

Abstract
The prevalence of the metabolic syndrome, a cluster of cardiovascular risk factors associated with obesity and insulin resistance, is dramatically increasing in Western and developing countries. This disorder consists of a cluster of metabolic conditions, such as hypertriglyceridemia, hyper-low-density lipoproteins, hypo-high-density lipoproteins, insulin resistance, abnormal glucose tolerance and hypertension, that-in combination with genetic susceptibility and abdominal obesity-are risk factors for type 2 diabetes, vascular inflammation, atherosclerosis, and renal, liver and heart diseases. One of the defects in metabolic syndrome and its associated diseases is excess of reactive oxygen species. Reactive oxygen species generated by mitochondria, or from other sites within or outside the cell, cause damage to mitochondrial components and initiate degradative processes. Such toxic reactions contribute significantly to the aging process. In this article we review current understandings of oxidative stress in metabolic syndrome related disease and its possible contribution to accelerated senescence.
Keywords: metabolic syndrome, aging, accelerated senescence, oxidative stress
Metabolic syndrome
Looking back upon the time when the humankind had to spent tremendous trouble to gather food for survival, there is no doubt that the industrialization has succeeded at making many choices of calorie-rich food easily available with little physical efforts. However, this over-alimentation has led to an opposite pattern of diseases and medical problems by introducing a worldwide outbreak of over nutrition related diseases such as obesity, type 2 diabetes (T2D), and cardiovascular diseases (CVDs) [1-7]. While this feature of social economic environment overnutrition will continue to exist, the health consequences are substantial and can afflict the fundamental welfare of modern human species [8-12]. From the physiological perspective, these health problems are often preceded by a cluster of pathophysiological abnormalities including obesity, insulin resistance, impaired glucose tolerance, dyslipidemia and high blood pressure, which are interconnected and called metabolic syndrome [13-20].
The metabolic syndrome is characterized by a group of metabolic risk factors in the same person. The first unified agreement about the definition of the Metabolic Syndrome was drawn up during a meeting organized by the International Diabetes Federation (IDF) in 2005. As stated in this the major contributing factor is obesity measured by waist circumference and Body Mass Index (BMI). In addition to central obesity, the other factors that have to be considered for diagnoses of metabolic syndrome involve to estimation of triglycerides, reduced high-density lipoprotein (HDL) cholesterol, blood pressure, or fasting plasma glucose of human [21]. The pathophysiology is very complex and not yet clear. Majority of patients are obese, older, sedentary, and have insulin resistance. The determining factors in order are weight, genetics, aging and lifestyle and excess caloric intake [22]. However, despite the importance of obesity, sometimes patients who are normal weight may be insulin-resistant and have the syndrome [23].
According to the most used definition, the revised Adult Treatment Panel-III (ATP-III), the metabolic syndrome is diagnosed when at least three of five of the following alterations are present: visceral obesity (waist circumference > 102 cm in men or > 88 cm in women); raised arterial blood pressure (> 130/85 mm Hg); dysglycemia (fasting plasma glucose >100 mg dL); raised triglyceride concentrations (> 150 mg dL); low HDL cholesterol (< 40 mg dL in men or < 50 mg dL in women) [24]. Even if it is not clear if a diagnosis of metabolic syndrome could help in clinical practice [25], it is recognized that this syndrome represents an important pathophysiological combination to study metabolism in humans and in animal models. The presence of metabolic syndrome leads to an increased risk of type 2 diabetes and cardiovascular disease, in the form of coronary or peripheral atherosclerosis and heart failure. Moreover, metabolic syndrome is associated with some other systemic complications that affect different organs and systems, such as fatty liver disease, respiratory disease, osteoarticular disease, and cancer. Subsequently, metabolic syndrome patients have increased all-cause mortality and a shortened lifespan compared with the general population [26; 27]. Thus, it is consecutively recognized that metabolic syndrome is associated with early aging [28], which is of predominant importance considering the worldwide growing epidemic of metabolic syndrome [29]. With this background, the description of biochemical mechanisms linking metabolic syndrome alterations to lifespan is very interesting.
Aging and oxidative stress
Aging is considered as a biological process characterized by a progressive deterioration in physiological functions and metabolic processes that drive to morbidity and mortality. In agreement with the free radical theory of aging, reactive oxygen species (ROS), generated as by-products of biological oxidations, induce casual and cumulative oxidative damage to macromolecules inducing to cellular dysfunction with age and eventually cell death [30]. Mitochondria seem to be closely involved in the aging process because these organelles are considered the main intracellular source of superoxide anion (O2•−), as well as the major target of free radical attack. ROS produced by the mitochondrial respiratory chain damage mitochondrial constituents, including proteins, lipids, and mitochondrial DNA (mtDNA) [31-33]. Progressive accumulation of oxidant-induced somatic mutations in mtDNA during an individual’s lifetime leads to a deterioration in the bioenergetic function of mitochondria and contributes to the aging process.
Low levels of ROS are generated during mitochondrial respiration under physiological conditions. Progressive oxidative damage to mtDNA with age may induce to DNA strand breaks and to the phenomenon of somatic mtDNA mutations [34-35]. Accumulation of these mtDNA alterations may lead to impairment of the respiratory chain complexes, leading to a vicious cycle with an increase in mitochondrial ROS production and a subsequent accumulation of more mitochondrial DNA mutations. This chain reaction has been proposed to be involved in the increased oxidative damage during aging that induces to the progressive decline in cellular and tissue function as a result of insufficient supply of energy and/or increased susceptibility to apoptosis [36-38]. Age-related increase in oxidative damage to DNA, lipids, and proteins has been well documented [39-40], as well as evidence supporting increased mtDNA deletions and mitochondrial dysfunction with aging [41-46].
Oxidative Stress and Metabolic syndrome
In the last years the potential role of oxidative stress in metabolic syndrome is rapidly evolving. Although it is generally accepted that the main pathogenic mechanism underlying the first level of metabolic changes in patients with the metabolic syndrome relies on insulin resistance, an abundance of evidence demonstrating a close link among the metabolic syndrome, a state of chronic low-level inflammation and oxidative stress as second-level abnormalities had emerged [47]. In fact, oxidative stress plays an important role in the pathogenesis of vascular alterations by either triggering or exacerbating the biochemical processes accompanying the metabolic syndrome [48]. In addition, experimental and clinical observations indicate oxidative stress as an important mechanism in obesity-associated metabolic syndrome, in the development of diabetes and its complications and in “satellite” conditions such as nonalcoholic steatohepatitis (NASH) [49-52].
Oxidative stress is a well-recognized mechanism playing important roles in many pathological conditions [53], and several human diseases have been closely related to oxidative stress [54]. A number of cell functions appear to be regulated by free radical molecules, which may also act as intracellular and intercellular signals [55-56]. Also, the protein redox state is implicated in the regulation of several cellular activities, including cell differentiation and activation of specific metabolic pathways [57-58].
Oxidative stress has been associated with all the individual components and with the onset of cardiovascular complications in subjects with the metabolic syndrome [48-49-59-60]. In a recent study [61], the role of oxidative stress in the pathophysiologic interactions among the constituent factors of the metabolic syndrome has been remarked. However, although some of the constituent characteristics of the metabolic syndrome are known to share common pathogenic mechanisms of damage, the impact of hereditary predisposition and the regulation of gene expression as well as the role of environment and dietary habit in determining inflammatory process-triggered oxidation are still unclear. These aspects of the problem deserve special attention since it is hypothesized that in patients with the metabolic syndrome, oxidative stress may be amplified by a concomitant antioxidant deficiency that may favor the propagation of oxidative alterations from intra- to extracellular spaces and from confined to distant sites, thus realizing a systemic oxidative stress state [62-63].
