

Graphical abstract
Keywords: Organocatalysis, Cinchona alkaloids, Hypervalent iodine, Phase-transfer catalysis
Abstract
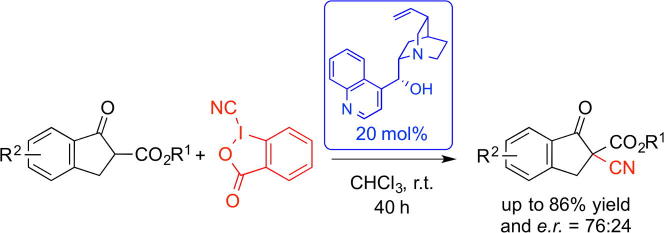
This communication describes the first proof of concept for an asymmetric α-cyanation of β-ketoesters using a hypervalent iodine-based electrophilic cyanide-transfer reagent. A series of different organocatalysts has been investigated and it was found that the use of naturally occurring Cinchona alkaloids allows obtaining the target products in good yields and with moderate enantioselectivities up to er = 76:24 under operationally simple conditions.
Introduction
The construction of quaternary stereogenic centres is an important but challenging task.1 The use of hypervalent iodine compounds has emerged as a powerful method for the synthesis of highly functionalized target molecules over the last years.2 Some of the most important applications of hypervalent iodine reagents include the transfer of carbon electrophiles like aryl,3,4 vinyl,5 alkynyl,6 or trifluoromethyl groups7 to prochiral nucleophiles. However, despite the spectacular progress made using these highly electrophilic reagents, their application in asymmetric reactions is still rather challenging, especially when it comes to the synthesis of quaternary stereogenic centres.2–7 The main challenge that arises is to ensure stereocontrol in the C–C bond forming step with a suitable asymmetric catalyst. For example, it is nowadays well accepted that the α-arylation of β-ketoesters with diaryliodonium salts proceeds via enolate O-attack to the hypervalent iodine reagent first, followed by a [2,3]-rearrangement to create the α-stereogenic centre.3b However, the hereby formed primary addition product is a neutral species and therefore it is difficult to control the subsequent stereo-defining rearrangement with those asymmetric catalysts that are commonly used to control prochiral enolates (e.g., asymmetric ammonium salt catalysts8). The groups of MacMillan and Gaunt addressed this challenge by controlling the addition of silylenolates to diaryliodonium salts using chiral Cu(I)-based catalysts.4c,d One approach that allows for the successful combination of hypervalent iodine reagents and asymmetric organocatalysts9 is the use of benziodoxole derivatives as shown by the groups of Waser, Maruoka and Vesely for the asymmetric α-alkynylation of β-ketoesters6a,b or α-fluoro phenylsulfonyl nitromethane6c under phase-transfer catalysis. Hereby the primary addition product (O–I bond formation) is charged and therefore the chiral ammonium salt catalyst can control the subsequent rearrangement.10
Inspired by these reports and based on our own interest in asymmetric phase-transfer catalysis11 we became interested in developing a protocol for the asymmetric α-cyanation of β-ketoesters 1 using the well-known cyano benziodoxole 212–14 as an electrophilic cyanide transfer reagent (Scheme 1).
Scheme 1.
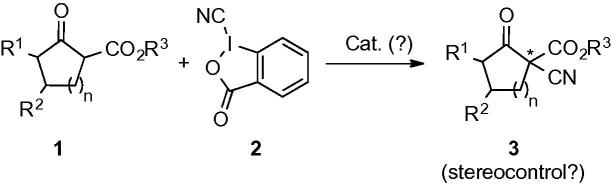
Targeted asymmetric α-cyanation of ketoesters 1.
