

Abstract
The S100A8 and epidermal growth factor receptor (EGFR) proteins are proto-oncogenes that are strongly expressed in a number of cancer types. EGFR promotes cellular proliferation, differentiation, migration and survival by activating molecular pathways. Involvement of proinflammatory S100A8 in tumor cell differentiation and progression is largely unclear and not studied in kidney cancer (KC). S100A8 and EGFR are potential therapeutic biomarkers and anticancer drug targets for KC. In this study, we explored molecular mechanisms of interaction profiles of both molecules with potential anticancer drugs. We undertook transcriptional profiling in Saudi KCs using Affymetrix HuGene 1.0 ST arrays. We identified 1478 significantly expressed genes, including S100A8 and EGFR overexpression, using cut-off p value <0.05 and fold change ≥2. Additionally, we compared and confirmed our findings with expression data available at NCBI’s GEO database. A significant number of genes associated with cancer showed involvement in cell cycle progression, DNA repair, tumor morphology, tissue development, and cell survival. Atherosclerosis signaling, leukocyte extravasation signaling, notch signaling, and IL-12 signaling were the most significantly disrupted signaling pathways. The present study provides an initial transcriptional profiling of Saudi KC patients. Our analysis suggests distinct transcriptomic signatures and pathways underlying molecular mechanisms of KC progression. Molecular docking analysis revealed that the kinase inhibitor "midostaurin" has amongst the selected drug targets, the best ligand properties to S100A8 and EGFR, with the implication that its binding inhibits downstream signaling in KC. This is the first structure-based docking study for the selected protein targets and anticancer drug, and the results indicate S100A8 and EGFR as attractive anticancer targets and midostaurin with effective drug properties for therapeutic intervention in KC.
Introduction
Cancer is a global major health problem. Dysregulation in molecular signaling pathways is a hallmark of cancer initiation and progression [1–3]. Kidney cancer (KC) accounts for approximately 1.5 percent of all cancer deaths. In particular, KC is common in obese male population [4]. Surgical tumor resection is the standard curative treatment. Metastatic KC is almost nonresponsive to conventional systemic treatments and nearly all patients die of metastasis. Lack of promising biomarkers for effective targeted chemotherapy poses a big challenge in KC management. Better understanding of the molecular mechanisms effective in KC have widened the window for development of effective targeted therapies [5,6]. High-throughput microarray platforms are well suited for identification of the novel induced or suppressed disease-related culprit genes [7]. Molecules showing direct involvement in a biochemical or regulatory pathway leading to disease are potential anticancer target. Drug/molecule interaction involving these targets can either be investigated by co-crystallization or tested by docking simulation to indentify molecular interactions required for rational drug designing [8]. High-throughput docking is the key entrance for drug discovery [9,10]. Transcriptomic profiling and functional pathway analysis in KC have identified several significantly differentially expressed genes, including S100A8 and EGFR. We tried to evaluate their potential as KC drug target by in silico docking with known protein kinase inhibitors. Overall, this study illustrates structure-based virtual screening and ligand-protein docking of anticancer drugs e.g. midostaurin, enzastaurin, and gefitinib, with anticancer targets, S100A8 and EGFR.
S100A8 is a small (10 kDa) proinflammatory protein of S100 family, which tends to form heterodimeric complexes with S100A9 (S100A8/A9) [9], that undergo conformational changes upon Ca2+ binding and function as intracellular Ca2+ sensors [10]. Under physiological conditions, these Ca2+ binding EF hand type proteins are constitutively expressed by myeloid cells [11–13]. However, under pathological conditions like inflammation and cancer, an increased expression of S100A8 is seen in epithelial cells [14,15]. Early stage death of S100A8 knock-out mice proves the essentiality of this gene for survival [16]. Enhanced level of S100A8 is found in different carcinomas including breast [15], prostate [17,18], lung [19], gastric [20], hepatic [21], pancreatic [22] and colorectal cancer [23,24]. A recent study shows cell growth-promoting activity and binding to receptor for advanced glycation endproducts (RAGE) at low S100A8 concentrations [15]; however, its direct role in tumorogenesis is ambiguous and has to be elucidated yet. It has been reported that primary tumors secrete soluble factors, which induce expression of S100A8 in the endothelial cells prior to tumor metastasis [19]. By activating the p38 MAPK pathway, it increases the motility of circulating cancer cells [25]. Therefore targeting S100A8 could be used to prevent the tumor cell migration and growth. Several lines of evidence point to vital functions of S100A8 during tumorigenesis and, although it’s exact role within the tumor microenvironment is still not clear, different tumor-promoting effects have been proposed. Though, research is limited on its expression pattern in cancer, its involvement in oncogenesis and its potential as therapeutic biomarker. To the best of our knowledge, no study has reported yet its expression pattern or its role in KC progression.
EGFR is one of the four member of the ErbB family of receptor tyrosine kinases. The receptor is overexpressed or mutated in many cancers, highlighting its role as therapeutic cancer biomarker. It is involved in diverse cellular functions such as proliferation, angiogenesis and suppression of cell death. Being a transmembrane protein, EGFR passes crucial signals from epithelial cell surface to the intracellular domain for controlled cell proliferation, migration and adhesion. Overexpressed EGFR transmits multiple signals to cell for accelerated growth and cellular longevity, and plays a key role in the carcinogenesis of different types of cancer [26–29]. Improved perception of the molecular signaling pathways has opened new strategies and ways for cancer treatment. Thus, targeting EGFR to turn off its signal transduction is envisaged to block growth and survival of cancer cells.
S100A8 and EGFR do play an important role in malignancy and thus considered as potential drug targets for cancer therapeutics development [26–30]. The reason for choosing midostaurin, enzastaurin, and gefitinib as inhibitor for S100A8 and EGFR is the recent research showing that EGFR-targeted therapies using kinase inhibitors are effective in many cancer patients [31–36]. Gefitinib, an inhibitor of EGFR kinase function, was approved by the U.S. FDA for lung cancer treatment [37]. S100A8 does not own any kinase domain or ATP binding pocket, however it participates in protein phosphorylation [38] and is an upstream pathway molecule of EGFR. Thus, a drug capable of inhibiting both EGFR and S100A bears prospective therapeutic impact.
After the discovery of protein kinase activity in 1954, a lot of kinase inhibitors have been identified [39]. Because of high degree of similarity in their structure and function within the “kinome” [40], identification of effective protein kinase inhibitors (PKIs) is a great challenge. At present kinase inhibitors comprise more than 30% of drug-discovery programs resulting in approval of dozens of kinase inhibitors as anti-cancer drugs or at least testing in clinical trials [41]. In this study, we determined by docking simulation the potential efficiency of three such drugs named midostaurin, enzastaurin, and gefitinib.