Altogether, these considerations would suggest a unifying hypothesis to explain the mechanisms underlying the onset and development of metabolic syndrome-associated risk factors. As the following subsections report, excessive free radical production and oxidative damages are supported by several experimental demonstrations and human observations. Therefore, oxidative stress appears to possess, at least in part, the credentials to mechanistically explain the perpetuation of insulin resistance, the altered energy production, the endothelial dysfunction and the appearance of vascular complications in this condition [64].
Oxidative Stress and Insulin Resistance
High concentrations of H2O2 promote insulin signaling and induce typical metabolic actions of insulin. This result could be considered as the first documentation on the link between ROS and insulin [65]. In particular, H2O2 uses the same pathway of insulin and causes downstream propagation of the signal producing typical metabolic actions of insulin. H2O2 induces an increase in glucose uptake by adipocytes and muscles [66], and also stimulates GLUT4 translocation and lipid synthesis in adipocytes [67].
Insulin receptor substrate (IRS) proteins are effectors for tyrosine kinase activity of the insulin receptor (IR) upon insulin binding. These proteins are involved in a critical step of insulin signalling. In normal physiological state, insulin signalling molecules are distributed between the cytosol and internal membrane pools. Whereas, after insulin stimulation, tyrosine residues on IR and IRS are phosphorylated through activated insulin receptor kinase. This leads to the enrolment of PI 3-kinase in the plasma membrane and in internal membrane pools. Subsequently, the activation of small GTPase Rac induces cytoskeletal reorganization that propagates the insulin signals. Finally, this induces to typical metabolic effects of insulin such as increased glucose uptake. In conditions of increased oxidative stress, stress-responsive signalling cascades are activated, such as the MAP kinase cascades. This induces to increase Ser/Thr phosphorylation of IRS molecules. Modified IRS molecules are released from internal membrane pools and are subjected to increased protein degradation. In these conditions, insulin fails to gain normal metabolic effects. This happens because IRS molecules are decreased in content and cannot be normally tyrosine phosphorylated when hyperphosphorylated on certain Ser/Thr residues [68]. It is important to note that, receptor substrates are able to directly modify the expression of glucose/metabolic genes such as GLUT4 [69] and adiponectin [70].
In animal models of obese mice, an increased H2O2 generation by adipose tissue could be observed prior to diabetes onset [71]. This event came with decreased mRNA levels of SOD, catalase, and glutathione peroxidase. Developing diabetes in these mice increased these alterations, which remained unobservable in other tissues. Obesity and the related insulin resistance are frequently related with increased accumulation of lipids (triglycerides) in the liver. Increased numbers of lipid peroxidation markers have been observed in the liver, in animal models of diabetes and obesity. Oxidation-induced disruption of cellular redistributed signalling molecules in response to insulin stimulation was associated with impaired insulin action. An animal model of oxidative stress provided support for this notion. In this in vivo model, oxidative stress was induced in rats using an inhibitor of glutathione biosynthesis enzyme. The reduction in tissue levels of glutathione, a cellular antioxidant, increased markers of oxidative stress and impaired glucose homeostasis [72].
Some studies in humans have evidenced the pivotal role of oxidative stress in insulin resistance states such as metabolic syndrome, obesity, and type 2 diabetes [73-76].
The drop of the antioxidant capacity, enhanced production of ROS with oxidation products of lipids, DNA, and proteins have been reported in plasma, urine, and various tissues, suggesting systemic and organ-specific oxidative stress. Recent evidence for systemic oxidative stress includes the discovery of high circulating and urinary levels of the lipid peroxidation product F2-isoprostane (8-epi-prostaglandin F2α) in both type 1 and type 2 diabetes and in obese patients [77, 78]. Moreover, this marker is related to blood glucose levels and glucose variability [79] and responded to antidiabetic therapy [77]. In particular, Pitocco et al showed the role of oxidative stress in the early phases of type 1 diabetes, with an impaired balance between NO, serum uric acid (a potent antioxidant), and asymmetric-dimethyl-L-arginine (ADMA) [80]. Furthermore ROS are able to directly modify the expression of adiponectin that, secreted from adipocytes, is inversely correlated with fat mass in obesity and with its associated cardiovascular risk. Notably, plasma and urinary lipid peroxidation markers, indicative of systemic oxidative stress, have been correlated with lower circulating adiponectin levels [81].
Oxidative Stress and Visceral Obesity
Obesity is closely associated with metabolic syndrome. Adipose tissue is now recognized as metabolically active, playing a role in the regulation of energy homeostasis and has significant pathological effects that result in many obesity-related diseases. The role of white adipose tissue in mediating systemic inflammation is an area of active investigation in both cardiac disease and diabetes. Adipose tissue is infiltrated by bone-marrow derived macrophages that secrete adipokines and cytokines in the systemic circulation resulting in a chronic inflammatory state [82, 83].
In cross-sectional studies, obese subjects have higher levels of oxidative stress biomarkers compared with their leaner counterparts [84]. Also, weight gain significantly increases the concentration of these biomarkers [85]. There are multiple sources for oxidative stress in relation to obesity. Some of them are inherently related to increased adiposity and fat distribution, whereas others are the result of comorbidities or behavioral changes associated with being obese. Increased adipose tissue and, in particular, visceral adiposity are significantly correlated with systemic levels of oxidative stress biomarkers [86, 87, 88].
Adipose tissue-mediated systemic oxidative stress and systemic inflammation may be secondary to increased leptin-to-adiponectin ratio and increased levels of other adipokines, such as tumor necrosis factor and plasminogen activator inhibitor-1 [89]. Obesity is associated with several comorbidities, including hypertension, insulin resistance, diabetes, and hyperlipidemia; each of these comorbidities alone can increase the oxidative stress burden. However, more often than not, these comorbidities occur simultaneously, as is the case of the metabolic syndrome that is characterized by insulin resistance, hypertension, and hyperlipidemia and is commonly present in adults in the US [90]. Compared with obese subjects without metabolic syndrome, those with metabolic syndrome have a greater degree of oxidative stress [84, 91].
Maintaining a healthy life style by eating a balanced diet rich in antioxidants and being physically active is associated with reduced oxidative stress. Unfortunately, this protection is less effective among obese subjects, who are more sedentary, having reduced intake of dietary antioxidants and lower serum vitamin levels [92].
Over time, chronic oxidative stress in obesity has a cumulative effect that favors the development of end-organ damage. This phenomenon has been mostly studied in the cardiovascular system and the liver in which chronic oxidative stress plays a critical role for the development of atherosclerosis and nonalcoholic hepatic steatosis [91, 93, 94, 95].
