

Abstract
PERK, PKR, HRI and GCN2 are the four mammalian kinases that phosphorylate the α subunit of the eukaryotic translation initiation factor 2 (eIF2α) on Ser51. This phosphorylation event is conserved among many species and attenuates protein synthesis in response to diverse stress conditions. In contrast, Saccharmyces cerevisiae expresses only the GCN2 kinase. It was demonstrated previously in S. cerevisiae that single point mutations in eIF2α’s N-terminus severely impaired phosphorylation at Ser51. To assess whether similar recognition patterns are present in mammalian eIF2α, we expressed human eIF2α’s with these mutations in mouse embryonic fibroblasts and assessed their phosphorylation under diverse stress conditions. Some of the mutations prevented the stress-induced phosphorylation of eIF2α by all mammalian kinases, thus defining amino acid residues in eIF2α (Gly 30, Leu 50, and Asp 83) that are required for substrate recognition. We also identified residues that were less critical or not required for recognition by the mammalian kinases (Ala 31, Met 44, Lys 79, and Tyr 81), even though they were essential for recognition of the yeast eIF2α by GCN2. We propose that mammalian eIF2α kinases evolved to maximize their interactions with the evolutionarily conserved Ser51 residue of eIF2α in response to diverse stress conditions, thus adding to the complex signaling pathways that mammalian cells have over simpler organisms.
Keywords: stress response, translational control, translation initiation, protein kinases
1. Introduction
The global inhibition of mRNA translation is a widespread cellular response to environmental stressors. By limiting protein synthesis during stress, this adaptation conserves energy and spares limited resources. It also fosters the selective translation of mRNAs encoding stress response proteins. Eukaryotic initiation factor 2 (eIF2) activity is pivotal to the initiation of translation since it delivers the charged initiator tRNA to the 40S ribosomal subunit. This activity is inhibited by the phosphorylation of eIF2’s α subunit on S51, which promotes the formation of an inactive eIF2-GDP-eIF2B complex (Kapp and Lorsch, 2004).
In Saccharomyces cerevisiae, eIF2α is phosphorylated by the GCN2 kinase, which is activated primarily by amino acid limitation, although other stresses can also elicit a response (Deng et al., 2002, Zaborske et al., 2009). In mammalian cells, three additional eIF2α kinases have been identified (Clemens, 2001, Donnelly et al., 2013, Zhang et al., 2002). PERK is activated by the accumulation of misfolded proteins within the endoplasmic reticulum (ER) during ER stress (Harding et al., 1999, Kaufman, 2004, Majumder et al., 2012). PKR is activated in response to viral infection and oxidative stress (Garcia et al., 2007, Harding, Zhang, 1999) while HRI is activated by the deficiency of heme (Han et al., 2001). PERK, PKR, and GCN2 are present in all mammalian cells (Donnelly, Gorman, 2013) whereas HRI is active primarily in erythroid precursor cells (Han, Yu, 2001). Remarkably, all four kinases phosphorylate the same S51 residue of eIF2α (Donnelly, Gorman, 2013).
Previously, it has been demonstrated that the N-terminal 180 amino acid residues of eIF2α are necessary for the phosphorylation of S51 (Dey et al., 2005). A screen for mutants in S. cerevisiae identified key residues in this region that are required for phosphorylation of S51 by endogenous GCN2 as well as by mammalian PKR and HRI, and C. elegans PERK (Dey, Trieselmann, 2005, Vazquez de Aldana et al., 1993). The N-terminal region of yeast eIF2α has 56% homology with its human counterpart and the first 100 residues have 75% homology. Notably, there is perfect conservation of this region between human and mouse. Of particular significance is the conserved K79GYID83 sequence, which is thought to facilitate the interaction between PKR and human eIF2α in vitro (Sharp et al., 1997). One model proposed that the removal or alteration of this sequence may alter eIF2α’s tertiary structure in a way that impedes kinase-substrate interaction (Dar et al., 2005).