(i) Midostaurin (CID 104937; also known as PKC412 and benzoylstaurosporine) is a multi-target kinase inhibitor used for acute myeloid leukemia treatment. It is a semi-synthetic alkaloid derived from bacterial staurosporine used to treat patients with CD135 (FMS-like tyrosine kinase 3 receptor) mutations [42]. Midostaurin inhibits growth or induces apoptosis in several types of cancer, blocks angiogenesis and sensitizes cancer cells to ionizing radiation, justifying its use in cancer treatment [43]. Preclinical studies have shown that midostaurin works synergistically with chemotherapy agents, reinforcing each other's effect against cancer [44]. (ii) Enzastaurin is a synthetic acyclic bisindolylmaleimide showing antineoplastic activity. It is a small oral serine/threonine kinase inhibitor of PKCβ and AKT pathways. It inhibits protein kinase Cβ that is known stimulator of neo-angiogenesis through induction of vascular endothelial growth factor (VEGF). Binding of protein kinase Cβ to the ATP-binding site of VEGF probably decreases tumor blood supply and prevents growth. Apart from its anti—VEGF factor effects, low concentration of enzastaurin suppresses proliferation in cancer cell lines [45]. Loss of GSK3β and AKT phosphorylation has been reported in myeloma cells treatment with enzastaurin [46]. (iii) Gefitinib (Iressa) selectively targets the ATP cleft within EGFR tyrosine kinase domain [47]. It interrupts EGFR signaling of target cells and therefore is effective only in cancer cells with mutated and overactive EGFR.
Molecular docking forecasts an optimized conformation and relative orientation for both the protein and ligand molecule. In this study we were aiming to evaluate the potential of S100A8 and EGFR as KC drug target by in silico molecular docking with the known protein kinase inhibitors midostaurin, enzastaurin, and gefitinib to test the effectiveness of these anticancer drugs under these constellations.
Materials and Methods
Patients and samples
The study was performed on patients from Saudi Arabia diagnosed with renal cell carcinoma. The samples were collected from King Abdulaziz University Hospital, Bakhsh Hospital and King Faisal Specialist Hospital & Research Center, Jeddah during 2010–2012. For gene expression analysis, fresh tumor and normal tissue specimens were taken from surgical resections of tumor and normal kidney tissues, respectively and were immediately placed in RNALater (Invitrogen—Life Technologies, Grand Island, NY, USA). Out of 18 tumor specimen, only two passed the criteria (RNA integrity number (RIN) >5) to be used for array expression analysis. One patient was 61 year old Saudi male, diagnosed with clear cell renal cell carcinoma of nuclear grade II and tumor size 4.5 x 3 x 4 cm. The second patient was 47 year old Saudi female, diagnosed with chromophobe renal cell carcinoma of Fuhrman’s grade II.
Ethical approval
All patients included in the study provided written informed consent. The study was reviewed and approved by the Center of Excellence in Genomic Medicine Research (CEGMR) local ethical committee (approval number 08-CEGMR-02-ETH).
RNA extraction and array processing
Total RNA was extracted from freshly preserved kidney tissue specimens with the Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany) including an on-column DNAse treatment according to manufacturer’s recommendations. Quality of the purified RNA was verified on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Mean value of RIN for processed samples was 8.0 only for two cancer samples and four normal kidney tissues. RNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA samples (250 ng) were processed according to the manufacturer’s recommendations (Life Technology, Grand Island, NY). After fragmentation and labeling, the samples were hybridized at 45°C for 17 hours to Human Gene 1.0 ST GeneChip arrays (Affymetrix, Santa Clara, CA, USA). These arrays are conceptually based on the Human Genome sequence assembly UCSC hg18, NCBI Build 36 and interrogated with a set of 764,885 probes 28,869 annotated genes.
Gene Expression Analysis
We conducted expression profiling of two renal cell carcinomas and four normal kidney tissues. Because of our limited number of samples, independent datasets available in public domain were incorporated in the gene expression analysis process. We used selected KC expression data from NCBI's GEO database (Accession no: GSE781, GSE7023, and GSE6344) for comparative analysis and confirmation. Sample size for GSE781, GSE7023, and GSE6344 were 34, 47, and 40 respectively. Affymetrix. CEL files were imported to Partek Genomics Suite version 6.6 (Partek Inc., MO, USA). The data was normalized using RMA normalization. Analysis of Variance (ANOVA) was applied on the complete data set and the list of differentially expressed genes was then generated using false discovery rate (FDR) of 0.05 with 2-fold change cut off. Unsupervised two-dimensional average linkage hierarchical clustering was performed using Spearman’s correlation as a similarity matrix.
Functional and Pathway analysis
To define biological networks, interaction and functional analysis among the differentially regulated genes in KC, pathway analyses were performed using Ingenuity Pathways Analysis software (IPA) (Ingenuity Systems, Redwood City, CA). Statistically differentially expressed datasets with probesets ID, Gene symbol and Entrez gene ID as clone identifier, p-value and fold change values were uploaded into IPA. The functional/pathway analysis of IPA identifies the biological functions and/or diseases and pathways that are most significantly altered for the differentially expressed gene set. The significance of the connection between the expression data and the canonical pathways were calculated by ratio and/or Fisher’s exact test.
Molecular Docking
We performed molecular docking studies (i) to investigate the role of S100A8 as inflammatory mediators and to establish their involvement in inflammation-associated cancers at molecular level and (ii) to investigate the role of EGFR signaling in proliferation, angiogenesis and suppression of cell death causing cancer. The 3-D structures of identified cancer drug targets were retrieved from Protein Data Bank (PDB ID: S100A8 dimer, 1MR8 and EGFR tyrosine kinase domain, 2GS2). The molecular structures of inhibitors were retrieved from PubChem compound database (midostaurin with CID 104937; enzastaurin with CID 176167 and gefitinib with CID 123631) (Fig. 1). The bound ligand was used as probe for the binding site grid generation. Structure visualization and identification of drug binding site was done using PyMol [48] (Fig. 2). PyMOL was used to analyze and generate an illustration of whole protein-ligand complex.
Fig 1. Two dimensional molecular structure of the three anti-cancer drugs: midostaurin, enzastaurin and gefitinib.
Fig 2. Structure visualization of cancer signaling target proteins S100A8 (1MR8), and EGFR tyrosine kinase domain (2GS2) retrieved from Protein Data Bank.
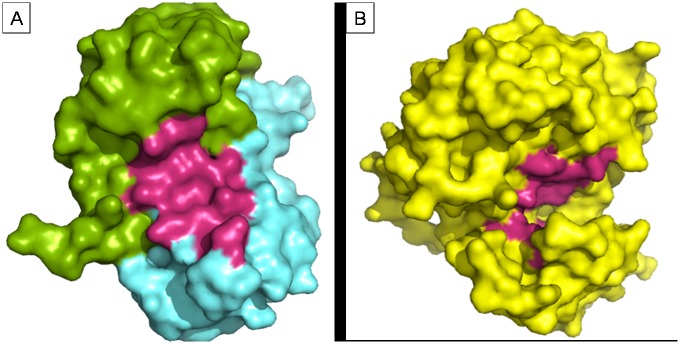
Surface representation of the two PDB structures used for docking analysis. Figure made using PyMol. (1) MR8 chain A (green) + chain B (cyan) (2) 2GS2 chain (yellow), drug binding cavity in magenta.