Oxidative Stress and hypertension
Oxidative stress, with an imbalance between ROS and antioxidant defense mechanisms, contributes to the etiology of hypertension in animals [92] and humans [93-98]. ROS are generated by multiple cellular sources, including NADPH oxidase, mitochondria, xanthine oxidase, uncoupled endothelium-derived nitric oxide synthase (eNOS), cyclo-oxygenase, and lipo-oxygenase [99]. Superoxide anion is produced by stimulation of the angiotensin II/angiotensin II type I receptor (AT1R) and NADPH oxidase by Angiotensin II. Superoxide anion is the predominant ROS species produced by these tissues, neutralizes nitric oxide (NO), and leads to downstream production of other ROS such as hydrogen peroxide, hydroxyl radicals, and peri-oxynitrite. Hypertensive patients have impaired endogenous and exogenous anti-oxidant defense mechanisms [95], increased plasma oxidative stress, and an exaggerated oxidative stress response to various stimuli [95-96]. Hypertensive patients also have lower plasma ferric-reducing ability of plasma (FRAP), lower vitamin C levels, and increased plasma 8-isoprostanes, which correlate with both systolic and diastolic blood pressure (BP). Various single-nucleotide polymorphisms (SNPs) in genes that codify for antioxidant enzymes are directly related to hypertension [97], including NADPH oxidase, xanthine oxidase, superoxide dismutase (SOD 3), catalase, glutathione peroxidase (GPx 1), and thioredoxin. ROS directly damage endothelial cells and degrade NO [93-96, 100]. Various neurohormonal systems including the renin-angiotensin-aldosterone system and sympathetic nervous system also contribute to oxidative stress, inflammation, and vascular immune dysfunctions. The increased oxidative stress, inflammation, and autoimmune vascular dysfunction in human hypertension results from a combination of increased generation of ROS, an exacerbated response to ROS, and decreased antioxidant reserve [95-98, 101, 102]. There are also direct interactions of the central nervous system, inflammation, and BP. Increased oxidative stress in the rostral ventrolateral medulla (RVLM) enhances glutamatergic excitatory inputs and attenuates GABAergic inhibitory inputs to the RVLM, which contributes to increased sympathetic nervous system (SNS) activity from the paraventricular nucleus [103]. Activation of the AT1R in the RVLM increases NADPH oxidase and increases oxidative stress and superoxide anion, increases SNS outflow causing an imbalance of SNS/periferic nervous system (PNS) activity with elevation of BP, and increases heart rate, alterations in heart rate variability, and heart rate recovery time, which can be blocked by AT1R blockers [103, 104, 105].
Oxidative Stress and dysglicemia
Dysglycemia has two components: chronic sustained hyperglycemia (including fasting and chronic postprandial hyperglycemia) and acute glycemic fluctuations from peaks to nadirs. Both components lead to diabetic complications through two main mechanisms: excessive protein glycation and activation of oxidative stress. These two mechanisms were unified in an elegant theory that suggested that the glycemic disorders observed in diabetic patients result in an activation of oxidative stress with an overproduction of superoxide by the mitochondrial electron-transfer chain. This activation in turn produces a cascade of such deleterious metabolic events as enhanced polyol activity, increased formation of advanced glycation end-products, activation of protein kinase C and nuclear factor κB, and increased hexosamine pathway flux [106]. It is now well established that hyperglycemia both at fasting and during postprandial periods results in exaggerated and accelerated glycation. For instance, all the studies conducted in type 1 and type 2 diabetes have clearly shown a strong positive relationship between HbA1c levels and plasma glucose levels at fasting and over postprandial periods [107, 108], the strongest correlation being observed between HbA1c and mean plasma glucose levels [109]. The latter relationship was considered sufficiently demonstrative to serve as a reference in the recent Standards of Medical Care in Diabetes that are published every year by the American Diabetes Association [110]. To day, glycated hemoglobin (HbA1c) is unanimously recognized as a reliable marker for the overall glucose exposure and its direct consequence, an excessive rate of glycation [111, 112]. The simplicity of this concept masks more complex phenomena because HbA1c is an integrator of both fasting and postprandial glycemic disorders. As a consequence, it is not surprising that either fasting or postprandial hyperglycemia were identified separately or concomitantly as major risk factors for diabetic complications. Furthermore, even though we consider only glycemia alterations, fasting and postprandial hyperglycemia may be not the only components of diabetic complications. Another risk factor is probably the glucose variability within a day, especially the acute glucose fluctuations.
Several markers have been used to assess oxidative stress and the antioxidant status in patients with diabetes. The short plasma half-life of these markers is one of the limiting factors for the assessment of oxidative stress in plasma samples. Thus, when available, urinary determinations provide a more reliable estimation of the activation of oxidative stress than plasma measurements [113, 114]. Accordingly, the determination of such specific isoprostane isomers as the 8-iso-PGF2α in urine has been proposed. Isoprostanes are collectively formed from free radical-mediated oxidation of arachidonic acid [115]. As this fatty acid is ubiquitously distributed in cell membranes, measurements of urinary isoprostanes most likely provide an excellent reflection of the activation of oxidative stress in the whole body. In different studies that have been conducted in diabetes, plasma and urinary metabolites have been alternatively or simultaneously used as oxidative stress markers. As free radical production has been reported to be increased in patients with diabetes mellitus, it has been suggested that hyperglycemia may directly contribute to the generation of oxidative stress. There is cogent evidence from several studies that hyperglycemia is associated with an increased formation and urinary excretion rate of 8-iso-PGF2α [116]. The urinary excretion rate of 8-iso-PGF2α was found to be significantly increased in type 2 diabetic patients as compared with age-matched healthy subjects. Furthermore, significant correlations were observed between blood glucose and urinary 8-iso-PGF2α suggesting that the activation of oxidative stress may be, at least in part, related to the determinants of diabetic control. These results has been recently confirmed by a study that showing the mean urinary excretion rates of 8-iso-PGF2α were significantly higher (p<0.01) in patients with type 2 diabetes (mean±SD=482±206 pg/mg of creatinine) than in nondiabetic healthy subjects (275±85 pg/mg of creatinine) [117]. Improved metabolic control has been shown to be associated with a significant reduction in urinary excretion rates of both 8-iso-PGF2α and 11-dehydrothromboxane (TXM) by 32 and 48%, respectively, in 21 patients with type 2 diabetes. Vitamin E supplementation led to complete normalization of 8-iso-PGF2α excretion rates [77]. Moreover, changes in 8-iso-PGF2α excretion were accompanied by similar reduction in TXM excretion, consistent with a cause-and-effect relation between enhanced lipid peroxidation and persistent platelet activation. Increase of plasma 8-iso-PGF2α is an early event in the evolution of type 2 diabetes and could precede the development of endothelial dysfunction [118].
Oxidative Stress and Dyslipidemia
Atherogenic dyslipidemia is an important component of the cluster of abnormalities characteristic of the metabolic syndrome. There are three major components of dyslipidemia that occur in metabolic syndrome: an increase in triglyceride-rich lipoproteins (TRLs) both fasting and postprandial, a reduction in high-density lipoprotein (HDL) and elevated small, dense low density lipoproteins (LDL) particles. Because the metabolism of all lipoproteins is highly interrelated, it is believable that a common fundamental metabolic defect explicates all of the lipoprotein changes in the dyslipidemia related to insulin resistance. It is indeed rare that they are found separately in insulin resistant individuals [119].
During the postprandial state, dietary fatty acids are transfered from the intestine to peripheral tissues as chylomicron triglycerides. In blood of the peripheral tissues, chylomicron triglycerides are lipolyzed by lipoprotein lipase (LPL), conceding the delivery of non-esterified fatty acids to cells and resulting in production of smaller, cholesteryl ester-enriched chylomicron remnants. These particles are fast removed.