Although the N-terminal region of mammalian eIF2α is highly similar to the yeast protein, it is not clear whether the important residues identified in yeast (Dey, Trieselmann, 2005) are also important for the function of the mammalian protein. In addition, because mammals have four different eIF2α kinases, it is not known whether the same residues of eIF2α are required for interaction with all the kinases. To investigate these questions, we prepared a set of expression vectors for WT human eIF2α and seven of these mutants (Dey, Trieselmann, 2005) and examined the stress-induced phosphorylation of these mutant eIF2α’s in mouse embryonic fibroblast (MEF) cell lines. We show that three of the seven amino acids in eIF2α that are required for phosphorylation in yeast are also required in the mammalian protein. However, two amino acids required for phosphorylation of yeast eIF2α are not required in the human protein. In addition several other mutations have differential effects on phosphorylation of eIF2α by different kinases. These demonstrate the importance of the N-terminal region of mammalian eIF2α in translational regulation and provide clues to the specificity of the interactions with the kinases that regulate the activity of this protein.
2. Materials and Methods
2.1 Cell Culture
Wild type, S51A mutant eIF2α (A/A), PERK−/−, and GCN2−/− MEF cell lines were a generous gift from Dr. R. Kaufman (Sanford-Burnham Medical Research Institute, La Jolla, CA). Human embryonic kidney 293A cells were from Dr A. Koromilas, McGill University. Cell lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum (FBS; Life Technologies), 2 mM glutamine, 100 U/ml penicillin, and 0.1 μg/ml streptomycin at 37°C and 5% CO2.
2.2 eIF2α adenoviral expression vectors
A plasmid with a cDNA encoding the open reading frame of human eIF2α with an N-terminal HA tag was used as a template for site-directed mutagenesis. Plasmids with the G30R, A31T, and D83A mutations were produced by polymerase chain reaction (PCR) using PfuUltra II Fusion HS DNA Polymerase (Agilent Technologies) and the primers in Supplemental Table I. The M44K, L50P, S51A, S51D and K79D mutants were from Mutagenex, Inc. Each plasmid was verified by DNA sequencing.
TOPO vector entry clones containing the eIF2α inserts were prepared using the pENTR Directional TOPO Cloning Kit from Invitrogen. Adenoviral expression vectors containing the eIF2α cDNA were prepared using the pAd/CMV/V5-DEST Gateway Vector Kit from Invitrogen. PCR primers were obtained from Integrated DNA Technologies, and PCR experiments were performed using either Expand High Fidelity polymerase or Platinum Pfx polymerase using the manufacturers’ protocols.
Plasmid DNAs were transfected into 293A cells with Lipofectamine 2000 (Invitrogen) and transfected cultures were followed until plaques were observed. Crude lysates were prepared and virus was amplified in 293A cells, all following the manufacturer’s instructions. Titers were determined using 293A cells by plaque assay as described before (Guan et al., 2012).
For experiments, MEF cell lines were grown to 50% confluency in 10 cm dishes, the medium was replaced, and ~1.5 × 107 plaque-forming units of adenoviral vector were added. Cells were used for experiments after overnight culture.
2.3 Stress conditions
Oxidative stress was induced with sodium arsenite (500 μM) (Saikia et al., 2012) and ER stress was induced with thapsigargin (400 nM; Tg) (Guan et al., 2014). For hypertonic treatment, cells were incubated in DMEM supplemented with 300 mM sucrose for a final osmolality of 600 mosmol/kg as described (Bevilacqua et al., 2010). Cells were starved for amino acids by culturing in Krebs Ringer bicarbonate buffer with 10% dialyzed FBS (Fernandez et al., 2001). In all cases, cells were harvested after 1 h. Cells were transfected with polyinosinic:polycytidylic acid (poly(I:C), Sigma-Aldrich, 1 μg/ml) using Lipofectamine 2000 in Opti-MEM medium for 4 h and harvested after 2 h of culture in growth medium.
2.4 Molecular modeling
The crystal structure of the catalytic domain of human PKR bound to the N-terminal region of yeast eIF2α (PDB: 2A19) (Dar, Dever, 2005) was used to guide the construction of molecular models of mammalian and yeast kinase:eIF2α complexes. Four complexes were modeled: mouse PKR (mPKR), GCN2 (mGCN2), and PERK (mPERK) bound to human eIF2α (heIF2α), and yeast GCN2 (yGCN2) bound to yeast eIF2α (yeIF2α).