Results and discussion
Initial experiments were carried out using Cinchona alkaloid-based phase-transfer catalysts A for the reaction of t-butyl ester 1a with cyanide 2. During these initial investigations we obtained cyanation product 3a accompanied by the formation of the α-brominated product 4 (see Table 1, entries 1–7 for representative results). The formation of α-halogenated products by reacting 1,3-dicarbonyl compounds and quaternary ammonium halides in the presence of hypervalent iodine reagents is a known transformation.15 While addressing this issue by screening different organocatalysts under different conditions we became aware of a very detailed and illustrative report by Chen et al. reporting the racemic cyanation of β-ketoesters 1 and the analogous amides using 2 as the cyanide transfer reagent under base-free conditions in different solvents.13 Interestingly, this group found that the α-hydroxy ketoester 5 is the only product under catalyst- and base-free conditions in solvents like toluene or CH2Cl2, whereas the use of DMF allowed them to totally suppress the formation of 5, giving the α-cyano products 3 in excellent yields of around 90% within minutes at room temperature.13 In addition, they also observed that the presence of an achiral Cu(II) Lewis acid favours cyanation over hydroxylation to some extent when carrying out the reaction in CH2Cl2 (still <20% yield). Accordingly, the presence of a catalyst/additive significantly influences the product ratio in apolar solvents. This is also supported by the fact that 3a was formed in every experiment when an organocatalyst was present but in neither case formation of 5 was observed (by 1H NMR of the crude mixture).
Table 1.

| Entry | Cat. (mol %) | Solvent | Base | T (°C) | t (h) | 3aa (%) | 4b (%) | erc (±) |
|---|---|---|---|---|---|---|---|---|
| 1 | A1 (20%) | CH2Cl2 | K2CO3 (5 equiv) | 25 | 24d | 58 | 10–15 | 48:52 |
| 2 | A1 (20%) | CH2Cl2 | — | 25 | 24 | 60 | 15–20 | 48:52 |
| 3 | A2 (20%) | CH2Cl2 | — | 25 | 24d | 30 | 15–20 | 55:45 |
| 4 | A3 (20%) | CH2Cl2 | — | 25 | 24 | 58 | 15–20 | 50:50 |
| 5 | A4 (20%) | CH2Cl2 | — | 25 | 24d | 20b | 15–20 | 50:50 |
| 6 | A5 (20%) | CH2Cl2 | — | 25 | 24d | 39 | 20 (17)a | 42:58 |
| 7 | A5 (20%) | CH2Cl2 | K2CO3 (5 equiv) | 25 | 24d | 47 | 15–20 (15)a | 44:56 |
| 8 | B1 (10%) | CH2Cl2 | — | 25 | 40d | 39 | n.d. | 37:63 |
| 9 | C1 (20%) | Toluene | — | 25 | 40d | 25 | n.d. | 34:66 |
| 10 | D1 (20%) | CH2Cl2 | — | 25 | 40 | 80 | n.d. | 64:36 |
| 11 | D2 (20%) | CH2Cl2 | — | 25 | 40 | 78 | n.d. | 66:34 |
| 12 | E1 (20%) | CH2Cl2 | — | 25 | 40 | 80 | n.d. | 38:62 |
| 13 | E2 (20%) | CH2Cl2 | — | 25 | 40 | 83 | n.d. | 30:70 |
| 14 | E3 (20%) | CH2Cl2 | — | 25 | 40 | 79 | n.d. | 50:50 |
| 15 | E4 (20%) | CH2Cl2 | — | 25 | 40 | 70 | n.d. | 42:58 |
| 16 | F1 (20%) | CH2Cl2 | — | 25 | 40d | 66 | n.d. | 50:50 |
| 17 | F2 (20%) | CH2Cl2 | — | 25 | 40d | 67 | n.d. | 47:53 |
| 18 | E2 (20%) | Toluene | — | 25 | 40d | 62 | n.d. | 33:67 |
| 19 | E2 (20%) | MTBE | — | 25 | 40 | 78 | n.d. | 41:59 |
| 20 | E2 (20%) | CHCl3 | — | 25 | 40 | 70e | n.d. | 26:74 |
| 21 | E2 (10%) | CHCl3 | — | 25 | 40d | 34 | n.d. | 26:74 |
| 22 | E2 (5%) | CHCl3 | — | 25 | 40d | 32 | n.d. | 34:66 |
| 23 | E2 (40%) | CHCl3 | — | 25 | 40 | 85 | n.d. | 30:70 |
| 24 | E2 (20%) | CHCl3 | — | 0 | 72d | 52 | n.d. | 25:75 |
| 25 | E2 (20%) | CHCl3 | — | 40 | 40 | 57 | n.d. | 27:73 |
Isolated yield.
Determined by 1H NMR of the crude reaction product.
Determined by HPLC using a chiral stationary phase.
Less than 90% conversion of 1a.