Some studies have examined the relation between postprandial lipemia and insulin resistance, plasma glucose, and insulin response to a dinner in healthy nondiabetic subjects [120]. Postprandial triglyceride levels, as an indirect measurement of chylomicron remnant particles, were found to be significantly related to insulin action. A significant relation of triglyceride levels to postheparin plasma LPL activity was also demonstrated. Because LPL is an insulin-sensitive enzyme, which is suppressed in insulin resistant individuals, its deficiency might contribute to the abnormal levels of remnant particles in insulin resistance [119].
A correlation between elevated LDL and low HDL and oxidative stress in animal models is well established. LDL receptor-deficient mice fed a cholesterol-enriched diet developed elevated LDL levels and consequently oxidative stress [121]. These observations extend to human studies. High plasma oxidative stress markers positively correlated with elevated plasma triglycerides and inversely correlated with low HDL [122] in a group of metabolic syndrome patients with end-stage renal disease, after all other factors (presence of obesity, hypertension, and/or type II diabetes) were adjusted for. Lipid peroxidation, as an index of oxidative stress, correlated with low HDL levels, irrespective of age, gender, and presence of the other metabolic syndrome components [123]. It is also now accepted that the numerous positive effects of some statins in the cardiovascular system are independent of their lipid-lowering effect and a consequence of a direct decrease in oxidative stress. For example, short-term pravastatin treatment reduced myocardial infarct (MI) size in hypercholesterolemic rabbits through reduction in peroxynitrate and nitrotyrosine formation [124]. Similar results, with regards to the atherogenic index, were achieved with rosuvastatin, which lowered oxidative stress by elevating the expression of antioxidant enzymes (SOD, catalase, glutathione, glutathione peroxidase), LDL, triglycerides, and C-reactive protein (CRP) and elevated HDL [125, 126].
Aging and methabolic syndrome
Metabolic syndrome is a major health challenge of the twenty-first century, threatening to reverse historic trends towards ever increasing life- and healthspans in the developed world. We are on the cusp of a molecular understanding of ageing itself, and how it is regulated by diet. Aging is defined as a series of morphological and functional changes which take place over time. The term also refers to the deterioration of the biological functions after an organism has attained its maximum reproductive potential. We do not know yet whether organisms start to grow old from the moment of conception or from maturity, or if aging constitutes a process of evolution or involution [127]. Stress response genes and nutrient sensors regulate energy directed to cell protection, maintenance and longevity; when food is plentiful and stress levels are low, genes support growth and reproduction, in contrast harsh conditions favor a shift in gene activity towards cell protection and maintenance extending lifespan. Important genes in extending lifespan include kinase mammalian target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), sirtuins and insulin/insulin like growth factor 1 (IGF-1) signaling. These genes integrate longevity pathways and metabolic signals in a complex interplay in which lifespan appears to be strictly dependent on substrate and energy bioavailability [128].
IGF-1 mediated signaling is determining for longevity. Abnormalities in the insulin signaling pathway generate age-related diseases and increased mortality, whereas the growth hormone (GH)/IGF-1 axis could potentially modulate longevity in many species. Moreover in humans, an age-related decline in IGF-1 levels occurs, and at old age, low IGF-1 levels are associated with frailty, poor nutrition and cognitive decline and an increased risk of death [129,130].
The aging process is altered or accelerated when metabolic and cardiovascular diseases are present and the risk of diseases increases with age. Many predisposing conditions which increase in prevalence during aging, such as obesity, insulin resistance, inflammation, changes in the activity of the hypothalamus-hypophysis suprarenal axis, stress and hypertension also contribute to increase prevalence of metabolic syndrome. Aging, the development of insulin resistance and cardiovascular disease seem to be accelerated in the metabolic syndrome [131-133].
It is generally considered that metabolic syndrome induces precocious aging although the mechanisms that account for this are incompletely known. It is becoming clear that longevity genes might be involved. Experiments with overactivation or disruption of key lifespan determinant pathways, such as silent information regulator sirtuins, p66Shc, and mTOR, lead to development of features of the MS in mice. Additional pathways are involved in linking nutrient availability and longevity, certainly including insulin and IGF-1 signaling, as well as FOXO transcription factors [134,135, 136]. This review demonstated that in metabolic disease there is an incessant production of free radicals that therefore thought to create conditions under which oxidative modifications of cellular constituents arise, which in turn leads to mitochondrial dysfunction and eventual loss of cellular homeostasis. This argument has been strongly used as a basis for age-related deterioration in physiological systems, and led to a proposal supporting mitochondria as the “biological clock” of cellular aging [31]. According with this theory a study by Passos et al. 2007 demonstrated that the senescent cells had higher ROS levels, dysfunctional mitochondria, more DNA double-strand breaks and shorter telomeres and it was shown that mitochondrial ROS enhanced telomere-dependent senescence [137]. Recently, some authors showed the link between telomere length and metabolic disease suggesting increased cellular turnover and therefore accelerated cell aging [138, 139].
Conclusions
A role for mitochondrial oxidative stress in the development and maintenance of the metabolic syndrome has been suggested. Here, we have proposed that elevated ROS production from the mitochondrial respiratory chain acts as a signal for accelerated senescence. Under the conditions of overnutrition and physical inactivity, typical of those that foster the metabolic syndrome, chronic overactivation of the redox signalling pathways may contribute to senescence (Fig. 1). Further exploration of this proposal for the role of oxidative stress in the metabolic syndrome and the related accelerated senescence could contribute to the development of further therapies.
Figure 1.
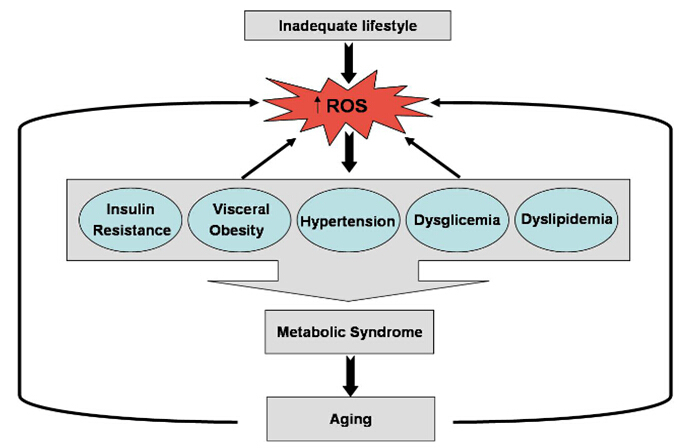
Schematic relationship between ROS production, accelerated senescence and metabolic syndrome.
