

Abstract
Objective:
To gain insight into different cerebral amyloid angiopathy (CAA) phenotypes and mechanisms, we investigated cortical superficial siderosis (CSS), a new imaging marker of the disease, and its relation with APOE genotype in patients with pathologically proven CAA, who presented with and without intracerebral hemorrhage (ICH).
Methods:
MRI scans of 105 patients with CAA pathologic confirmation and MRI were analyzed for CSS (focal, ≤3 sulci; disseminates, ≥4 sulci) and other imaging markers. We compared pathologic, imaging, and APOE genotype data between subjects with vs without ICH, and investigated associations between CSS and APOE genotype.
Results:
Our cohort consisted of 54 patients with CAA with symptomatic lobar ICH and 51 without ICH. APOE genotype was available in 53 patients. More than 90% of pathology samples in both groups had neuritic plaques, whereas neurofibrillary tangles were more commonly present in the patients without ICH (87% vs 42%, p < 0.0001). There was a trend for patients with CAA with ICH to more commonly have APOE ε2 (48.7% vs 21.4%, p = 0.075), whereas patients without ICH were more likely to be APOE ε4 carriers (85.7% vs 53.9%, p = 0.035). Disseminated CSS was considerably commoner in patients with ICH (33.3% vs 5.9%, p < 0.0001). In logistic regression, disseminated CSS was associated with APOE ε2 (but not APOE ε4) (odds ratio 5.83; 95% confidence interval 1.49–22.82, p = 0.011).
Conclusions:
This neuropathologically defined CAA cohort suggests that CSS and APOE ε2 are related to the hemorrhagic expression of the disease; APOE ε4 is enriched in nonhemorrhagic CAA. Our study emphasizes the concept of different CAA phenotypes, suggesting divergent pathophysiologic mechanisms.
Sporadic cerebral amyloid angiopathy (CAA) is a small vessel disease that preferentially involves small cortical and leptomeningeal arteries due to progressive amyloid-β deposition in their walls.1,2 CAA occurs frequently in elderly people, and is a common and important cause of symptomatic lobar intracerebral hemorrhage (ICH).1–3 However, CAA might present without major lobar ICH, but instead with cognitive impairment (either chronic or rapidly progressive), or transient focal neurologic symptoms.4 CAA is almost invariably found in Alzheimer disease (AD), but in most cases is relatively mild.5
CAA is also associated with characteristic MRI biomarkers, including strictly lobar cerebral microbleeds (CMBs), cortical superficial siderosis (CSS),6 centrum semiovale (CSO) perivascular spaces (PVS), and white matter hyperintensities (WMH).1,7 These neuroimaging markers probably reflect related but distinct aspects of CAA pathophysiology. CSS in particular is an interesting recently recognized form of CAA-related hemorrhage, which likely reflects repeated episodes of blood leaking into the subarachnoid space from brittle and fragile CAA-affected vessels. CSS has been shown to carry a high risk of future symptomatic lobar ICH.6,8,9
Genetic factors, such as APOE genotype, are important in the pathophysiology of CAA.10,11 APOE ε4 appears to enhance vascular amyloid-β deposition in a dose-dependent fashion,12 while APOE ε2 promotes vasculopathic changes that can lead to vessel rupture.13 To gain further insights into different CAA phenotypes and potential mechanisms,14 we investigated associations between neuroimaging markers of the disease, APOE genotype, and pathologic findings in patients with CAA presenting with and without symptomatic ICH. We hypothesized that (1) hemorrhagic markers of CAA (CSS and lobar CMBs) would be more strongly associated with APOE ε2 genotype; (2) hemorrhagic markers of disease severity in CAA (CSS and lobar CMBs) and APOE ε2 genotype would be more common in patients presenting with symptomatic ICH than in those presenting without ICH; and (3) neurofibrillary tangles (NFTs) will be more common in nonhemorrhagic compared to hemorrhagic CAA, whereas amyloid plaques might be invariably present in the 2 groups due to their close molecular pathogenesis.
METHODS
Case selection and clinical data collection.