For the mPKR:heIF2α complex, a homology model of mPKR was constructed using the crystal structure of hPKR as a template (70% amino acid sequence identity for modeled region), and the crystal structure of heIF2α (PDB: 1KL9) (Nonato et al., 2002) was superimposed onto yeIF2α (r.m.s.d. of 1.5 Å for 241 superimposed Cα atoms). A model of mGCN2:heIF2α was similarly constructed by superimposing the crystal structure of yGCN2 (PDB: 1ZYD) (Padyana et al., 2005) onto hPKR (r.m.s.d. of 2.0 Å for 228 superimposed Cα atoms) before constructing a homology model of mGCN2 (49% sequence identity for modeled region), and superimposing the crystal structure of heIF2α onto yeIF2α. For the mPERK:heIF2α complex, the crystal structure of mPERK (PDB: 3QD2) (Cui et al., 2011) was superimposed onto hPKR (r.m.s.d. of 1.4 Å for 234 superimposed Cα atoms), and the crystal structure of heIF2α onto yeIF2α. Finally, a model of the yGCN2:yeIF2α complex was constructed as described for the mGCN2:heIF2α model. The crystal structures of free yeIF2α (PDB: 1Q46) (Dhaliwal and Hoffman, 2003) and free hPKR (PDB: 3UIU) were also used to identify regions that may be involved in conformational change upon complex formation or in additional interactions. Homology and molecular modeling were carried out with the computer program Coot (Emsley et al., 2010), and molecular figures prepared with Molscript (Kraulis, 1991).
2.5 Other Methods
Cell extracts were prepared and analyzed by SDS gel electrophoresis and Western blotting as previously described (Krokowski et al., 2013) using rabbit antibodies to phosphorylated eIF2α, PERK, and PKR (Cell Signaling), and mouse monoclonal antibodies to eIF2α and PKR (Cell Signaling), tubulin (Sigma-Aldrich) and the HA epitope (Roche). Horseradish peroxidase-labeled secondary antibodies were used and the signals were visualized by chemiluminescence using Western Lightning Plus-ECL (Perkin Elmer).
3. Results
3.1 Identification of critical residues in the N-terminal region of mammalian eIF2α that influence its phosphorylation under diverse cellular stress conditions
Dey et al (Dey, Trieselmann, 2005) identified mutations in the N-terminal domain of S. cerevisiae eIF2α that abolished recognition by the GCN2 kinase. We wished to study the effects of these mutations in a mammalian eIF2α. This was accomplished by expressing human eIF2α in A/A cells from a replication-incompetent adenoviral vector. This system allowed us to assess the phosphorylation of the expressed eIF2α’s because the endogenous protein contains an S51A mutation at the phosphorylation site that prevents its phosphorylation. WT eIF2α, seven of the mutants identified by Dey et al. (Dey, Trieselmann, 2005), as well as two mutants that lack the phosphorylation site (S51A and S51D) were studied. All the mutant proteins were expressed at levels comparable to WT, based on Western blotting against the HA epitope tag, with the exception of S51D which was expressed at lower levels (Fig. 1).
Fig. 1.

Mutations in eIF2α affect its phosphorylation during ER stress, oxidative stress, and amino acid starvation. WT and mutant eIF2α’s were expressed in A/A cells using adenoviral vectors. After the cells were treated with Tg, Ars, or cultured in amino-acid deficient medium for 1 h, lysates were prepared and analyzed by Western blotting for the indicated proteins.
In higher eukaryotes, specific stresses activate four kinases that phosphorylate eIF2α to cause translational reprogramming. We examined the effects of five stress conditions in our experimental system that activate these kinases. We first studied the effects of thapsigargin (Tg), which activates the PERK kinase as part of the unfolded protein response in the ER. As expected, Tg caused a large increase in the phosphorylation of WT eIF2α, whereas no phosphorylation was seen with the S51A or S51D mutants that abolish the phosphorylation target (Fig. 1A–B). Among the mutants identified by Dey at al. (Dey, Trieselmann, 2005), Tg-induced phosphorylation of the G30R, L50P, and D83A eIF2α mutants was much less than WT (Fig. 1A–C), similar to the findings in yeast. However, the A31T, M44K, K79D and Y81D mutants showed increases in S51 phosphorylation similar to WT, as compared to their untreated counterparts (Fig. 1A–C), in contrast to the finding in yeast.