References
- [1].Humphreys MH (2011). The brain splits obesity and hypertension. Nat Med, 17:782–3. [DOI] [PubMed] [Google Scholar]
- [2].Kahn SE,Hull RL,Utzschneider KM (2006). Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature, 444: 840–6. [DOI] [PubMed] [Google Scholar]
- [3].Giovannucci E,Michaud D (2007). The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology, 132:2208–25. [DOI] [PubMed] [Google Scholar]
- [4].Parekh S,Anania FA (2007). Abnormal lipid and glucose metabolism in obesity: implications for nonalcoholic fatty liver disease. Gastroenterology, 132:2191–207. [DOI] [PubMed] [Google Scholar]
- [5].Bamba V,Rader DJ (2007). Obesity and atherogenic dyslipidemia. Gastroenterology, 132:2181–90. [DOI] [PubMed] [Google Scholar]
- [6].Van Gaal LF,Mertens IL,De Block CE (2006). Mechanisms linking obesity with cardiovascular disease. Nature, 444:875–80. [DOI] [PubMed] [Google Scholar]
- [7].Semenkovich CF (2006). Insulin resistance and atherosclerosis. J Clin Invest, 116:1813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Friedman JM (2009). Obesity: Causes and control of excess body fat. Nature, 459:340–2. [DOI] [PubMed] [Google Scholar]
- [9].Ogden CL,Yanovski SZ,Carroll MD,Flegal KM (2007). The epidemiology of obesity. Gastroenterology, 132:2087–102. [DOI] [PubMed] [Google Scholar]
- [10].Yoon KH,Lee JH,Kim JW,Cho JH,Choi YH,Ko SH et al. (2006). Epidemic obesity and type 2 diabetes in Asia. Lancet, 368:1681–8. [DOI] [PubMed] [Google Scholar]
- [11].Abelson P,Kennedy D (2004). The obesity epidemic. Science, 304:1413. [DOI] [PubMed] [Google Scholar]
- [12].Zimmet P,Alberti KG,Shaw J (2001). Global and societal implications of the diabetes epidemic. Nature, 414:782–7. [DOI] [PubMed] [Google Scholar]
- [13].Despres JP,Lemieux I (2006). Abdominal obesity and metabolic syndrome. Nature, 444:881–7. [DOI] [PubMed] [Google Scholar]
- [14].Eckel RH,Grundy SM,Zimmet PZ (2005). The metabolic syndrome. Lancet, 365:1415–28. [DOI] [PubMed] [Google Scholar]
- [15].Guarente L (2006). Sirtuins as potential targets for metabolic syndrome. Nature, 444:868–74. [DOI] [PubMed] [Google Scholar]
- [16].Duvnjak L,Duvnjak M (2009). The metabolic syndrome - an ongoing story. J. Physiol. Pharmacol, 60(Suppl 7):19–24. [PubMed] [Google Scholar]
- [17].Gaddam KK,Ventura HO,Lavie CJ (2011). Metabolic syndrome and heart failure-the risk, paradox, and treatment. Curr Hypertens Rep, 13:142–8. [DOI] [PubMed] [Google Scholar]
- [18].Rodriguez A,Muller DC,Metter EJ,Maggio M,Harman SM,Blackman MR et al. (2007). Aging, androgens, and the metabolic syndrome in a longitudinal study of aging. J Clin Endocrinol Metab, 92:3568–72. [DOI] [PubMed] [Google Scholar]
- [19].Moulana M,Lima R,Reckelhoff JF (2011). Metabolic syndrome, androgens, and hypertension. Curr Hypertens Rep, 13:158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cai D,Liu T (2012). Inflammatory cause of metabolic syndrome via brain stress and NF-κB. Aging (Albany NY), 4(2):98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wilson PW,D’Agostino RB,Parise H,Sullivan L,Meigs JB (2005). Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation, 112:3066–72. [DOI] [PubMed] [Google Scholar]
- [22].Katzmaryk PT,Leon AS,Wilmore JH,Skinner JS,Rao DC,Rankinen T, et al. (2003). Targeting the metabolic syndrome with exercise: evidence from the heritage family study. Medial Sci Sports Exerc, 35(10):1703–9. [DOI] [PubMed] [Google Scholar]
- [23].Fauci AS (2008). Harrison’s principles of internal medicine. McGraw-Hill Medical; ISBN 0-07-147692-X. [Google Scholar]
- [24].Grundy SM,Cleeman JI,Daniels SR,Donato KA,Eckel RH,Franklin BA,Gordon DJ,Krauss RM,Savage PJ,Smith SC Jr,Spertus JA,Costa F; American Heart Association; National Heart, Lung, and Blood Institute (2005) Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation, 112: 2735–2752. [DOI] [PubMed] [Google Scholar]
- [25].Borch-Johnsen K,Wareham N (2010). The rise and fall of the metabolic syndrome. Diabetologia, 53: 597–599. [DOI] [PubMed] [Google Scholar]
- [26].Guize L,Thomas F,Pannier B,Bean K,Jego B,Benetos A (2007). All-cause mortality associated with specific combinations of the metabolic syndrome according to recent definitions. Diabetes Care, 30: 2381–2387. [DOI] [PubMed] [Google Scholar]
- [27].Zambon S,Zanoni S,Romanato G,Corti MC,Noale M,Sartori L et al. (2009). Metabolic syndrome and all-cause and cardiovascular mortality in an Italian elderly population: the Progetto Veneto Anziani (Pro.V.A.) study. Diabetes Care, 32:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nunn AV,Bell JD,Guy GW (2009) Lifestyle-induced metabolic inflexibility and accelerated ageing syndrome: insulin resistance, friend or foe?. Nutr Metab (Lond.), 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Haffner S,Taegtmeyer H (2003) Epidemic obesity and the metabolic syndrome. Circulation, 108: 1541–1545. [DOI] [PubMed] [Google Scholar]
- [30].Harman D (1956). Aging: a theory based on free radical and radiation chemistry. J Gerontol, 11:298–300. [DOI] [PubMed] [Google Scholar]
- [31].Harman D (1972). The biologic clock: the mitochondria?. J. Am. Geriatr. Soc., 20: 145–147. [DOI] [PubMed] [Google Scholar]
- [32].Miquel J,Economos AC,Fleming J,Johnson Jr JE (1980). Mitochondrial role in cell aging. Exp Gerontol, 15:575–591. [DOI] [PubMed] [Google Scholar]
- [33].Pak JW,Herbst A,Bua E,Gokey N,McKenzie D,Aiken JM (2003). Mitochondrial DNA mutations as a fundamental mechanism in physiological declines associated with aging. Aging Cell, 2:1–7. [DOI] [PubMed] [Google Scholar]
- [34].Wei YH (1992). Mitochondrial DNA alterations as ageing-associated molecular events. Mutat Res, 275:145–155. [DOI] [PubMed] [Google Scholar]
- [35].Richter C (1995). Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol, 27:647–653. [DOI] [PubMed] [Google Scholar]
- [36].Linnane AW,Marzuki S,Ozawa T,Tanaka M (1989). Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet, 1: 642–645. [DOI] [PubMed] [Google Scholar]
- [37].Judge S,Leeuwenburgh C (2007). Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol, 292:C1983–C1992. [DOI] [PubMed] [Google Scholar]
- [38].Sohal RS,Weindruch R (1996). Oxidative stress, caloric restriction, and aging. Science: 5: 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Beckman KB,Ames BN (1998). The free radical theory of aging matures. Physiol Rev, 78: 547–581. [DOI] [PubMed] [Google Scholar]
- [40].Cortopassi GA,Shibata D,Soong NW,Arnheim NA (1992). A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci USA, 89:7370–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hayakawa M,Hattori K,Sugiyama S,Ozawa T (1992). Age-associated oxygen damage and mutations in mitochondrial DNA in human hearts. Biochem Biophys Res Commun, 189(2):979–85. [DOI] [PubMed] [Google Scholar]