Docking calculations were done with Molecular Docking Server [49]. Merck molecular force field 94 (MMFF94) [50] was used for energy minimization of drug molecules used as ligand: midostaurin, enzastaurin and gefitinib. Gasteiger partial charges were added to ligand atoms. Non-polar hydrogen atoms were combined, and rotatable bonds were defined. Molecular docking of each ligand was performed individually with (S100A8)2 homo-dimmer (1MR8), and EGFR tyrosine kinase domain (2GS2) protein models, to predict the binding orientation and interaction. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools [51]. Autogrid program was used to generate affinity (grid) maps of 20×20×20 Å grid points and 0.375 Å binding site grid generation spacing [52]. AutoDock parameter set- and distance-dependent dielectric functions were used in van der Waals and the electrostatic terms calculation, respectively. Docking simulations were achieved using the Lamarckian genetic algorithm (LGA) and the Solis & Wets local search method [53]. Orientation, initial position, and torsion angles of ligand molecules were set randomly. Each docking experiment was the resultant of 10 different runs that were set to cease after a maximum of 250000 energy evaluations. Population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of 5 were applied in the current series of docking analysis.
Availability of supporting data
The data sets supporting the results of this study are available in the NCBI's Gene Expression Omnibus (GEO), identified as GSE781, GSE7023, and GSE6344 (http://www.ncbi.nlm.nih.gov/gds/?term=GSE781).
Results
The main focus of this study was to discover novel anticancer drug target by transcriptomic profiling and to identify possible protein-drug interactions by molecular docking analysis. We identified S100A8 and EGFR as important proteins of KC and made an attempt to demonstrate their anticancer drug target potential.
Identification of differentially expressed genes
We profiled fresh kidney tissue specimens and compared them with normal control samples. Clear differences were also observed between the tumors and normal tissues revealing distinct expression profiles for each tissue types. Comparison of the genome-wide expression of KC revealed 1478 differentially expressed genes, 943 up-regulated and 535 down-regulated, with a ≥ 2 fold change and false discovery rate of p < 0.05 (Fig. 3, Table 1, S1 Table). Identified differentially expressed genes in our dataset were compared with re-analyzed dataset (GSE-781, -6344 and-7023) retrieved from GEO database. 876, 1200 and 1258 differentially expressed genes were found significant for GSE781, GSE6344 and GSE7023 dataset respectively at cut off value of fold change >2 and p value <0.05. As expected, there were hundreds of genes showing similar expression pattern including S100A8 and EGFR, both gene were over-expressed in all dataset under study except GSE7023, where fold change were 1.948 for S100A8 and 1.893 for EGFR. Elevated expression of S100A8 and EGFR in our CEGMR dataset and GEO dataset is confirming our finding (Table 2, S2 Table).
Fig 3. Hierarchical clustering and functional analysis of significantly differentially expressed genes in kidney cancer using Affymetrix Human ST 1.0 array and Partek Genomics suite (ver 6.6).

Table 1. Differentially expressed significant genes in Kidney cancer.
| Gene Symbol | Gene_assignment | Transcript ID | RefSeq | p-value | Fold-Change |
|---|---|---|---|---|---|
| TOPBP1 | topoisomerase (DNA) II binding protein 1 | 8090772 | NM_007027 | 1.11E-06 | 2.05612 |
| TDO2 | tryptophan 2,3-dioxygenase | 8097991 | NM_005651 | 7.91E-06 | 3.30054 |
| FOXM1 | forkhead box M1 | 7960340 | NM_202002 | 8.08E-06 | 2.89359 |
| NPHS2 | nephrosis 2, idiopathic, steroid-resistant (podocin) | 7922627 | NM_014625 | 1.56E-05 | -41.0848 |
| C3orf58 | chromosome 3 open reading frame 58 | 8083223 | NM_173552 | 1.65E-05 | 2.13922 |
| UMOD | uromodulin | 7999936 | NM_003361 | 1.66E-05 | -150.524 |
| ANKRD13A | ankyrin repeat domain 13A | 7958600 | NM_033121 | 3.83E-05 | 2.78943 |
| KCNJ1 | potassium inwardly-rectifying channel, subfamily J, member 1 | 7952617 | NM_153767 | 4.93E-05 | -24.6069 |
| ANKRD2 | ankyrin repeat domain 2 (stretch responsive muscle) | 7929653 | NM_020349 | 4.95E-05 | -3.11353 |
| CALB1 | calbindin 1, 28kDa | 8151730 | NM_004929 | 5.79E-05 | -158.598 |
| PRSS42 | protease, serine, 42 | 8086683 | NM_182702 | 5.84E-05 | -2.01485 |
| GLTPD2 | glycolipid transfer protein domain containing 2 | 8003948 | NM_001014985 | 5.90E-05 | -2.68064 |
| ESCO1 | establishment of cohesion 1 homolog 1 (S. cerevisiae) | 8022473 | NM_052911 | 5.99E-05 | 2.10289 |
| NTRK2 | neurotrophic tyrosine kinase, receptor, type 2 | 8156134 | NM_006180 | 6.46E-05 | -2.76027 |
| BLM | Bloom syndrome, RecQ helicase-like | 7986068 | NM_000057 | 7.64E-05 | 2.14272 |
| SLC12A3 | solute carrier family 12 (sodium/chloride transporters), member | 7995868 | NM_000339 | 8.17E-05 | -61.4283 |
| ITGA6 | integrin, alpha 6 | 8046380 | NM_000210 | 8.51E-05 | 2.82412 |
| SPN | sialophorin | 7994603 | NM_001030288 | 0.00010918 | 2.01423 |
| S100A8 | S100 calcium binding protein A8 | 7920244 | NM_002964 | 0.0159132 | 2.66364 |
| EGFR | epidermal growth factor receptor (erythroblastic leukemia viral (v) | 8132860 | NM_005228 | 0.0410945 | 3.40122 |
Negative fold change value indicates the downregulation.