We included all eligible patients from Massachusetts General Hospital (MGH) identified retrospectively by a systematic keyword search of pathology reports and prospective clinical databases. Cases were defined as subjects with both pathology-proven CAA (from routinely collected brain biopsy, biopsy at hematoma evacuation, or autopsy) and adequate brain MRI sequences for the study. Data search covered patients seen at the hospital between 1997 and 2012. An additional overlapping search through established prospective datasets of patients with lobar hemorrhages (ICH or CMB) was performed to confirm identification of all potential eligible cases for the study. Among more than 3,200 retrieved cases, we initially included those having (1) a pathology report containing explicit information regarding CAA assessment and (2) available brain MRI sequences of adequate quality including T2-weighted, T2*-weighted gradient-recalled echo (T2*-GRE), or susceptibility-weighted imaging (SWI) and fluid-attenuated inversion recovery (FLAIR) sequences. After reviewing all neuroimaging, pathologic, and clinical data available, we excluded subjects with (1) small brain biopsy samples (greater diameter <1 cm) or samples from clot evacuations not containing any assessable vessels; (2) autopsy studies not grading CAA; and (3) known alternative causes for lobar ICH. A total of 192 patients were eligible for analysis. Patients with no pathologic evidence of CAA (n = 54), clinical/imaging presentations not characteristic of CAA (3 cases with deep ICH and 9 with ischemic stroke at baseline), no T2 or T2*-GRE MRI (n = 18), T2/T2*-GRE MRI of insufficient quality (n = 2), and irretrievable sequences (n = 1) were excluded.
Demographic and clinical information was obtained from prospective databases and medical records using standardized data collection forms. Variables of interest were age, sex, history of hypertension, antithrombotic drug use, and clinical presentation at baseline. APOE genotype was determined in a subset of patients who provided blood samples and consented to genetic testing as previously described,15 and without knowledge of clinical or neuroimaging data.
The original clinical presentation of patients was ascertained from all available neuroimaging, pathologic, and clinical data, and was determined as either symptomatic lobar ICH confirmed on neuroimaging or nonhemorrhagic (including cognitive impairment, transient focal neurologic episodes, or other neurologic symptoms). Cases of inflammatory CAA were included in the analysis when an MRI outside the acute phase (≥1 month) was available. To investigate the stability of the non-ICH CAA clinical phenotypes, we extrapolated follow-up information from prospective databases and medical records on incident symptomatic ICH.
Standard protocol approvals, registrations, and patient consents.
The study received ethical approval by the institutional review board of MGH.
Pathologic data collection.
Morphologic assessment was performed in routine hematoxylin & eosin staining and the presence or absence and severity of vascular amyloid-β deposition was confirmed by immunohistochemical detection or Congo red staining. CAA presence and severity was assessed in all available vessels. Cases were considered positive for CAA when they had at least 1 leptomeningeal or cortical vessel with amyloid-β reported, providing enough information to reliably classify CAA severity using the Vonsattel grading system16,17 and were classified as mild (Vonsattel grade 1) or moderate to severe (Vonsattel grades 2–4). Where available from neuropathology reports, we also systematically extracted information on neuritic plaques and NFTs assessed in routinely immunostained sections for amyloid-β and phosphorylated tau and recorded as present or absent.
Neuroimaging data and analysis.
Imaging for all patients included T2-weighted, FLAIR, T2*-GRE, or SWI. MRI were reviewed blinded to clinical, histopathologic, and genetic data by trained observers, according to Standards for Reporting Vascular Changes on Neuroimaging (STRIVE).18
The presence and number of CMBs was evaluated on axial T2*-GRE or SWI images according to current consensus criteria7 and categorized as lobar (i.e., cortical-subcortical), deep (i.e., basal ganglia [BG], thalami, brainstem), or cerebellar. The presence and number of macro ICHs (>5 mm in diameter on T2*-GRE/SWI)19 was also noted.