We tested 3 additional stresses that lead to increased eIF2α-P: (i) A/A cells were cultured in amino acid-deficient medium, which induces GCN2 kinase activity (Fig. 1D–E) (Zhang, McGrath, 2002). (ii) Oxidative stress was induced by treating cells with sodium arsenite (Fig. 1B–D) (Saikia, Krokowski, 2012). (iii) Cells were transfected with poly(I:C) (Fig. 2A–B). The last two stressors activate the PKR kinase (Fernandez et al., 2002). The activation of the PKR kinase by poly(I:C) is demonstrated by the shift of the PKR to a slower mobility due to its autophosphorylation upon binding of the double stranded poly(I:C). Most of the eIF2α mutants showed responses to these stresses that were similar to the effects of Tg: the G30R, L50P, and D83A mutants showed little or no increase in S51 phosphorylation, while A31T and M44K showed a significant increase in eIF2α phosphorylation as compared to their corresponding untreated controls. The K79D and Y81D mutants also showed increased phosphorylation by these stresses. In conclusion, all four stresses tested showed similar patterns of mutant eIF2α phosphorylation.
Fig. 2.

Mutations in eIF2α affect its phosphorylation in response to hypertonic stress and poly(I:C) treatment. WT and mutant eIF2α’s were expressed in A/A cells, the cells were subjected to stresses, and lysates were analyzed by Western blotting as in Fig. 1.
Finally, we tested the effects of hypertonic stress by culturing the A/A cells in medium with about twice the osmolality of normal medium (Bevilacqua, Wang, 2010, Saikia, Krokowski, 2012). This stress is unique because it is not well documented which kinase phosphorylates eIF2α under these conditions (Burg et al., 2007). The effects of these mutations on eIF2α phosphorylation in response to hypertonic stress were similar to Tg: the G30R, L50P, and D83A mutants were not phosphorylated whereas the A31T and M44K mutants showed increased phosphorylation (Fig. 2C–E). In contrast, the K79D and Y81D mutants, showed little or no increase of eIF2α-P during hypertonic stress. The latter is in contrast to the increased eIF2α phosphorylation that we observed with the K79D and Y81D mutants in all other stress conditions. An explanation for this finding could be that macromolecular crowding in response to hypertonic stress may affect the binding of these eIF2α mutants with cytosolic chaperones, thus affecting its proper folding into a tertiary structure.
Collectively, we conclude that the G30R, L50P, and D83A mutations prevented eIF2α phosphorylation in response to all the stresses tested, while the A31T and M44K mutations did not affect stress-induced phosphorylation. Moreover, K79D and Y81D showed variable phosphorylation under the different stresses, despite their position within the strongly conserved K79GYID83 region of eIF2α. Because all of these residues are important for the stress-induced phosphorylation of yeast eIF2α, our results suggest that the recognition of human eIF2α by its cognate kinases requires different protein-protein interactions than the ones important in yeast. It also suggests that the recognition and phosphorylation of human eIF2α requires different interactions with the various kinases. Finally, we observed that four of the mutant eIF2α’s, M44K, L50P, K79D, and Y81D, had higher basal phosphorylation than WT in unstressed cells in the majority of our experiments (Fig. 1). This can be explained if these mutations make the eIF2α protein more accessible to sporadic low activation of the eIF2α kinases in normal cells. These data further suggest that certain residues of the mammalian eIF2α may contribute to the maintenance of the protein in a conformation that cannot be phosphorylated under basal conditions.
3.2 Verification of kinase specificity during ER stress and amino acid starvation
To verify which kinases are activated by some of the stress conditions used in our experiments the phosphorylation of eIF2α in cell lines defective in PERK (PERK−/−) and GCN2 (GCN2−/−) was studied. Previous reports have established that PERK−/− cells do not show increased eIF2α phosphorylation when treated with Tg (Mounir et al., 2011). Similarly, GCN2−/− cells do not increase eIF2α phosphorylation under conditions of amino acid starvation (Zhang, McGrath, 2002). We therefore reasoned that if transfection of mutant eIF2α’s results in increased eIF2α phosphorylation in the kinase-deficient cells under the corresponding stress conditions, it should derive from the mutant and not the endogenous WT eIF2α. WT and GCN2−/− MEFs were infected with eIF2α’s and assayed for eIF2α phosphorylation in response to amino acid deprivation (Fig. 3A). As expected, the phosphorylation of WT eIF2α was stimulated by amino acid deprivation in WT cells, but not in cells without GCN2. The shift of the GCN2 protein to a higher molecular weight further supported its activation by amino acid starvation. The absence of increased eIF2α phosphorylation in GCN2−/− cells transfected with the A31T mutant of eIF2α suggested that the transfected mutant eIF2α is not phosphorylated in GCN2−/− cells during amino acid starvation, similar to endogenous eIF2α. These findings confirm that GCN2 is the kinase that phosphorylates WT and mutant eIF2α’s during amino acid deprivation in our cells. A similar experiment was performed with Tg-treated WT and PERK−/− cells (Fig. 3B). As expected PERK was activated (notice the shifted band) and phosphorylation of eIF2α was induced by Tg treatment in WT but not in PERK−/− cells, confirming the role of this kinase in the phosphorylation of WT and mutant eIF2α’s during ER stress.