Table 2. Expression of S100A8 and EGFR in kidney cancer among Saudi patients (CEGMR dataset) and GEO dataset (GSE781, GSE6344 and GSE7023).
| Gene Symbol | CEGMR (own data) Sample size = 6 | GSE781 Sample size = 34 | GSE6344 Sample size = 40 | GSE7023 Sample size = 47 | ||||
|---|---|---|---|---|---|---|---|---|
| Fold Change | p-value | Fold Change | p-value | Fold Change | p-value | Fold Change | p-value | |
| S100A8 | 2.663 | 0.0159 | 2.759 | 0.0109 | 3.107 | 0.0019 | 1.948 | 0.0105 |
| EGFR | 3.401 | 0.0410 | 5.563 | 8.54E-05 | 3.32472 | 6.00E-08 | 1.893 | 0.0026 |
Pathways and networks underlying kidney cancer
To understand the mechanisms by which the genes alter a wide range of physiological processes, we examined biofunctions, molecular network and pathways associated with KC. Interestingly, the biological process, cellular movement was significantly overrepresented in both down-regulated and up-regulated gene lists pointing that the metastasis is probably linked to a different equilibrium of switching on and off. Functional analysis of KC-associated genes found an over expression of genes involved in cell cycle progression, DNA repair, cell death, tumor morphology and tissue developments. Pathway analysis showed significant disruption in certain signaling pathways including atherosclerosis signaling, LXR/RXR activation, leukocyte extravasation signaling, hepatic fibrosis / hepatic stellate cell activation, notch signaling, and IL-12 signaling and production in macrophages (Fig. 4, Table 3). Extensive pathway analysis of differentially regulated genes may provide novel hypotheses underlying tumor invasion and metastatic progression of KC.
Fig 4. Leukocyte Extravasation Signaling: Transcriptomic signatures of kidney cancer showed a significant activation in leukocyte extravasation signaling pathway.

Red represents overexpression and green underexpression.
Table 3. Canonical pathways predicted by Ingenuity Pathway Analysis for significant genes differentially expressed in kidney cancer.
| Ingenuity Canonical Pathways | -log (p-value) | Down-regulated | Upregulated | Molecules |
|---|---|---|---|---|
| Atherosclerosis Signaling | 3.14E00 | 5/136 (4%) | 13/136 (10%) | APOE,APOM,MSR1,PLA2R1,PLA2G7,SELPLG,COL1A2,APOC1,APOL1,COL1A1,IL18,ALB, LYZ,CCL2, S100A8,PDGFD,RBP4,COL3A1 |
| LXR/RXR Activation | 2.93E00 | 8/139 (6%) | 10/139 (7%) | KNG1,SCD,APOE,APOM,ECHS1,MSR1,AMBP,ABCG1,APOC1,APOL1,IL18,ALB,LYZ, LY96, CCL2, S100A8,HADH,RBP4 |
| Leukocyte Extravasation Signaling | 2.65E00 | 4/205 (2%) | 20/205 (10%) | CLDN10,ARHGAP6,SPN,PIK3C2A,CLDN19,JAM2,ITGA6,RAPGEF4,SELPLG,PIK3R3,BTK,ROCK1,NCF1,WIPF1,ITGAM,CDH5,JAM3, CLDN16,RASGRP1,NCF2,PRKCH,RASSF5, ACTN1,CLDN3 |
| Hepatic Fibrosis / Hepatic Stellate Cell Activation | 2.52E00 | 3/153 (2%) | 15/153 (10%) | FN1,CXCL9,FGFR1,EGF,FGF1,BCL2,COL1A2,COL1A1,LY96,IGF1,CCL2,TGFB2,IL10RA, MYH9, EDNRA,ECE1,MYL3,COL3A1 |
| Maturity Onset Diabetes of Young (MODY) Signaling | 2.46E00 | 5/29 (17%) | 1/29 (3%) | HNF1B,PKLR,ALDOB,SLC2A2,CACNA1C, FABP1 |
| Coagulation System | 2.21E00 | 5/38 (13%) | 2/38 (5%) | F11,KNG1,PLG,SERPINA5,PROC,VWF, PLAUR |
| Valine Degradation I | 2.21E00 | 6/35 (17%) | 1/35 (3%) | HIBCH,BCAT1,ECHS1,ABAT,ACADSB,EHHADH,ALDH6A1 |
| Notch Signaling | 2.07E00 | 1/42 (2%) | 6/42 (14%) | DLL1,ADAM17,DTX1,JAG2,MAML3,DLL4, HEY1 |
| Intrinsic Prothrombin Activation Pathway | 2.03E00 | 3/36 (8%) | 3/36 (8%) | F11,KNG1,COL1A2,COL1A1,PROC,COL3A1 |
| IL-12 Signaling and Production in Macrophages | 1.88E00 | 5/154 (3%) | 11/154 (7%) | PPARG,APOE,APOM,PIK3C2A,MST1,APOL1,PIK3R3,APOC1,ALB,LYZ,IL18,TGFB2,MAP3K8, S100A8,PRKCH,RBP4 |
Docking studies
Molecular docking studies predicted potential interactions of our proposed protein drug target with the selected drug molecules. This is a structural modeling approach to study possible binding for cancer therapeutics. To understand the molecular interaction between drugs and S100A8, series of molecular docking analysis were performed using three dimensional structure available (PDBID: 1MR8) with three anti-cancer drugs i.e. midostaurin, enzastaurin and gefitinib. The ligand binding site was a hinge region containing two EF-hand motifs. In similar way, we also simulated docking of the above-mentioned drugs with the tyrosine kinase domain of EGFR. Based on their size, stereochemistry and structural differences the ligands exhibited varied intensity in binding with the protein target molecules. The predicted parameters of estimated binding free energy, inhibition constant (Ki), total energy of vdW + Hbond + desolvation + electrostatic energy, total intermolecular energy and interacting surface area were evaluated to estimate the favorable binding of ligand drug molecules to the target protein. Molecular visualization was performed using PyMol. Complete interaction profile (H-bonds, polar, hydrophobic, pi-pi, cation-pi and other contacts), and hydrogen bonding (HB plot) interactions were studied (Figs. 5 and 6, Table 4).
Fig 5. 2D plot of inhibitors with S100A8 and EGFR tyrosine kinase domain proteins interaction profile by DockingServer.

Ligand bond, non-ligand bond, hydrogen bond and their lengths are marked for midostaurin, enzastaurin and gefitinib. Where A, B, C shows interaction of S100A8 (1MR8) with the drugs, and E, F shows interaction of EGFR (2GS2) with midostaurin and gefitinib respectively.
Fig 6. Interactions of ligand with the protein.

Red represents protein as cartoon; grey represents interacting side chain as cylinder; and green represents drug as ball and stick model.