CSS was defined as linear residues of chronic blood products in the superficial layers of the cerebral cortex showing a characteristic gyriform pattern of low signal on T2*-GRE/SWI; T1-weighted and FLAIR images were used for anatomical confirmation of the gyral location of these signal hypointensities.20 The distribution and severity of CSS was classified as focal (restricted to ≤3 sulci) or disseminated (≥4 sulci).6 Areas of CSS were ≥2 unaffected sulci away from any lobar ICH, at multiple axial levels; CSS contiguous or potentially anatomically connected with any lobar ICH were not included in these categories.20
PVS were assessed in line with STRIVE definitions18 and rated on axial T2-weighted MRI, using a validated 4-point visual rating scale in the BG and CSO as previously described.21,22
Periventricular and deep WMH were visually assessed on axial FLAIR images on the 4-point Fazekas rating scale for each, adding up to a total score on a 7-point scale.23
Statistical analysis.
Categorical variables were analyzed using Pearson χ2 or Fisher exact test, and continuous variables by the 2-sample t test (for normal distributions) and Wilcoxon rank sum (for non-normal distributions). We compared demographic, genetic, pathologic, and imaging characteristics of CAA patients with and without ICH. Variables for APOE ε2 and ε4 were each coded as the number of alleles per participant (0, 1, or 2). Separate logistic regression models were used to assess the relationship between APOE genotype and CSS (presence or burden), as well as CMB count (linear regression). As sensitivity analyses, these models were predetermined to adjust for age and clinical presentation of CAA. Significance level was set at 0.05. Stata software (version 11.2, StataCorp., College Station, TX) was used. The manuscript was prepared with reference to the STROBE guidelines.24
RESULTS
Our final cohort consisted of 105 patients with pathologic evidence of CAA: 52 from autopsies, 22 from brain biopsies, and 31 with pathologic samples from hematoma evacuations. Fifty-four patients were admitted with symptomatic, spontaneous lobar ICH, while 51 patients were admitted without any symptomatic ICH. Patients without ICH presented with cognitive impairment (n = 42), transient focal neurologic episodes (n = 3), or a combination of other symptoms (n = 6, including altered mental status, or seizures, with findings consistent with inflammatory CAA). The median Clinical Dementia Rating score of patients without ICH and available data (n = 27) was 1 (interquartile range [IQR] 0.5–2).
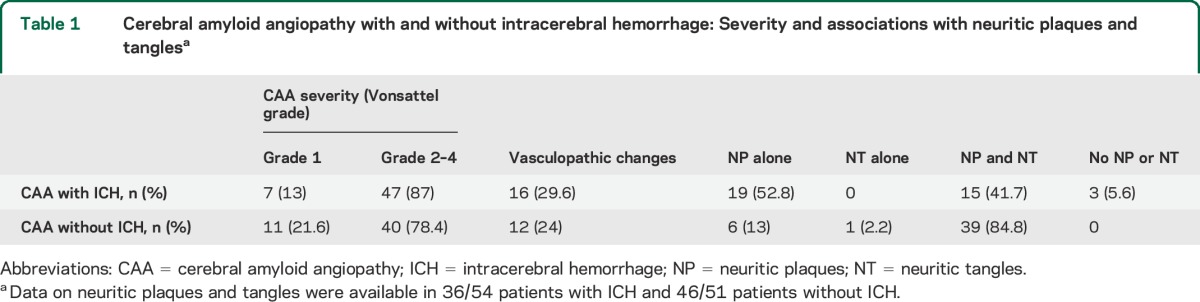
Table 1 indicates the severity of CAA and the presence or absence of neuritic plaques and NFTs in the 2 groups. In general, mild (Vonsattel grade 1) or moderate to severe (Vonsattel grades 2–4) CAA were equally represented in the cohorts (p > 0.2 for both comparisons). There was no difference in the presence of vasculopathic changes (vessel-within-vessel appearance and vessel wall necrosis) in the 2 groups (29.6% vs 24%, p = 0.336). Although the prevalence of neuritic plaques was similar in the 2 groups, neuritic plaques in isolation (i.e., with no tangles) were much more frequent in the ICH patients than in the patients with CAA without ICH (53% vs 13%, p < 0.0001). More than 90% of pathology samples in both groups had neuritic plaques, whereas NFTs were more commonly present in the patients without ICH (87% vs 42%, p < 0.0001). These associations remained consistent in logistic regression models controlling for age.
Table 1.