- [42].Paradies G,Ruggiero FM,Quagliariello E (1992).Age-dependent changes in the activity of anion carriers and in the lipid composition in rat heart mitochondria. Ann NY Acad Sci, 26:160–164. [DOI] [PubMed] [Google Scholar]
- [43].Paradies G,uggiero FM,Quagliariello E (1992). Age-dependent changes in the activity of anion carriers and in the lipid composition in rat heart mitochondria. Ann NY Acad Sci, 26:160–164. [DOI] [PubMed] [Google Scholar]
- [44].Shigenaga MK,Hagen TM Ames BN (1994). Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA, 91:10771–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lesnefsky EJ,Moghaddas S,Tandler B,Kerner J,Hoppel CL (2001). Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol 33:1065–1089. [DOI] [PubMed] [Google Scholar]
- [46].Paradies G,Petrosillo G,Paradies V,Ruggiero FM (2010). Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic Biol Med, 48(10):1286–95. [DOI] [PubMed] [Google Scholar]
- [47].Wellen KE,Hotamisligil GS (2005). Inflammation, stress, and diabetes. J Clin Invest, 115:1111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Stocker R,Keaney Jr JF (2004). Role of oxidative modifications in atherosclerosis. Physiol Rev, 84:1381–478. [DOI] [PubMed] [Google Scholar]
- [49].Furukawa S,Fujita T,Shimabukuro M,Iwaki M,Yamada Y,Nakajima Y et al. (2004). Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest, 114:1752–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Grattagliano I,Vendemiale G,Boscia F,Micelli-Ferrari T,Cardia L,Altomare E (1998). Oxidative retinal products and ocular damages in diabetic patients. Free Radic Biol Med, 25:369–72. [DOI] [PubMed] [Google Scholar]
- [51].Grattagliano I,Vendemiale G,Caraceni P,Domenicali M,Nardo B,Cavallari A et al. (2000). Starvation impairs antioxidant defense in fatty livers of rats fed a choline-deficient diet. J Nutr, 130(9):2131–6. [DOI] [PubMed] [Google Scholar]
- [52].Portincasa P.,Grattagliano I.,Palmieri V.O.,Palasciano G (2005). Nonalcoholic steatohepatitis: recent advances from experimental models to clinical management. Clin Biochem, 38:203–217. [DOI] [PubMed] [Google Scholar]
- [53].Sies H (1997). Oxidative stress: oxidants and antioxidants. Exp Physiol, 82:291–295. [DOI] [PubMed] [Google Scholar]
- [54].Vendemiale G.,Grattagliano I.,Altomare E (1999). An update on the role of free radicals and antioxidant defense in human disease. Int J Clin Lab Res, 29: 49–55. [DOI] [PubMed] [Google Scholar]
- [55].Knopp RH,Paramsothy P (2006). Oxidized LDL and abdominal obesity: a key to understanding the metabolic syndrome. Am J Clin Nutr, 83(1):1–2. [DOI] [PubMed] [Google Scholar]
- [56].Sáez GT,Tormos C,Giner V,Chaves J,Lozano JV,Iradi A, et al. (2004). Factors related to the impact of antihypertensive treatment in antioxidant activities and oxidative stress by-products in human hypertension. Am J Hypertens, 17:809–16. [DOI] [PubMed] [Google Scholar]
- [57].Palmieri VO,Grattagliano I,Portincasa P,Palasciano G (2006). Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome. J Nutr, 136:3022–6. [DOI] [PubMed] [Google Scholar]
- [58].Couillard C,Ruel G,Archer WR,Pomerleau S,Bergeron J,Couture P et al. (2005). Circulating levels of oxidative stress markers and endothelial adhesion molecules in men with abdominal obesity. J Clin Endocrinol Metab, 90:6454–9. [DOI] [PubMed] [Google Scholar]
- [59].Keaney JF Jr,Larson MG,Vasan RS,Wilson PW,Lipinska I,Corey D et al. (2003). Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol, 23:434–9. [DOI] [PubMed] [Google Scholar]
- [60].Pinzani M,Marra F,Carloni V (1998). Signal transduction in hepatic stellate cells. Liver, 18:2–13. [DOI] [PubMed] [Google Scholar]
- [61].Suthanthiran M.,Anderson M.E.,Sharma V.K.,Meister A (1990). Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc Natl Acad Sci U S A, 87:3343–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sahaf B,Heydari K,Herzenberg LA,Herzenberg LA (2005). The extracellular microenvironment plays a key role in regulating the redox status of cell surface proteins in HIV-infected subjects. Arch Biochem Biophys, 434:26–32. [DOI] [PubMed] [Google Scholar]
- [63].Vendemiale G,Guerrieri F,Grattagliano I,Didonna D,Muolo L,Altomare E (1995). Mitochondrial oxidative phosphorylation and intracellular glutathione compartmentation during rat liver regeneration. Hepatology, 21:1450–1454. [PubMed] [Google Scholar]
- [64].Grattagliano I,Palmieri VO,Portincasa P,Moschetta A,Palasciano G (2008). Oxidative stress-induced risk factors associated with the metabolic syndrome: a unifying hypothesis. J Nutr Biochem, 19(8):491–504. [DOI] [PubMed] [Google Scholar]
- [65].Czech MP,Lawrence JC Jr,Lynn WS (1974). Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proc Natl Acad Sci SA, 71:4173–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Higaki Y,Mikami T,Fujii N,Hirshman MF,Koyama K,Seino T et al. (2008). Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol-3-kinase dependent pathway. Am J Physiol Endocrinol Metab, 294:E889–E897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].May JM,de Haen C (1979). The insulin-like effect of hydrogen peroxide on pathways of lipid synthesis in rat adipocytes. J Biol Chem, 254:9017–9021. [PubMed] [Google Scholar]
- [68].Powell DJ,Hajduch E,Kular G,Hundal HS (2003). Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol, 23:7794–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pessler D,Rudich A,Bashan N (2001). Oxidative stress impairs nuclear proteins binding to the insulin responsive element in the GLUT4 promoter. Diabetologia, 44:2156–2164. [DOI] [PubMed] [Google Scholar]
- [70].Kamigaki M,Sakaue S,Tsujino I,Ohira H,Ikeda D,Itoh N,Ishimaru S,Ohtsuka Y,Nishimura M (2006). Oxidative stress provokes atherogenic changes in adipokine gene expression in 3T3-L1 adipocytes. Biochem Biophys Res Commun, 339:624–632. [DOI] [PubMed] [Google Scholar]
- [71].Furukawa S,Fujita T,Shimabukuro M,Iwaki M,Yamada Y,Nakajima Y et al. (2004) Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest, 114:1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Svegliati-Baroni G,Candelaresi C,Saccomanno S,Ferretti G,Bachetti T,Marzioni M et al. (2006). A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol, 169:846–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Schaffer SW,Jong CJ,Mozaffari M (2012). Role of oxidative stress in diabetes-mediated vasculardysfunction: Unifying hypothesis of diabetes revisited. Vascul Pharmacol, 57:139–149. [DOI] [PubMed] [Google Scholar]
- [74].Campia U,Tesauro M,Cardillo C (2012). Human obesity and endothelium-dependent responsiveness. Br J Pharmacol, 165:561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tesauro M,Cardillo C (2011). Obesity, blood vessels and metabolic syndrome. Acta Physiol (Oxf), 203:279–286. [DOI] [PubMed] [Google Scholar]