Table 4. Binding and interaction values for docking of S100A8 dimer and EGFR kinase domain with inhibitors (midostaurin, enzastaurin and gefitinib).
| (S100A8)2 (1MR8) | Midostaurin | Enzastaurin | Gefitinib |
|---|---|---|---|
| Est. Free Energy of Binding | -9.77 kcal/mol | -4.19 kcal/mol | -4.21 kcal/mol |
| Est. Inhibition Constant, Ki | 68.48 nM | 844.18 μM | 813.89 μM |
| vdW + Hbond + desolv Energy | -7.65 kcal/mol | -6.33 kcal/mol | -6.11 kcal/mol |
| Electrostatic Energy | -0.03 kcal/mol | -0.02 kcal/mol | -0.07 kcal/mol |
| Total Intermolec. Energy | -7.68 kcal/mol | -6.35 kcal/mol | -6.18 kcal/mol |
| Interact. Surface | 754.992 | 849.622 | 667.469 |
| EGFR kinase domain (2GS2) | Midostaurin | Enzastaurin | Gefitinib |
| Est. Free Energy of Binding | -6.58 kcal/mol | +7.10 kcal/mol | -4.15 kcal/mol |
| Est. Inhibition Constant, Ki | 15.13 μM | ----- | 905.30 μM |
| vdW + Hbond + desolv Energy | -1.49 kcal/mol | +0.44 kcal/mol | -6.07 kcal/mol |
| Electrostatic Energy | -0.16 kcal/mol | -0.88 kcal/mol | -0.17 kcal/mol |
| Total Intermolec. Energy | -1.65 kcal/mol | -0.44 kcal/mol | -6.24 kcal/mol |
| Interact. Surface | 911.021 | 895.39 | 795.106 |
The docking studies reveal the presence of one midostaurin molecule at the substrate-binding site of S100A8 dimer (PDB code 1MR8). It is observed that the drug molecule is buried in the substrate-binding hydrophobic channel of the S100A8 ligand binding domain. Most of the interacting residues are hydrophobic in nature. Interaction and accessible surface area analysis reveal that the cavity involved in the binding site of drugs has a molecular accessible surface area of 123.017 Å2 and a solvent accessible surface area of 509.834 Å2. The conformations of the binding site in S100A8 as well as that in drugs were not altered upon binding as rigid docking simulation was carried out. The binding cavity is the same for all three drugs.
We selected the crystal structure of the active EGFR kinase domain having a length of 330 residues (PDB code 2GS2) for docking study. The drug binding cavity of the EGFR tyrosine kinase domain is more like a channel and is deeper as compared to the S100A8 case and has a molecular and solvent accessible surface area of 181.409 Å2 and 875.519 Å2 respectively. The cavity is lined with mostly polar as well as few non-polar residues. Enzastaurin did not exhibit any binding with the selected tyrosine kinase domain. Gefitinib is a previously known inhibitor of EGFR and its co-crystal structure has been determined and presented in PDB (3UG2), however docking studies revealed that midostaurin is the more suitable binding partner of EGFR than gefitinib.
(S100A8)2 (1MR8) with midostaurin
Midostaurin binds at the ligand binding site and forms an H-bond with a critical amino acid residue in the ligand binding domain site of S100A8 i.e. Gln 69 (B) formed between N1 of ligand and the terminal O of Gln. The residues involved in ligand cavity formation are two polar residues Gln 69 (B) and Glu 70 (B) and other hydrophobic residues Ile 60 (B), Ile 73 (A), Ile 73 (B), Ile 76 (A), Ile 76 (B), Val 80 (A) and Leu 72 (B) as shown in Fig. 2. More than 20 hydrophobic contacts and van der Waals interactions are also noted. The inhibitor exhibits favorable binding characteristics with the predicted free binding energy-9.77 kcal/mol and estimated inhibition constant, Ki of 68.48 nM. These results are promising and are superior to other docking results done in this particular study.
(S100A8)2 (1MR8) with enzastaurin
The drug binds to the protein molecule satisfactorily with an estimated free binding energy of-4.19 kcal/mol and Ki of 844.18 μM. It shows binding with the S100A8 dimer but without the formation of neither any H-bonds nor polar contacts. It displays five hydrophobic contacts mediated by aliphatic non-polar amino acids Leu 72 (B), Ile 73 (B), Ile 76 (A), Ile 76 (B), and Val 80 (A). Other noticeable interactions are with Lys 77 (A) and Gln 69 (B).
(S100A8)2 (1MR8) with gefitinib
Binding ability of gefitinib is good but there are no H-bonds formed and the free energy of bound structure is-4.21 kcal/mol. One noticeable polar interaction is with Lys 77 of protein chain A. There are around ten hydrophobic interactions with the drug binding site lining hydrophobic residues—Ile 73 (A), Ile 73 (B), Ile 76 (A) and Val 80 (A). Halogen atoms F and Cl of the drug show non-covalent interaction. For protein-ligand complexes, halogen bonds are energetically and geometrically comparably equal to that of hydrogen bonding if the donor-acceptor directionality remains consistent. This intermolecular interaction has been shown to be stabilizing and a conformational determinant in complexes [54]. F displays a water mediated bond and Cl shows interaction with Gln 69 (A). A couple of other weak interactions was noticed too.
EGFR kinase domain (2GS2) with midostaurin
Midostaurin being primarily a kinase inhibitor docks well with the EGFR tyrosine kinase domain with free binding energy of-6.58 kcal/mol and inhibition constant of 15.13 μM. Four H-bonds are possibly involved between the protein and ligand molecule. Cys 773 Sγ shows H-bonding with N4 of drug at a interatomic distance of 3.28 Å. In addition, atom 0δ2 of Cys 773 likely forms another H-bond with H atom of ligand. Similar pattern of H-bond is seen with Asp 776. Residues Arg 817, Thr 830 and Asp 831 are involved in polar interactions with the ligand. Hydrophobic interactions are mediated by Cys 773 and Leu 820. Other interactions with the drug are mediated through Lys 721, Glu 738, Asp776, Arg 817, Leu 820, Thr 830 and Asp 831.
EGFR kinase domain (2GS2) with enzastaurin
The docked structure shows positive free energy of binding implying that binding is not feasible as most of the decomposed interaction energies have positive value. The result can be partly explained by the fact that enzastaurin is mainly a serine/threonine kinase inhibitor but we have docked it with EGFR tyrosine kinase domain. Otherwise, enhanced docking properties can be possibly achieved by increasing the simulation box size.
EGFR kinase domain (2GS2) with gefitinib
In our docking analysis total of three H-bonds are seen, two with Thr 830 (interatomic distance of 2.99 and 3.89 Å) and one with Glu 738 (3.48 Å). Glu 738, Asn 818 and Asp 831 display polar interactions. Leu 764, Cys 773 and Leu 820 exhibit hydrophobic interactions. Two water-mediated halogen bonds are seen with F. Several other interactions are noticed with Val 702, Lys 721, Glu 738, Thr 766, Cys 773, Arg 817, Asn 818, Leu 820, Thr 830 and Asp 831. The monomeric (V948R) gefitinib/erlotinib resistant double mutant (L858R+T790M) EGFR kinase domain has been previously co-crystallized with gefitinib (PDB ID: 4I22) [55].