Using 2 equiv of 2 gave 3a in 75% yield and the same enantioselectivity.
Table 1 gives an overview of the most significant results obtained in a detailed screening of catalysts and reaction conditions. Initial experiments with phase-transfer catalysts A revealed that 3a can be obtained with low enantioselectivities when using free-OH containing catalysts A2 or A5 and it also turned out that base-free and basic conditions give almost the same selectivity (see entries 6 and 7). Cyanation was always the dominant reaction but it should be pointed out that conversion of starting material was usually not complete within 24 h and formation of racemic bromide side-product 4 was always observed (more or less quantitatively with respect to the amount of ammonium bromide catalyst used). As the presence of an H-bonding donor16 favoured selectivity, we next tested urea-containing ammonium salt B that was recently introduced by our group.17 The enantioselectivity could be slightly enhanced but the yield was not very high (entry 8). Thus, no further screening of quaternary ammonium salts was carried out but we tested different tert-amine containing bifunctional H-bonding donors next. Use of the thiourea-containing C1 resulted in a slightly higher selectivity of 66:34 (entry 9) but the yield was relatively low even after 40 h. In contrast, the use of simple Cinchona alkaloids D and E resulted in high yields and moderate enantioselectivities when using 20 mol% of catalyst in CH2Cl2 at room temperature (40 h reaction time ensured more than 90% conversion) (entries 10–13). Interestingly, cinchonidine (E2) gave 3a with the highest selectivity among the four tested naturally occurring Cinchona alkaloids (entry 13). Blocking of the 9-OH group totally suppressed chiral induction (E3, entry 14), whereas use of the 6′-OH containing catalyst E4 gave 3a with reduced selectivity compared to the use of E2. Testing the truncated aminoalcohols F resulted in racemic 3a only, indicating that the quinoline moiety plays an important role. Again, in neither case the formation of 5 was observed when using such different aminoalcohols as catalysts. Screening of different solvents showed that CHCl3 is best-suited to obtain enantioenriched 3a in reasonable yield and with moderate selectivity (er = 74:26; entry 20). Unfortunately neither a change in temperature, nor dilution of the reaction mixture (not given in the table) or a change in the stoichiometric ratio of 1 and 2 had a beneficial effect. Lowering the amount of catalyst resulted in a lower conversion rate and a lower selectivity when using 5 mol % catalyst (entries 21 and 22). On the other hand increasing the catalyst loading also caused a slight decrease of enantioselectivity (entry 23). By following the enantioselectivity in dependence of the reaction progress we found no noteworthy trend, thus degradation of the catalyst as well as formation of a more active catalyst species during the reaction can be excluded at the present stage of knowledge about this transformation.
Based on this detailed catalyst screening, which revealed cinchonidine (E2) to be the best-suited chiral aminoalcohol catalyst to obtain α-cyano ketoester 3a with a modest enantioselectivity of 74:26 in good yield, we next tested the scope of this protocol for some differently substituted β-ketoesters 1 (Scheme 2). The nature of the ester group had a strong influence on the observed enantioselectivity. While the methyl and benzylesters 3b and 3c could only be obtained with rather low selectivity, the adamantyl ester 3d could be accessed with an er of 76:24 under the standard conditions. Unfortunately, attempts to increase the enantiopurity by recrystallization were not very successful (best er for 3d was 81:19 but in low yield only). In contrast to the strong influence of the ester group, different aryl substituents did not significantly influence the outcome. On the other hand, tetralone derivative 3i could not be accessed under these reaction conditions (this compound is however accessible when carrying out the reaction under the racemic conditions reported previously by Chen et al.13).
Scheme 2.

Application scope of the asymmetric α-cyanation of 1 (in each case the (−)-enantiomer was the major stereoisomer).
Conclusion
A first proof of concept towards an asymmetric α-cyanation of β-ketoesters using a hypervalent iodine-based cyanide-transfer reagent has been made. The use of naturally occurring Cinchona alkaloids as chiral organocatalysts allowed us to obtain the target products in good yields and with moderate enantioselectivities (up to er = 76:24) under operationally simple conditions. Noteworthy, the presence of catalysts/additives significantly influences the product ratio (cyano vs hydroxyl transfer) in this approach (see also Ref. 13). Despite the obtained enantioselectivity is not very high yet, the herein reported protocol should be promising with respect to further development and future studies will aim on the identification of more selective catalysts (e.g., designed chiral aminoalcohols or asymmetric transition metal catalysts) and on the expansion of this concept towards other (prochiral) nucleophiles.
