem. 2013, 31, 67–71. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.