Discussion
Kidney cancer comprises heterogeneous tumors with diverse molecular and clinical characteristics as reflected by their response to specific treatments. To understand the mechanisms by which genes alter a wide range of physiological processes, we performed a transcriptional profiling study to identify significant genes and examined their biological functions, and to identify KC associated molecular network and pathways. Among hundreds of differentially expressed genes we identified S100A8 and EGFR as potential biomarker of KC and attempted to demonstrate their anticancer drug target potential. In silico docking analysis showed that midostaurin and gefitinib binds to S100A8 as well as to EGFR and predictably inhibits downstream signaling in KC. However, enzastaurin binds only to the S100A8 dimer. Our finding leads to the hypothesis that S100A8 and EGFR are promising anticancer drug targets and midostaurin can be a potential inhibitor. This hypothesis surely needs further validation.
S100 proteins participate in numerous functions including protein phosphorylation, enzymatic activation, calcium homeostasis, and interaction with cytoskeletal components [38]. Most genes encoding S100 proteins are clustered on a region of human chromosome 1q21 that is prone to chromosomal rearrangements, suggesting a link between S100A8/A9 proteins and metastasis and tumor formation [38,56]. S100A8 has cell growth-promoting activity at low concentrations by binding to RAGE. This binding enhances cell mesenchymal properties, induces epithelial-mesenchymal transition and plays important role in promoting cancer invasion and metastasis in cancer [15,22]. Abnormal expressions of S100A8 proteins were observed in a variety of cancers, such as gastric, lung, breast, liver, pancreatic and squamous esophageal carcinomas [15,17,20–23,57–60]. These studies indicate the potential of S100A8 as anticancer target. Despite showing the elevated expression and distinct role of S100A8 in different cancer types, less is known about the expression status or role of S100A8 in KC progression.
The EGFR family of RTPKs is an important cancer target because of the complex signaling through their configuration as homo- or hetrodimers [61,62]. The 4-anilino-quinazoline gefitinib (Iressa) targets the active conformation of EGFR kinase and has been approved for second- and third-line chemotherapy for advanced non small cell lung carcinoma (NSCLC) [63]. Interestingly, a subset of patients who have specific activating mutations in the EGFR tyrosine kinase domain and/or amplification of EGFR often display enhanced sensitivity, positive clinical responses and improved survival with these inhibitors [64,65]. However, the gatekeeper mutation T790M in EGFR enhances affinity for ATP and reduces affinity for gefitinib and thereby inducing resistance [66,67]. We, therefore, used another kinase inhibitor, midostaurin to determine their efficiencies. In our docking study, the best overall binding was exhibited in terms of estimated free energy of binding and Ki value by midostaurin followed by gefitinib and at least by enzastaurin. Interestingly, enzastaurin did not show any binding with EGFR. Enzastaurin has been previously evaluated both in vitro and in nude mice alone and in combination with the EGFR inhibitor gefitinib, where it showed a cooperative inhibitory effect with gefitinib in parental and in gefitinib-resistant cells [68].
This was a pioneering structure-based approach to study S100A8 and EGFR protein interactions with the chosen kinase inhibitors at the molecular level. The EGFR binding cavity is quite hydrophobic and is the most preferable binding site for compounds having compatible shape and stereochemistry. The computational results provide valuable insights into the binding modes of the three tested inhibitors to the S100A8 and EGFR targets. It has been demonstrated that the hydrophobic interactions and the hydrogen bonding with these targets have pivotal contributions to the binding structures and binding free energies, although the van der Waals and electrostatic interactions contributed to the stabilization of the binding structures. The calculated binding free energies comply with the available experimental activity data. The detailed structural insight, binding modes and the crucial factors affecting the binding free energies obtained from the present computational studies may provide valuable insights for future rational structure-based design of novel and potent inhibitors.
The size of a single amino acid in the ATP binding pocket—termed the gatekeeper residue—is a critical determinant of inhibitor sensitivity [67]. Kinases which possess a threonine at this position are sensitive to a range of inhibitors, whereas those having a larger amino acid side chain show significantly higher IC50 values than those having a threonine residue at this site and are broadly resistant [69]. For an ATP-competitive kinase inhibitor, a micromolar inhibition constant, Ki alone is compelling evidence that the proposed drug will be non-selective [41]. In our study, the most favourable docking result was determined for midostaurin with the S100A8 dimer with the estimated Ki as 68.48 nM and free energy of binding as-9.77 kcal/mol. There exists a strong logical correlation between inhibitor potency and selectivity [70] hence, more potent compounds are more selective because they can be applied at a lower dose.
Conclusion
Our analysis suggests distinct transcriptomic signatures for KC with significantly high levels of S100A8 and EGFR expression involved in KC progression. Although further validation on larger dataset is needed to corroborate these findings, analysis of KC tissue is a promising tool for identifying biomarkers of interest. Protein-ligand interaction studies play a vital role in the structure based computational drug design. Our docking based findings shed insight into S100A8 protein as an attractive anticancer target and midostaurin as potential anticancer drug for therapeutic intervention in KC. S100A8 gained importance as target for anticancer drug development due to its central role in mediating inflammatory pathways that facilitate cancer metastasis. The docking simulatio of midostaurin with EGFR is also promising. Further investigations like quantitative structure-activity relationship (QSAR) studies are required to study and identify more favorable interactions with S100A8, EGFR and their partners. Furthermore, better binding ligands with higher affinity and efficacy can be designed and validated using combinatorial chemistry and co-crystallization approaches.
Therapeutic inhibition of kinase signaling cascades and emergence of novel clinically validated potent and selective kinase inhibitors facilitated by rational drug design is a proven boon especially for oncology. Studies to evaluate on- and off-target pharmacology (side effects) of these inhibitors in relation to efficacy and toxicity should be carried using KC cell lines. This will help in holistic monitoring of the changes in phosphorylation resulting from kinase inhibition.