Cerebral amyloid angiopathy with and without intracerebral hemorrhage: Severity and associations with neuritic plaques and tanglesa
Comparisons of clinical and imaging characteristics between patients with CAA with vs without ICH are summarized in table 2. There was a trend for patients with CAA and ICH to more often have APOE ε2 (48.7% vs 21.4%, p = 0.075), whereas patients without ICH were more likely to be carriers of APOE ε4 (85.7% vs 53.9%, p = 0.035). The 2 groups were similar in imaging markers of cerebral small vessel disease, including WMH burden, high degree of CSO–enlarged PVS (EPVS) and BG-EPVS, and lobar CMB counts (table 2). However, the prevalence of CSS was higher in patients with ICH (51.9% vs 19.6%, p = 0.001), especially disseminated CSS (33.3% vs 5.9%, p < 0.0001). Representative MRIs are shown in the figure.
Table 2.
Comparison of clinical, imaging, and genetic characteristics between pathologically proven CAA with vs without ICH cohorts
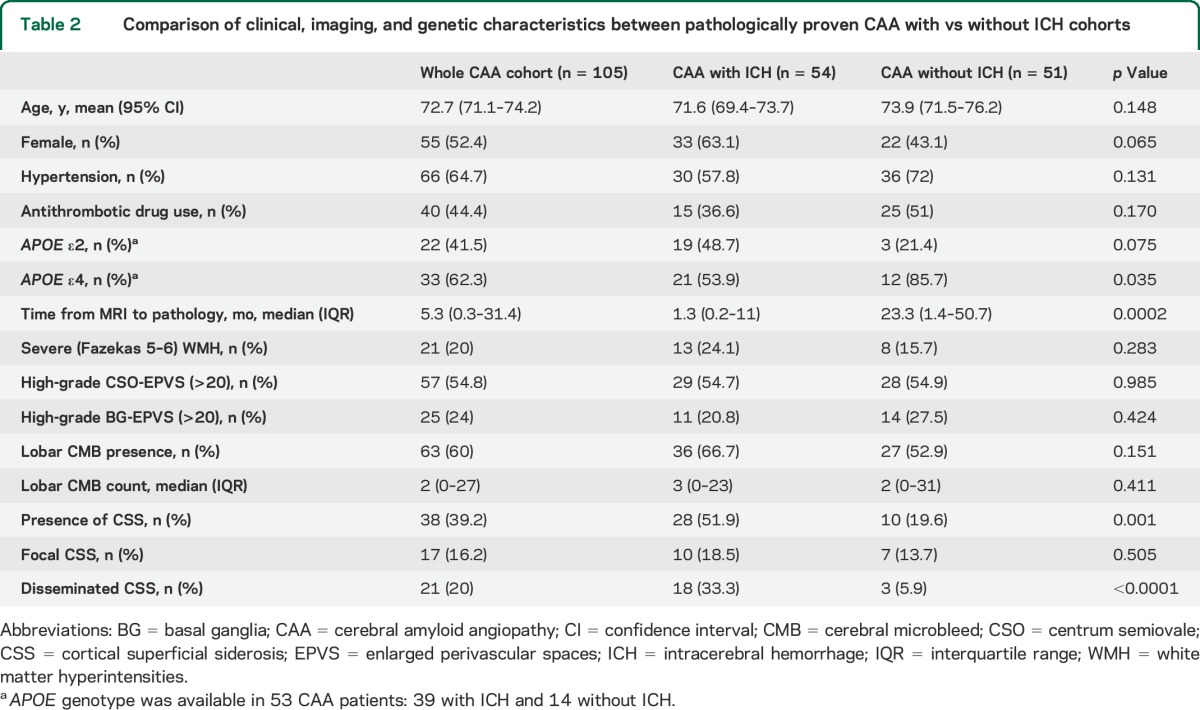
Figure. Representative MRIs of patients with pathologic evidence of cerebral amyloid angiopathy with and without symptomatic intracerebral hemorrhage.