- [76].Stadler K (2012). Oxidative stress in diabetes. Adv Exp Med Biol, 771: 272–287. [DOI] [PubMed] [Google Scholar]
- [77].Davi G,Ciabattoni G,Consoli A,Mezzetti A,Falco A,Santarone S et al. (1999). In vivo formation of 8-iso-prostaglandin F2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation, 99:224–229. [DOI] [PubMed] [Google Scholar]
- [78].Davi G,Chiarelli F,Santilli F,Pomilio M,Vigneri S,Falco A et al. (2003). Enhanced lipid peroxidation and platelet activation in the early phase of type 1 diabetes mellitus: role of interleukin-6 and disease duration. Circulation, 107:3199–3203. [DOI] [PubMed] [Google Scholar]
- [79].Monnier L,Mas E,Ginet C,Michel F,Villon L,Cristol JP et al. (2006). Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA, 295(14):1681–7. [DOI] [PubMed] [Google Scholar]
- [80].Pitocco D,Zaccardi F,Di Stasio E,Romitelli F,Martini F,Scaglione GL et al. (2009). Role of asymmetric-dimethyl-L-arginine (ADMA) and nitrite/nitrate (NOx) in the pathogenesis of oxidative stress in female subjects with uncomplicated type 1 diabetes mellitus. Diabetes Res Clin Pract, 86(3):173–6. [DOI] [PubMed] [Google Scholar]
- [81].Pitocco D,Zaccardi F,Di Stasio E,Romitelli F,Santini SA,Zuppi C et al. (2010). Oxidative stress, nitric oxide, and diabetes. Rev Diabet Stud, 7(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wellen KE,Hotamisligil GS (2003). Obesity-induced inflammatory changes in adipose tissue. J Clin Invest, 112:1785–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lugogo NL,Bappanad D,Kraft M (2011). Obesity, metabolic dysregulation and oxidative stress in asthma. Biochim Biophys Acta, 1810:1120–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Keaney JF Jr.,Larson MG,Vasan RS,Wilson P W,LIpinska I,Corey D et al. (2003). Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol, 23: 434–439. [DOI] [PubMed] [Google Scholar]
- [85].Vincent HK,Bourguignon CM,Taylor AG (2010). Relationship of the dietary phytochemical index to weight gain, oxidative stress and inflammation in overweight young adults. J Hum Nutr Diet, 23:20–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Couillard C,Ruel G,Archer WR,Pomerleau S,Bergeron J,Couture P et al. (2005). Circulating levels of oxidative stress markers and endothelial adhesion molecules in men with abdominal obesity. J Clin Endocrinol Metab, 90:6454–9. [DOI] [PubMed] [Google Scholar]
- [87].Fujita K,Nishizawa H,Funahashi T,Shimomura I,Shimabukuro M (2006). Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circ J, 70(11):1437–42. [DOI] [PubMed] [Google Scholar]
- [88].Steffes MW,Gross MD,Lee DH,Schreiner PJ,Jacobs DR Jr (2006). Adiponectin, visceral fat, oxidative stress, and early macrovascular disease: the Coronary Artery Risk Development in Young Adults Study. Obesity, 14(2):319–26. [DOI] [PubMed] [Google Scholar]
- [89].Wu B,Fukuo K,Suzuki K,Yoshino G,Kazumi T (2009). Relationships of systemic oxidative stress to body fat distribution, adipokines and inflammatory markers in healthy middle-aged women. Endocr J, 56(6):773–82. [DOI] [PubMed] [Google Scholar]
- [90].Shaheen M,Echeverry D,Oblad MG,Montoya MI,Teklehaimanot S,Akhtar AJ (2007). Hepatitis C, metabolic syndrome, and inflammatory markers: results from the Third National Health and Nutrition Examination Survey [NHANES III]. Diabetes Res Clin Pract, 75(3):320–6. [DOI] [PubMed] [Google Scholar]
- [91].Roberts CK,Sindhu KK (2009). Oxidative stress and metabolic syndrome. Life Sci, 84(21–22):705–12. [DOI] [PubMed] [Google Scholar]
- [92].Nayak DU,Karmen C,Frishman WH,Vakili BA (2001). Antioxidant vitamins and enzymatic and synthetic oxygen-derived free radical scavengers in the prevention and treatment of cardiovascular disease. Heart Dis, 3:28–45. [DOI] [PubMed] [Google Scholar]
- [93].Kizhakekuttu TJ,Widlansky ME (2010). Natural antioxidants and hypertension: promise and challenges. Cardiovasc Ther, 28:e20–e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kitiyakara C,Wilcox C (1998). Antioxidants for hypertension. Curr Opin Nephrol Hypertens, 7:531–538. [DOI] [PubMed] [Google Scholar]
- [95].Russo C,Olivieri O,Girelli D,Faccini G,Zenari ML,Lombardi S,Corrocher R (1998). Antioxidant status and lipid peroxidation in patients with essential hypertension. J Hypertens, 16:1267–1271. [DOI] [PubMed] [Google Scholar]
- [96].Tse WY,Maxwell SR,Thomason H,Blann A,Thorpe GH,Waite M et al. (1994). Antioxidant status in controlled and uncontrolled hypertension and its relationship to endothelial damage. J Hum Hypertens.;8:843–849. [PubMed] [Google Scholar]
- [97].Mansego ML,Solar Gde M,Alonso MP,Martínez F,Sáez GT,Escudero JC et al. (2011) Polymorphisms of antioxidant enzymes, blood pressure and risk of hypertension. J Hypertens, 29:492–500. [DOI] [PubMed] [Google Scholar]
- [98].Galley HF,Thornton J,Howdle PD,Walker BE,Webster NR (1997). Combination oral antioxidant supplementation reduces blood pressure. Clin Sci, 92:361–365. [DOI] [PubMed] [Google Scholar]
- [99].Broadhurst CL (1997). Balanced intakes of natural triglycerides for optimum nutrition: an evolutionary and phytochemical perspective. Med Hypotheses, 49:247–261. [DOI] [PubMed] [Google Scholar]
- [100].Houston MC (2007). Treatment of hypertension with nutraceuticals. vitamins, antioxidants and minerals. Expert Rev Cardiovasc Ther, 5:681–691. [DOI] [PubMed] [Google Scholar]
- [101].Dhalla NS,Temsah RM,Netticadam T (2000). The role of oxidative stress in cardiovascular diseases. J Hypertens, 18:655–673. [DOI] [PubMed] [Google Scholar]
- [102].Touyz RM (2004). Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension, 44(3):248–52. [DOI] [PubMed] [Google Scholar]
- [103].Nishihara M,Hirooka Y,Matsukawa R,Kishi T,Sunagawa K (2012). Oxidative stress in the rostral ventrolateral medulla modulates excitatory and inhibitory inputs in spontaneously hypertensive rats. J. Hypertension, 30: 97–106. [DOI] [PubMed] [Google Scholar]
- [104].Konno S,Hirooka Y,Kishi T,Sunagawa K (2012). Sympathoinhibitory effects of telmisartan through the reduction of oxidative stress in the rostral ventrolateral medulla of obesity-induced hypertensive rats. J Hypertens, 30:1992–1999. [DOI] [PubMed] [Google Scholar]
- [105].Houston M (2013). Nutrition and nutraceutical supplements for the treatment of hypertension: part I. J Clin Hypertens, 15(10):752–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Brownlee M (2001). Biochemistry and molecular cell biology of diabetic complications. Nature, 414: 813–820. [DOI] [PubMed] [Google Scholar]