Supporting Information
(XLS)
(XLS)
Acknowledgments
The authors would like to thank Nuha Alansari, Alaa Albogmi, Manal Shaabad, Mona AlKharraz, Amal Noor and Manar Ata for samples collection, performing RNA extraction, bioanalyzer assays and microarray experiments. We thank the patients, physicians, nurses, and pathologists of the King Abdulaziz University Hospital and King Faisal Specialist Hospital and Research Center, Jeddah, Saudi Arabia.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by King Abdullah City for Science and Technology, Riyadh, Saudi Arabia (KACST, Strategic Project ID. 10-BIO1258-03 and 10-BIO1073-03) and Deanship of Research, King Abdulaziz University (Project ID: HiCi-1434-117-2). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013; 63: 11–30. 10.3322/caac.21166 [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 4. Chow W-H, Gridley G, Fraumeni JF Jr, Järvholm B. Obesity, hypertension, and the risk of kidney cancer in men. New England Journal of Medicine. 2000; 343: 1305–1311. [DOI] [PubMed] [Google Scholar]
- 5. Pu YS, Huang CY, Kuo YZ, Kang WY, Liu GY, Huang AM, et al. Characterization of membranous and cytoplasmic EGFR expression in human normal renal cortex and renal cell carcinoma. J Biomed Sci. 2009; 16: 82 10.1186/1423-0127-16-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Motzer RJ, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol. 2006; 24: 5601–5608. [DOI] [PubMed] [Google Scholar]
- 7. Debouck C, Goodfellow PN. DNA microarrays in drug discovery and development. Nature genetics. 1999; 21: 48–50. [DOI] [PubMed] [Google Scholar]
- 8. Drews J. Drug discovery: a historical perspective. Science. 2000; 287: 1960–1964. [DOI] [PubMed] [Google Scholar]
- 9. Vogl T, Leukert N, Barczyk K, Strupat K, Roth J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim Biophys Acta. 2006; 1763: 1298–1306. [DOI] [PubMed] [Google Scholar]
- 10. Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. Calcium-dependent and-independent interactions of the S100 protein family. Biochem J. 2006; 396: 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donato R, R Cannon B, Sorci G, Riuzzi F, Hsu K, J Weber D, et al. Functions of S100 proteins. Current Molecular Medicine. 2013; 13: 24–57. [PMC free article] [PubMed] [Google Scholar]
- 12. Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochemical and biophysical research communications. 2004; 322: 1111–1122. [DOI] [PubMed] [Google Scholar]
- 13. Hessian PA, Edgeworth J, Hogg N. MRP-8 and MRP-14, two abundant Ca(2+)-binding proteins of neutrophils and monocytes. J Leukoc Biol. 1993; 53: 197–204. [PubMed] [Google Scholar]
- 14. Kerkhoff C, Voss A, Scholzen TE, Averill MM, Zänker KS, Bornfeldt KE. Novel insights into the role of S100A8/A9 in skin biology. Experimental dermatology. 2012; 21: 822–826. 10.1111/j.1600-0625.2012.01571.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yin C, Li H, Zhang B, Liu Y, Lu G, Lu S, et al. RAGE-binding S100A8/A9 promotes the migration and invasion of human breast cancer cells through actin polymerization and epithelial-mesenchymal transition. Breast cancer research and treatment. 2013; 142: 297–309. 10.1007/s10549-013-2737-1 [DOI] [PubMed] [Google Scholar]
- 16. Passey RJ, Williams E, Lichanska AM, Wells C, Hu S, Geczy CL, et al. A null mutation in the inflammation-associated S100 protein S100A8 causes early resorption of the mouse embryo. The Journal of Immunology. 1999; 163: 2209–2216. [PubMed] [Google Scholar]
- 17. Grebhardt S, Muller-Decker K, Bestvater F, Hershfinkel M, Mayer D. Impact of S100A8/A9 Expression on Prostate Cancer Progression In Vitro and In Vivo. J Cell Physiol. 2014; 229: 661–671. 10.1002/jcp.24489 [DOI] [PubMed] [Google Scholar]
- 18. Hermani A, De Servi B, Medunjanin S, Tessier PA, Mayer D. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res. 2006; 312: 184–197. [DOI] [PubMed] [Google Scholar]
- 19. Rafii S, Lyden D. S100 chemokines mediate bookmarking of premetastatic niches. Nat Cell Biol. 2006; 8: 1321–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yong HY, Moon A. Roles of calcium-binding proteins, S100A8 and S100A9, in invasive phenotype of human gastric cancer cells. Arch Pharm Res. 2007; 30: 75–81. [DOI] [PubMed] [Google Scholar]
- 21. Nemeth J, Stein I, Haag D, Riehl A, Longerich T, Horwitz E, et al. S100A8 and S100A9 are novel nuclear factor kappa B target genes during malignant progression of murine and human liver carcinogenesis. Hepatology. 2009; 50: 1251–1262. 10.1002/hep.23099 [DOI] [PubMed] [Google Scholar]
- 22. Chen KT, Kim PD, Jones KA, Devarajan K, Patel BB, Hoffman JP, et al. Potential prognostic biomarkers of pancreatic cancer. Pancreas. 2014; 43: 22–27. 10.1097/MPA.0b013e3182a6867e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008; 29: 2035–2043. 10.1093/carcin/bgn188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stulik J, Osterreicher J, Koupilova K, Knizek, Macela A, Bures J, et al. The analysis of S100A9 and S100A8 expression in matched sets of macroscopically normal colon mucosa and colorectal carcinoma: the S100A9 and S100A8 positive cells underlie and invade tumor mass. Electrophoresis. 1999; 20: 1047–1054. [DOI] [PubMed] [Google Scholar]
- 25. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006; 8: 1369–1375. [DOI] [PubMed] [Google Scholar]
- 26. Higgins RJ, Dickinson PJ, LeCouteur RA, Bollen AW, Wang H, Wang H, et al. Spontaneous canine gliomas: overexpression of EGFR, PDGFRalpha and IGFBP2 demonstrated by tissue microarray immunophenotyping. J Neurooncol. 2010; 98: 49–55. 10.1007/s11060-009-0072-5 [DOI] [PubMed] [Google Scholar]
- 27. Shiomitsu K, Johnson CL, Malarkey DE, Pruitt AF, Thrall DE. Expression of epidermal growth factor receptor and vascular endothelial growth factor in malignant canine epithelial nasal tumours. Vet Comp Oncol. 2009; 7: 106–114. 10.1111/j.1476-5829.2009.00178.x [DOI] [PubMed] [Google Scholar]
- 28. Pomerantz RG, Grandis JR. The epidermal growth factor receptor signaling network in head and neck carcinogenesis and implications for targeted therapy. Semin Oncol. 2004; 31: 734–743. [DOI] [PubMed] [Google Scholar]
- 29. Lee HJ, Xu X, Choe G, Chung DH, Seo JW, Lee JH, et al. Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas: Comparison of four commercially available antibodies by immunohistochemistry and fluorescence in situ hybridization study. Lung Cancer. 2010; 68: 375–382. 10.1016/j.lungcan.2009.07.014 [DOI] [PubMed] [Google Scholar]
- 30. Duan L, Wu R, Ye L, Wang H, Yang X, Zhang Y, et al. S100A8 and S100A9 are associated with colorectal carcinoma progression and contribute to colorectal carcinoma cell survival and migration via Wnt/beta-catenin pathway. PLoS One. 2013; 8: e62092 10.1371/journal.pone.0062092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fukihara J, Watanabe N, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, et al. Clinical Predictors of Response to EGFR Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Non-Small Cell Lung Cancer. Oncology. 2014; 86: 86–93. 10.1159/000357129 [DOI] [PubMed] [Google Scholar]