Experimental section
General procedure for the α-cyanation of β-ketoesters 1
Cyano benziodoxole 2 (1.2 equiv) was added to a stirred solution of the corresponding β-ketoester 1 and cinchonidine (E2, 20 mol %) in chloroform (5 mL per mmol 1) at room temperature. The reaction mixture was stirred at this temperature for 40 h. The crude product was directly transferred to a silica-gel column and eluted with a gradient of heptane and EtOAc to give the products 3 in the reported yields and enantiopurities.
Cyanide 3a: Obtained in 70% (0.7 mmol scale) as an oil that crystallizes upon prolonged storage in the refrigerator. Mp: 31–32 °C; [α]D20 (c 1.2, DCM, er = 74:26) = −15; 1H NMR (300 MHz, δ, CDCl3, 298 K): 7.83 (d, J = 7.8 Hz, 1H), 7.68–7.73 (m, 1H), 7.44–7.53 (m, 2H), 3.87 (d, J = 17.4 Hz, 1H), 3.63 (d, J = 17.4 Hz, 1H), 1.48 (s, 9H) ppm; 13C NMR (75 MHz, δ, CDCl3, 298 K): 191.3, 162.8, 151.7, 136.8, 132.3, 128.8, 126.4, 126.2, 116.1, 85.9, 55.2, 37.5, 27.6 (3C) ppm; IR (film): = 2977, 2935, 2245, 1723, 1148, 835, 735 cm−1; HRMS (ESI): m/z calcd for C15H15NO3: 280.0960 [M+Na]+; found: 280.0946. The enantioselectivity was determined by HPLC (Chiralcel OD-H, eluent: hexane:i-PrOH = 90:10, 0.5 mL/min, 10 °C, retention times: tminor = 15.7 min, tmajor = 16.7 min).
Acknowledgments
Generous support of the Austrian Science Funds (FWF) by a Lise-Meitner fellowship to Ragunath Chowdury is gratefully acknowledged (Project No. M1602). The used NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ programme (project M00146, ‘RERI-uasb’).
Contributor Information
Raghunath Chowdhury, Email: raghuch14@gmail.com.
Mario Waser, Email: mario.waser@jku.at.
Supplementary data
General procedure and spectral data.
References and notes
- 1.Christoffers J., Baro A., editors. Quaternary Stereocenters. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 2.(a) Zhdankin V.V., Stang P.J. Chem. Rev. 2008;108:5299–5358. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhdankin V.V. Wiley; Chichester: 2013. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds. [Google Scholar]; (c) Brand J.P., Fernandez Gonzalez D., Nicolai S., Waser J. Chem. Commun. 2011:102–115. doi: 10.1039/c0cc02265a. [DOI] [PubMed] [Google Scholar]; (d) Dong D.-Q., Hao S.-H., Wang Z.-L., Chen C. Org. Biomol. Chem. 2014;12:4278–4289. doi: 10.1039/c4ob00318g. [DOI] [PubMed] [Google Scholar]
- 3.Oh C.H., Kim J.S., Jung H.H. J. Org. Chem. 1999;64:1338–1340. For selected racemic approaches generating quaternary stereogenic centers see: (a) [Google Scholar]; (b) Norrby P.-O., Petersen T.B., Bielawski M., Olofsson B. Chem. Eur. J. 2010;16:8251–8254. doi: 10.1002/chem.201001110. [DOI] [PubMed] [Google Scholar]
- 4.Ochiai M., Kitagawa Y., Takayama N., Takaoka Y., Shiro M. J. Am. Chem. Soc. 1999;121:9233–9234. For asymmetric arylation reactions generating quaternary or tertiary stereogenic centers see: (a) [Google Scholar]; (b) Allen A.E., MacMillan D.W.C. J. Am. Chem. Soc. 2011;133:4260–4263. doi: 10.1021/ja2008906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bigot A., Williamson A.E., Gaunt M.J. J. Am. Chem. Soc. 2011;133:13778–13781. doi: 10.1021/ja206047h. [DOI] [PubMed] [Google Scholar]; (d) Harvey J.S., Simonovich S.P., Jamison C.R., MacMillan D.W.C. J. Am. Chem. Soc. 2011;133:13782–13785. doi: 10.1021/ja206050b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhu S., MacMillan D.W.C. J. Am. Chem. Soc. 2012;134:10815–10818. doi: 10.1021/ja305100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skucas E., MacMillan D.W.C. J. Am. Chem. Soc. 2012;134:9090–9093. doi: 10.1021/ja303116v. For a highly selective protocol generating tertiary stereogenic centers see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez D., Brand J.P., Mondiere R., Waser J. Adv. Synth. Catal. 