- [107].Avignon A,Radauceanu A,Monnier L (1997). Nonfasting plasma glucose is a better marker of diabetic control than fasting plasma glucose in type 2 diabetes. Diabetes Care, 20:1822–1826. [DOI] [PubMed] [Google Scholar]
- [108].El-Kebbi IM,Ziemer DC,Cook CB,Gallina DL,Barnes CS,Philipps LS (2004). Utility of casual postprandial glucose levels in type 2 diabetes management. Diabetes Care, 27:335–339. [DOI] [PubMed] [Google Scholar]
- [109].Rohlfing CL,Wiedmeyer HS,Little RR,England JD,Tennill A,Goldstein DE (2002). Defining the relationship between plasma glucose and HbA1c. Diabetes Care, 25:275–278. [DOI] [PubMed] [Google Scholar]
- [110].American Diabetes Association (2007). Standards of medical care in diabetes-2007. Diabetes Care, 30(S1):S4–S41. [DOI] [PubMed] [Google Scholar]
- [111].Sacks DB,Bruns DE,Goldstein DE,Mac Laren NK,Mac Donald JM,Parrott M (2002). Guidelines and recommendations for laboratory analysis in the diagnosis and management of diabetes mellitus (position statement). Diabetes Care, 25:750–786. [Google Scholar]
- [112].Gorus F,Mathieu C,Gerlo E (2006). How should HbA1 measurements be reported? Diabetologia, 49:7–10. [DOI] [PubMed] [Google Scholar]
- [113].Morrow JD,Hill KE,Burk RF,Nammour TM,Badr KF,Roberts LJ (1991). A series of prostaglandin F2-like compounds are produced in vivo in humans by non-cyclooxygenase free radical-catalyzed mechanism. Proc Natl Acad Sci USA, 87:9383–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Roberts LJ,Morrow JD (2000). Measurement of F (2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Bio Med, 28:505–513. [DOI] [PubMed] [Google Scholar]
- [115].Basu S (1998). Metabolism of 8-iso-prostaglandin F2α. FEBS Lett, 428:32–36. [DOI] [PubMed] [Google Scholar]
- [116].Davi G,Falco A,Patrono C (2005). Lipid peroxidation in diabetes mellitus. Antioxid Redox Signal, 7:256–258. [DOI] [PubMed] [Google Scholar]
- [117].Monnier L,Mas E,Ginet C,Michel F,Villon L,Cristol J-P et al. (2006). Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA, 295:1681–1687. [DOI] [PubMed] [Google Scholar]
- [118].Colette C,Monnier L (2007). Acute glucose fluctuations and chronic sustained hyperglycemia as risk factors for cardiovascular diseases in patients with type 2 diabetes. Horm Metab Res, 39(9):683–6. [DOI] [PubMed] [Google Scholar]
- [119].Ruotolo G,Howard BV (2002). Dyslipidemia of the metabolic syndrome. Curr Cardiol Rep, 4:494–500. [DOI] [PubMed] [Google Scholar]
- [120].Jeppesen J,Hollenbeck CB,Zhou MY,Coulston AM,Jones C,Chen YD et al. (1995). Relation between insulin resistance, hyperinsulinemia, postheparin plasma lipoprotein lipase activity, and postprandial lipemia. Arterioscler Thromb Vasc Biol, 15:320–4. [DOI] [PubMed] [Google Scholar]
- [121].de Oliveira J,Hort MA,Moreira EL,Glaser V,Ribeiro-do-Valle RM,Prediger RD et al. (2011). Positive correlation between elevated plasma cholesterol levels and cognitive impairments in LDL receptor knockout mice: relevance of cortico-cerebral mitochondrial dysfunction and oxidative stress. Neuroscience, 197:99–106. [DOI] [PubMed] [Google Scholar]
- [122].Marques de Mattos A,Marino LV,Ovidio PP,Jordão AA,Almeida CC,Chiarello PG (2012). Protein oxidative stress and dyslipidemia in dialysis patients. Ther Apher Dial, 16:68–74. [DOI] [PubMed] [Google Scholar]
- [123].Zelzer S,Fuchs N,Almer G,Raggam RB,Prüller F,Truschnig-Wilders M et al. (2011). High density lipoprotein cholesterol level is a robust predictor of lipid peroxidation irrespective of gender, age, obesity, and inflammatory or metabolic biomarkers. Clin Chim Acta, 412:1345–9. [DOI] [PubMed] [Google Scholar]
- [124].Andreadou I,Farmakis D,Prokovas E,Sigala F,Zoga A,Spyridaki K et al. (2012). Short-term statin administration in hypercholesterolaemic rabbits resistant to postconditioning: effects on infarct size, endothelial nitric oxide synthase, and nitro-oxidative stress. Cardiovasc Res, 94:501–9. [DOI] [PubMed] [Google Scholar]
- [125].Ansari JA,Bhandari U,Pillai KK,Haque SE (2012). Effect of rosuvastatin on obesity-induced cardiac oxidative stress in Wistar rats--a preliminary study. Indian J Exp Biol, 50(3):216–22. [PubMed] [Google Scholar]
- [126].Hutcheson R,Rocic P (2012). The metabolic syndrome, oxidative stress,environment, and cardiovascular disease: the great exploration. Exp Diabetes Res, 2012: 271028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Guarner V,Carbó R,Rubio ME, Baños de MacCarthy G (2005). Aging of the cardiovascular system. In:Benhagen EF, editor. Hypertension: New research. Nova Biomedical Books; USA, 47–68. [Google Scholar]
- [128].Kenyon CJ (2010). The genetics of aging. Nature, 464:504–512. [DOI] [PubMed] [Google Scholar]
- [129].Rincon M,Rudin E,Barzilai N (2005). The insulin/IGF-1 signaling in mammals and its relevance to human longevity. Exp Gerontol, 40:873–877. [DOI] [PubMed] [Google Scholar]
- [130].Rozing MP,Westendorp RGJ,Frölich M,de Craen AJM,Beekman M,Heijmans BT et al. (2009). Human insulin/IGF-1 and familial longevity at middle age. Aging, 1:714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Guarner V,Carbó R,Rubio ME,Baños de MacCarthy G (2005). Aging of the cardiovascular system. In:Benhagen EF, editor. Hypertension: New Research. USA: Nova Biomedical Books Publishers, 47–68. [Google Scholar]
- [132].Baños G,El Hafidi M,Pérez-Torres I,Guarner V (2009). Insulin resistance and the metabolic syndrome. In:Yao EB, editor. Insulin Resistance: New Research. USA: Nova Biomedical Books Publishers, 49–97. [Google Scholar]
- [133].Guarner V,Rubio-Ruiz ME,Perez-Torres I,Baños de McCarthy G (2011). Relation of aging and sex hormones to metabolic syndrome and cardiovascular disease. Exp Gerontol, 46:517–523. [DOI] [PubMed] [Google Scholar]
- [134].Fadini GP,Ceolotto G,Pagnin E,de Kreutzenberg S,Avogaro A (2011). At the crossroads of longevity and metabolism: the metabolic syndrome and lifespan determinant pathways. Aging Cell, 10:10–17. [DOI] [PubMed] [Google Scholar]
- [135].Ren J,Pulakat L,Whaley-Connell A,Sowers JR (2010). Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. Mol Med, 88:993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Veronica G,Esther RR (2012). Aging, metabolic syndrome and the heart. Aging Dis, 3:269–79. [PMC free article] [PubMed] [Google Scholar]
- [137].Passos JF,Saretzki G,Ahmed S,Nelson G,Richter T,Peters H et al. (2007). Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol, 5:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Bonomini F,Rodella LF,Moghadasian M,Lonati C,Coleman R,Rezzani R (2011). Role of apolipoprotein E in renal damage protection. Histochem Cell Biol, 135(6):571–9. [DOI] [PubMed] [Google Scholar]
- [139].Kong CM,Lee XW,Wang X (2013). Telomere shortening in human diseases. FEBS J, 280:3180–93. [DOI] [PubMed] [Google Scholar]