Fig. 3.
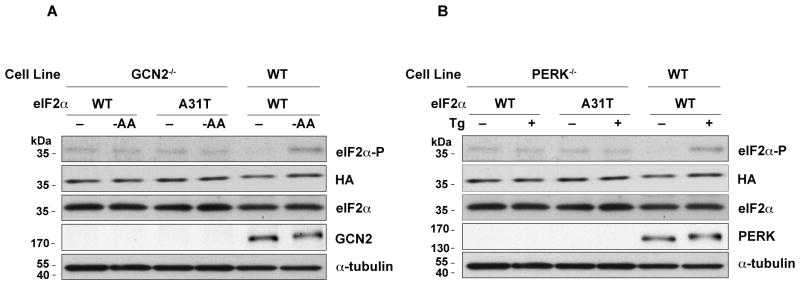
Verification of kinase specificity during ER stress and amino acid starvation. The indicated eIF2α species were expressed in WT cells and GCN2−/− (A) or PERK−/− (B) cells. Cells were subjected to amino acid deprivation (A) or Tg treatment (B) for 1 h, lysates prepared and analyzed by Western blotting.
4. Discussion
Prior studies have discovered that residues distant from S51 affect recognition of yeast eIF2α by the eIF2α kinase family (Dey, Trieselmann, 2005). In this study, we tested these patterns of eIF2α substrate recognition in mammalian cells under a wide variety of cellular stress conditions. The stress-induced phosphorylation of human eIF2α was prevented by the G30R, L50P, and D83A mutations in the N-terminal domain, which had similar effects on yeast eIF2α. However, the A31T, M44K, K79D, and Y81D mutations did not affect the phosphorylation of human eIF2α even though they prevented the phosphorylation of the yeast protein.
We examined homology models of human eIF2α and the murine kinases to explain these results. Crystal structures of the N-terminal ~175 residues of both mammalian and yeast eIF2α show a conserved protein fold that is predominantly β-sheet in the N-terminal half and α-helical in the C-terminal half (Fig. 4A). The S51 phosphorylation site is located in a surface loop between β-strands that is ordered in the crystal structure of free yeast eIF2α (Dhaliwal and Hoffman, 2003) but missing in the crystal structures of free human eIF2α (Nonato, Widom, 2002) and of yeast eIF2α bound to human PKR (Dar, Dever, 2005), presumably due to high flexibility (Fig. 4A). When superimposing the structure of free yeast eIF2α onto the yeast eIF2α in the human PKR:yeast eIF2α complex, the S51 side chain is >17 Å from the γ-phosphate of the ATP bound in the PKR active site, and thus at a distance too great, and oriented in a direction inappropriate, for phosphorylation. It is clear that the flexibility of the S51 loop is important in sampling a conformation that will bring the S51 side chain to the kinase active site for phosphorylation. The L50P mutation, which prevents phosphorylation, alters a residue adjacent to S51, and the relatively rigid proline main chain ring would reduce the flexibility of this loop. The likely consequence would be compromising eIF2α binding to kinases and reducing the accessibility of S51 to the kinase active sites.
Fig. 4.

Structural modeling of the PKR-eIF2α interactions. (A) Structural model of the mPKR-eIF2α complex. The mPKR homology model and heIF2α are shown as gray and green ribbon diagrams, respectively. The disordered loop in mPKR is represented as a dashed line; the loop in heIF2α containing the S51 phosphorylation site (red sphere) is disordered, and the ordered loop in yeIF2α is superimposed for reference (yellow). The K79-D83 motif constitutes a β-strand and preceding turn (yellow). Residues that yielded active mutants (cyan) and residues that gave inactive mutants (pink and red) are shown as spheres. (B) Modeling of the D83A mutant. The heIF2α D83 is located in the K79-D83 motif (yellow β-strand) and its side chain (ball-and-stick with yellow carbon atoms) forms hydrogen bonds (black dotted lines) with the main chain nitrogen atoms of mPKR E451 and S452, in addition to the side chain of heIF2α R74. The loss of the side chain carboxylate moiety in the D83A mutant (side chain as pink ball-and-stick) would remove these interactions. (C) Modeling of the K79D mutant. Also located in the K79-D83 motif, the heIF2α K79 side chain (ball-and-stick with yellow carbon atoms) forms a salt bridge with mPKR E453 (ball-and-stick with gray carbon atoms; interaction shown as black dotted lines). The K79D mutant (ball-and-stick with pink carbon atoms) can be modeled such that the direct K79-E453 interaction is replaced with a bridging water molecule (pink sphere) that forms hydrogen bonds (thin pink dotted lines).