(A) A 73-year-old woman with cerebral amyloid angiopathy (CAA)–intracerebral hemorrhage (ICH) and disseminated cortical superficial siderosis on T2*-weighted gradient-recalled echo (T2*-GRE) (left). Her APOE genotype was ε2/ε4. (B) Multiple strictly lobar cerebral microbleeds (but no siderosis) on T2*-GRE MRI (left) in a 73-year-old woman with cognitive impairment and ε4/ε4 APOE genotype. Note the comparable white matter hyperintensities burden on fluid-attenuated inversion recovery MRI (middle panels) and centrum semiovale perivascular spaces on T2-weighted images (right panels) in the 2 patients. Both cases had severe CAA with vasculopathic changes (vessel-within-vessel appearance) on pathology.
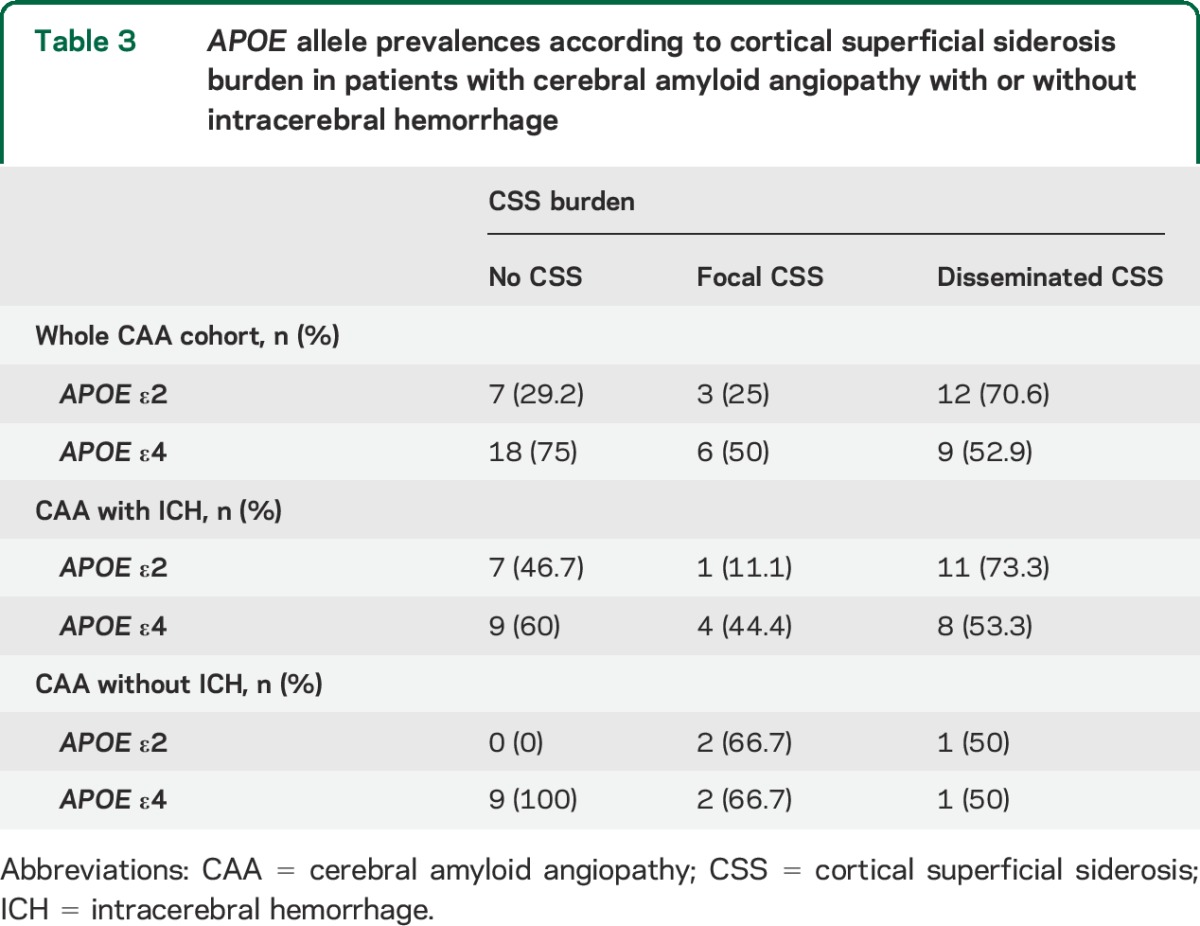
Among subjects with available genetic testing (n = 53), APOE ε2 (but not ε4) allele was overrepresented in cases with disseminated CSS, in the whole cohort (p = 0.013), and in the CAA subgroup with ICH (table 3). In logistic regression, disseminated CSS was associated with APOE ε2 (odds ratio [OR] 5.83; 95% confidence interval [CI] 1.49–22.82, p = 0.011). These results remained consisted and of similar effect size in a sensitivity analysis adjusting for age and clinical presentation with symptomatic ICH (OR 4.97; 95% CI 1.11–22.21, p = 0.036) and in models further adjusting for the presence of vasculopathic changes on pathology. There was no association between CSS (burden or presence) and APOE ε4. There was no association between CMB counts and APOE genotype.