2013;355:1631–1639. For asymmetric procedures see: (a) [Google Scholar]; (b) Wu X., Shirakawa S., Maruoka K. Org. Biomol. Chem. 2014;12:5388–5392. doi: 10.1039/c4ob00969j. [DOI] [PubMed] [Google Scholar]; (c) Kamlar M., Putaj P., Vesely J. Tetrahedron Lett. 2013;54:2097–2100. [Google Scholar]
- 7.(a) Matousek V., Togni A., Bizet V., Cahard D. Org. Lett. 2011;13:5762–5765. doi: 10.1021/ol2023328. [DOI] [PubMed] [Google Scholar]; (b) Deng Q.-H., Wadepohl H., Gade L.H. J. Am. Chem. Soc. 2012;134:10769–10772. doi: 10.1021/ja3039773. [DOI] [PubMed] [Google Scholar]
- 8.Ooi T., Maruoka K. Angew. Chem., Int. Ed. 2007;46:4222–4266. doi: 10.1002/anie.200601737. For selected reviews about asymmetric phase-transfer catalysis see: (a) [DOI] [PubMed] [Google Scholar]; (b) Shirakawa S., Maruoka K. Angew. Chem., Int. Ed. 2013;52:4312–4348. doi: 10.1002/anie.201206835. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez Gonzalez D., Benfatti F., Waser J. ChemCatChem. 2012;4:955–958. For an overview about the combination of asymmetric organocatalysis and hypervalent iodine reagents see: [Google Scholar]
- 10.Unfortunately this concept cannot be used for asymmetric α-arylation reactions using aryl benziodoxoles due to the lower reactivity of these compounds (unpublished results).
- 11.Waser M., Gratzer K., Herchl R., Müller N. Org. Biomol. Chem. 2012;10:251–254. doi: 10.1039/c1ob06573d. For selected examples from our group see: (a) [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gratzer K., Waser M. Synthesis. 2012;44:3661–3670. doi: 10.1055/s-0032-1316804. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Herchl R., Waser M. Tetrahedron Lett. 2013;54:2472–2475. doi: 10.1016/j.tetlet.2013.02.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Zhdankin V.V., Kuehl C.J., Krasutsky A.P., Bolz J.T., Mismash B., Woodward J.K., Simonsen A.J. Tetrahedron Lett. 1995;36:7975–7978. [Google Scholar]; (b) Akai S., Okuno T., Egi M., Takada T., Tohma H., Kita Y. Heterocycles. 1996;42:47–51. [Google Scholar]
- 13.Wang Y.-F., Qiu J., Kong D., Gao Y., Lu F., Karmaker P.G., Chen F.-X. Org. Biomol. Chem. 2015;13:365–368. doi: 10.1039/c4ob02032d. [DOI] [PubMed] [Google Scholar]
- 14.Frei R., Courant T., Wodrich M.D., Waser J. Chem. Eur. J. 2015;21:2662–2668. doi: 10.1002/chem.201406171. [DOI] [PubMed] [Google Scholar]
- 15.(a) Salgaonkar P.D., Shukla V.G., Akamanchi K.G. Synth. Commun. 2007;37:275–280. [Google Scholar]; (b) Galligan M.J., Akula R., Ibrahim H. Org. Lett. 2014;16:600–603. doi: 10.1021/ol403504z. [DOI] [PubMed] [Google Scholar]
- 16.Novacek J., Waser M. Eur. J. Org. Chem. 2013:637–648. For a review about bifunctional ammonium salt catalysts see: [Google Scholar]
- 17.Novacek J., Waser M. Eur. J. Org. Chem. 2014:802–809. doi: 10.1002/ejoc.201301594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General procedure and spectral data.