The G30R mutation is also predicted to alter eIF2α structure and thus its binding to kinases. In the yeast eIF2α:human PKR structure, G30 is in a β-strand and immediately adjacent to a tight turn whose two residues, M29 and E28, directly interact with PKR via hydrophobic packing and hydrogen bonds to main chain atoms, respectively (Dar, Dever, 2005). The E28-M29-G30 region is conserved in sequence and conformation in yeast eIF2α and human eIF2α, and the interactions involving the M29 and E28 side chains with mouse PKR, GCN2, and PERK are predicted to be conserved in the modeled complexes. The G30R substitution replaces the minimal G30 with a bulky, charged arginine, which can only be accommodated by altering the main chain conformation of both the β-strand containing the G30P and the adjacent E28-M29 turn. The predicted consequence is an alteration of the eIF2α structure that affects its recognition by eIF2α kinases.
D83 is in the conserved K79GYID83 motif that is located in a β-strand that forms part of the yeast eIF2α:human PKR binding site: the K79 and D83 side chains form electrostatic interactions with side chain and main chain atoms in PKR, respectively, while Y81 participates in hydrophobic interactions with PKR residues (Dar, Dever, 2005). The D83A mutation removes two hydrogen bonds to main chain nitrogen atoms in human PKR, as well as an internal salt bridge to R74 in an adjacent β-strand, which in turn forms stacking interactions with the human PKR residues (Fig. 4B). While the smaller alanine side chain can be easily spatially accommodated, the loss of the hydrogen bonds is predicted to compromise binding to the kinase. Homology models show that the interactions involving the K79GYID83 would be conserved in the mouse PKR, GCN2, and PERK complexes, and provide an explanation for how the D83A mutation would affect recognition by eIF2α kinases.
While computational modeling suggests that G30R, L50P, and D83A have structural consequences that affect kinase recognition and phosphorylation, similar calculations for A31T, M44K, K79D, and Y81D do not predict any significant structural changes that would affect kinase recognition. For example, while K79D and Y81D alter residues in the conserved K79GYID83 motif and are found at the kinase-binding site, the mutants are compatible with the crystallized and modeled structures. The K79 side chain is partially solvent-exposed, and thus the eIF2α protein fold can easily accommodate the substitution of the large hydrophilic lysine with the smaller hydrophilic aspartic acid. K79 is located in a tight turn at the beginning of a β-strand, and its side chain forms salt bridges with human PKR D486 and E490 side chains in the yeast eIF2α:human PKR crystal structure (Dar, Dever, 2005). Homology modeling indicates that the interaction with the human PKR D486 is not important since this aspartic acid is poorly conserved among the eIF2α kinase family. In addition, the human PKR E490 is conserved among the eIF2α kinases, suggesting that this interaction may be important for eIF2α recognition. However, it is possible to computationally replace this interaction with a bridging water between the eIF2α K79D and human PKR E490 (E453 in mouse PKR) residues (Fig. 4C).
Y81D is located in the β-strand portion of the conserved K79GYID83 motif, and packs against the side chains of human PKR residues T487, F489, E490, and K493 in the yeast eIF2α:human PKR crystal structure (Dar, Dever, 2005). Of these four human PKR residues, only T487 and E490 are conserved in the mouse PKR, PERK, and GCN2; however, the residues homologous to F489 and K493 can also be modeled to form hydrophobic interactions with eIF2α Y81. Since the Y81 side chain is partially solvent-exposed, the Y81D mutant can be modeled such that the aliphatic portion of the side chain retains hydrophobic interactions with PKR and the charged carboxylate moiety is directed out to the solvent. Computational molecular modeling thus shows that both K79D and Y81D mutants do not require significant conformational change in the eIF2α:kinase complex, consistent with the observation that these mutants can still be phosphorylated.