- 32. Yamaguchi NH, Mayer IA, Malzyner A, de Andrade CJ, Murad AM, Del Giglio A, et al. Gefitinib and celecoxib in advanced metastatic gastrointestinal tumors: a pilot feasibility study. J Gastrointest Oncol. 2014; 5: 57–66. 10.3978/j.issn.2078-6891.2013.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moody TW, Chan DC, Mantey SA, Moreno P, Jensen RT. SR48692 inhibits non-small cell lung cancer proliferation in an EGF receptor-dependent manner. Life Sci. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shiogama K, Wongsiri T, Mizutani Y, Inada K-i, Tsutsumi Y. High-sensitivity epidermal growth factor receptor immunostaining for colorectal carcinomas, compared with EGFR PharmDx: A study of diagnostic accuracy. International journal of clinical and experimental pathology. 2013; 6: 24 [PMC free article] [PubMed] [Google Scholar]
- 35. Baba Y, Fujii M, Tokumaru Y, Kato Y. Present and Future of EGFR Inhibitors for Head and Neck Squamous Cell Cancer. J Oncol. 2012; 2012: 986725 10.1155/2012/986725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ciardiello F, Tortora G. Epidermal growth factor receptor (EGFR) as a target in cancer therapy: understanding the role of receptor expression and other molecular determinants that could influence the response to anti-EGFR drugs. Eur J Cancer. 2003; 39: 1348–1354. [DOI] [PubMed] [Google Scholar]
- 37. Kim Y, Li Z, Apetri M, Luo B, Settleman JE, Anderson KS. Temporal resolution of autophosphorylation for normal and oncogenic forms of EGFR and differential effects of gefitinib. Biochemistry. 2012; 51: 5212–5222. 10.1021/bi300476v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heizmann CW, Fritz G, Schafer BW. S100 proteins: structure, functions and pathology. Front Biosci. 2002; 7: d1356–1368. [DOI] [PubMed] [Google Scholar]
- 39. Burnett G, Kennedy EP. The enzymatic phosphorylation of proteins. J Biol Chem. 1954; 211: 969–980. [PubMed] [Google Scholar]
- 40. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002; 298: 1912–1934. [DOI] [PubMed] [Google Scholar]
- 41. Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005; 12: 621–637. [DOI] [PubMed] [Google Scholar]
- 42. Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010; 28: 4339–4345. 10.1200/JCO.2010.28.9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. El Fitori J, Su Y, Büchler P, Ludwig R, Giese NA, Büchler MW, et al. PKC 412 small‐molecule tyrosine kinase inhibitor. Cancer. 2007; 110: 1457–1468. [DOI] [PubMed] [Google Scholar]
- 44. Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012; 26: 2061–2068. 10.1038/leu.2012.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005; 65: 7462–7469. [DOI] [PubMed] [Google Scholar]
- 46. Rizvi MA, Ghias K, Davies KM, Ma C, Weinberg F, Munshi HG, et al. Enzastaurin (LY317615), a protein kinase Cβ inhibitor, inhibits the AKT pathway and induces apoptosis in multiple myeloma cell lines. Molecular cancer therapeutics. 2006; 5: 1783–1789. [DOI] [PubMed] [Google Scholar]
- 47. Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, et al. (2002) ZD1839 (Iressa) an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. 5749–5754 p. [PubMed] [Google Scholar]
- 48. DeLano W (2010) The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. Accessed 6/25/2007. Available at http://www. pymol, org. [Google Scholar]
- 49. Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform. 2009; 1: 15 10.1186/1758-2946-1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. Journal of computational chemistry. 1996; 17: 490–519. [Google Scholar]
- 51. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry. 2009; 30: 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of computational chemistry. 1998; 19: 1639–1662. [Google Scholar]
- 53. Solis FJ, Wets RJ-B. Minimization by random search techniques. Mathematics of operations research. 1981; 6: 19–30. [Google Scholar]
- 54. Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Natl Acad Sci U S A. 2004; 101: 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gajiwala KS, Feng J, Ferre R, Ryan K, Brodsky O, Weinrich S, et al. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure. 2013; 21: 209–219. 10.1016/j.str.2012.11.014 [DOI] [PubMed] [Google Scholar]
- 56. Heizmann CW, Ackermann GE, Galichet A. Pathologies involving the S100 proteins and RAGE. Subcell Biochem. 2007; 45: 93–138. [DOI] [PubMed] [Google Scholar]
- 57. Seth A, Kitching R, Landberg G, Xu J, Zubovits J, Burger AM. Gene expression profiling of ductal carcinomas in situ and invasive breast tumors. Anticancer Res. 2003; 23: 2043–2051. [PubMed] [Google Scholar]
- 58. Luo A, Kong J, Hu G, Liew CC, Xiong M, Wang X, et al. Discovery of Ca2+-relevant and differentiation-associated genes downregulated in esophageal squamous cell carcinoma using cDNA microarray. Oncogene. 2004; 23: 1291–1299. [DOI] [PubMed] [Google Scholar]
- 59. Cross SS, Hamdy FC, Deloulme JC, Rehman I. Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology. 2005; 46: 256–269. [DOI] [PubMed] [Google Scholar]
- 60. El-Rifai W, Moskaluk CA, Abdrabbo MK, Harper J, Yoshida C, Riggins GJ, et al. Gastric cancers overexpress S100A calcium-binding proteins. Cancer Res. 2002; 62: 6823–6826. [PubMed] [Google Scholar]
- 61. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005; 5: 341–354. [DOI] [PubMed] [Google Scholar]
- 62. Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006; 7: 505–516. [DOI] [PubMed] [Google Scholar]
- 63. Muhsin M, Graham J, Kirkpatrick P. Gefitinib. Nat Rev Drug Discov. 2003; 2: 515–516. [DOI] [PubMed] [Google Scholar]
- 64. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non—small-cell lung cancer to gefitinib. New England Journal of Medicine. 2004; 350: 2129–2139. [DOI] [PubMed] [Google Scholar]
- 65. Fabbro D, Cowan-Jacob SW, Möbitz H, Martiny-Baron G (2012) Targeting cancer with small-molecular-weight kinase inhibitors Kinase Inhibitors: Springer; pp. 1–34. [DOI] [PubMed] [Google Scholar]
- 66. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005; 2: e73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horvath Z, et al. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004; 11: 691–701. [DOI] [PubMed] [Google Scholar]
- 68. Gelardi T, Caputo R, Damiano V, Daniele G, Pepe S, Ciardiello F, et al. Enzastaurin inhibits tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer. 2008; 99: 473–480. 10.1038/sj.bjc.6604493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Godl K, Wissing J, Kurtenbach A, Habenberger P, Blencke S, Gutbrod H, et al. An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc Natl Acad Sci U S A. 2003; 100: 15434–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Frye SV. Structure-activity relationship homology (SARAH): a conceptual framework for drug discovery in the genomic era. Chem Biol. 1999; 6: R3–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS)
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.
The data sets supporting the results of this study are available in the NCBI's Gene Expression Omnibus (GEO), identified as GSE781, GSE7023, and GSE6344 (http://www.ncbi.nlm.nih.gov/gds/?term=GSE781).