Computational modeling also shows that A31T and M44K mutants are compatible with the yeast eIF2α:human PKR crystal structure and homology models. A31 is in a β-strand and adjacent to the G30 residue which, when replaced in G30R, requires a conformational change that is predicted to compromise kinase recognition. In contrast, the A31 side chain is partially solvent-exposed and directed away from the PKR binding interface (Dar, Dever, 2005); the A31T mutant can be modeled in both yeast eIF2α and human eIF2α without introducing any unfavorable interactions or requiring any conformational change from the native protein structure. M44 is located in a β-strand and packs against human PKR residues A488, F489, and S492. While these three human PKR residues are not all conserved among mouse PKR, PERK, and GCN2, M44 forms similar hydrophobic interactions with the homologous kinase residues. In M44K, the lysine side chain is one atom longer than the wild-type methionine, and can be structurally modeled so that the aliphatic chain retains the hydrophobic interactions with the kinase residues and the amine group is directed into solvent. Computational modeling indicates that both A31T and M44K mutants are compatible with wild-type eIF2α conformation and kinase binding.
Since mammalian eIF2α kinases expressed in yeast showed substrate recognition patterns similar to yeast GCN2 (Dey, Trieselmann, 2005) and we found separate but equally consistent pattern for human eIF2α in mammalian cells, we propose that a related but distinct set of substrate recognition requirements exist for human and yeast eIF2α. Despite their dissimilarities, members of the eIF2α kinase family appear to recognize both yeast and mammalian eIF2α in vitro.
Exactly how stringent the substrate recognition pattern for mammalian eIF2α is compared to its yeast equivalent extends beyond the scope of this study. It is possible that residues whose alteration did not affect yeast eIF2α phosphorylation could, in a mammalian system, strongly impair phosphorylation under a wide range of mutations. Alternatively, there remains the possibility that more forgiving requirements for eIF2α kinase substrate recognition in mammalian systems have evolved, which could provide higher eukaryotes an increasingly robust stress signaling system more suitable for instigating a rapid adaptive response against a wider and more complicated array of threats.
Supplementary Material
Acknowledgments
This research was supported by grants R37-DK060596 and R01-DK053307 (to M. H.), and T32-DK007319 from NIH (to DM).
Glossary
- eIF2
eukaryotic initiation factor 2
- ER
endoplasmic reticulum
- heIF2α
human eIF2α
- MEF
mouse embryonic fibroblast
- mGCN2
murine GCN2 protein
- mPERK
murine PERK protein
- mPKR
murine PKR protein
- PCR
polymerase chain reaction
- poly(I:C)
polyinosinic:polycytidylic acid
- Tg
thapsigargin
- yeIF2α
Saccharomyces cerevisiae eIF2α
- yGCN2
Saccharomyces cerevisiae GCN2 protein
References
- Bevilacqua E, Wang X, Majumder M, Gaccioli F, Yuan CL, Wang C, et al. eIF2alpha phosphorylation tips the balance to apoptosis during osmotic stress. J Biol Chem. 2010;285:17098–111. doi: 10.1074/jbc.M110.109439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg MB, Ferraris JD, Dmitrieva NI. Cellular response to hyperosmotic stresses. Physiol Rev. 2007;87:1441–74. doi: 10.1152/physrev.00056.2006. [DOI] [PubMed] [Google Scholar]
- Clemens MJ. Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog Mol Subcell Biol. 2001;27:57–89. doi: 10.1007/978-3-662-09889-9_3. [DOI] [PubMed] [Google Scholar]
- Cui W, Li J, Ron D, Sha B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr D Biol Crystallogr. 2011;67:423–8. doi: 10.1107/S0907444911006445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, et al. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol. 2002;12:1279–86. doi: 10.1016/s0960-9822(02)01037-0. [DOI] [PubMed] [Google Scholar]
- Dey M, Trieselmann B, Locke EG, Lu J, Cao C, Dar AC, et al. PKR and GCN2 kinases and guanine nucleotide exchange factor eukaryotic translation initiation factor 2B (eIF2B) recognize overlapping surfaces on eIF2alpha. Mol Cell Biol. 2005;25:3063–75. doi: 10.1128/MCB.25.8.3063-3075.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaliwal S, Hoffman DW. The crystal structure of the N-terminal region of the alpha subunit of translation initiation factor 2 (eIF2alpha) from Saccharomyces cerevisiae provides a view of the loop containing serine 51, the target of the eIF2alpha-specific kinases. J Mol Biol. 2003;334:187–95. doi: 10.1016/j.jmb.2003.09.045. [DOI] [PubMed] [Google Scholar]
- Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci. 2013;70:3493–511. doi: 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez J, Yaman I, Mishra R, Merrick WC, Snider MD, Lamers WH, et al. Internal ribosome entry site-mediated translation of a mammalian mRNA is regulated by amino acid availability. J Biol Chem. 2001;276:12285–91. doi: 10.1074/jbc.M009714200. [DOI] [PubMed] [Google Scholar]
- Fernandez JM, Yaman I, Sarnow P, Snider MD, Hatzoglou M. Regulation of internal ribosomal entry site-mediated translation by phosphorylation of the translation initiation factor eIF2alpha. J Biol Chem. 2002;277:19198–205. doi: 10.1074/jbc.M201052200. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Meurs EF, Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Guan BJ, Krokowski D, Majumder M, Schmotzer CL, Kimball SR, Merrick WC, et al. Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2alpha. J Biol Chem. 2014;289:12593–611. doi: 10.1074/jbc.M113.543215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan BJ, Su YP, Wu HY, Brian DA. Genetic evidence of a long-range RNA-RNA interaction between the genomic 5′ untranslated region and the nonstructural protein 1 coding region in murine and bovine coronaviruses. J Virol. 2012;86:4631–43. doi: 10.1128/JVI.06265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–18. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Kapp LD, Lorsch JR. The molecular mechanics of eukaryotic translation. Annu Rev Biochem. 2004;73:657–704. doi: 10.1146/annurev.biochem.73.030403.080419. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci. 2004;29:152–8. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kraulis P. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. Journal of Applied Crystallography. 1991;24:946–50. [Google Scholar]
- Krokowski D, Han J, Saikia M, Majumder M, Yuan CL, Guan BJ, et al. A self-defeating anabolic program leads to beta-cell apoptosis in endoplasmic reticulum stress-induced diabetes via regulation of amino acid flux. J Biol Chem. 2013;288:17202–13. doi: 10.1074/jbc.M113.466920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder M, Huang C, Snider MD, Komar AA, Tanaka J, Kaufman RJ, et al. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Molecular and cellular biology. 2012;32:992–1003. doi: 10.1128/MCB.06665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounir Z, Krishnamoorthy JL, Wang S, Papadopoulou B, Campbell S, Muller WJ, et al. Akt determines cell fate through inhibition of the PERK-eIF2alpha phosphorylation pathway. Sci Signal. 2011;4:ra62. doi: 10.1126/scisignal.2001630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonato MC, Widom J, Clardy J. Crystal structure of the N-terminal segment of human eukaryotic translation initiation factor 2alpha. J Biol Chem. 2002;277:17057–61. doi: 10.1074/jbc.M111804200. [DOI] [PubMed] [Google Scholar]
- Padyana AK, Qiu H, Roll-Mecak A, Hinnebusch AG, Burley SK. Structural basis for autoinhibition and mutational activation of eukaryotic initiation factor 2alpha protein kinase GCN2. J Biol Chem. 2005;280:29289–99. doi: 10.1074/jbc.M504096200. [DOI] [PubMed] [Google Scholar]
- Saikia M, Krokowski D, Guan BJ, Ivanov P, Parisien M, Hu GF, et al. Genome-wide identification and quantitative analysis of cleaved tRNA fragments induced by cellular stress. The J Biol Chem. 2012;287:42708–25. doi: 10.1074/jbc.M112.371799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Witzel JE, Jagus R. Homologous regions of the alpha subunit of eukaryotic translational initiation factor 2 (eIF2alpha) and the vaccinia virus K3L gene product interact with the same domain within the dsRNA-activated protein kinase (PKR) Eur J Biochem. 1997;250:85–91. doi: 10.1111/j.1432-1033.1997.00085.x. [DOI] [PubMed] [Google Scholar]
- Vazquez de Aldana CR, Dever TE, Hinnebusch AG. Mutations in the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2 alpha) that overcome the inhibitory effect of eIF-2 alpha phosphorylation on translation initiation. Proc Natl Acad Sci U S A. 1993;90:7215–9. doi: 10.1073/pnas.90.15.7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborske JM, Narasimhan J, Jiang L, Wek SA, Dittmar KA, Freimoser F, et al. Genome-wide analysis of tRNA charging and activation of the eIF2 kinase Gcn2p. J Biol Chem. 2009;284:25254–67. doi: 10.1074/jbc.M109.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. 2002;22:6681–8. doi: 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.