Table 3.
APOE allele prevalences according to cortical superficial siderosis burden in patients with cerebral amyloid angiopathy with or without intracerebral hemorrhage
Follow-up data were available in all patients presenting without ICH at baseline. During a median follow-up time of 3 years (IQR 1.1–5.6 years), 2 of 51 patients (3.9%, 95% CI 0.5–13.5%) experienced a symptomatic lobar ICH. One of these 2 patients had focal CSS at baseline MRI.
DISCUSSION
The major findings from this study show that patients with CAA presenting with ICH are more likely to have CSS (particularly disseminated) and the APOE ε2 genotype compared to CAA patients presenting without ICH. By contrast, APOE ε4 is enriched in nonhemorrhagic CAA. In addition, there was an overall higher burden of NFT pathology in the non-hemorrhagic CAA group. APOE genotype might partly influence these relationships: APOE ε2 was found to be associated with both symptomatic ICH clinical phenotype and disseminated CSS.
Our results provide new insights into the clinical and imaging spectrum of sporadic CAA, pointing to different disease phenotypes.14 While sporadic CAA is commonly found in the elderly, it is currently unknown why only a fraction of people with CAA pathology develop symptomatic disease, and why some present with symptomatic ICH, while others only develop cognitive impairment or other clinical symptoms. A previous comparative postmortem histopathologic study found that the brain features from patients with CAA that are most consistently related to ICH are severe degree of vascular amyloid deposition and the presence of fibrinoid necrosis (with or without microaneurysms).16 However, cases of CAA with symptomatic ICH in our study had a fairly similar vascular amyloid burden and prevalence of CAA-associated vasculopathic changes compared to cases without ICH, although this could have been influenced by sampling bias. In addition, the 2 groups had a similar profile of putative neuroimaging biomarkers of CAA severity, including lobar CMBs, WMH, and CSO-EPVS. It thus seems likely that additional biological pathways from those involved in amyloid-β accumulation in cortical and leptomeningeal vessels play a role in determining clinical expression.
The most distinctive neuroimaging feature between the 2 groups was the much higher prevalence of disseminated CSS in patients with CAA and ICH. The prevalence of CSS in our histopathology-confirmed CAA-ICH group is in line with the reported prevalence in a previous imaging study of CAA-ICH.20 In addition, the presence of CSS in the group without ICH (10%) is more in line with recent imaging studies evaluating CSS in a memory clinic setting and AD.25
Although the pathophysiologic mechanisms underlying CSS in CAA are not yet fully understood, observational data suggest that CSS represents blood residues related to blood leaking episodes into the subarachnoid space from CAA-affected vessels.6,20,26 A secondary mechanism due to leakage or expansion of a lobar ICH into the subarachnoid space cannot be fully excluded; however, similar to other studies, CSS was mostly found distant from any ICH (and often in the opposite hemisphere), and was also detected even in cases without any ICH. Two recent studies have identified CSS as a particular risk factor for subsequent ICH in CAA.8,9 In a cohort of 51 patients with CAA-related CSS and a median follow-up of 35.3 months, new intracranial hemorrhages were observed in 24 patients (47.1%).8 A European multicenter study of patients with probable or possible CAA (n = 118) found that CSS was a significant predictor of time until ICH.9 The ICH risk at 4 years was 28.9% (95% CI 7.7–76.7%) and 74% (95% CI 44.1–95.7) for patients with focal and disseminated CSS, respectively, compared to 25% (95% CI 7.6–28.3%) for patients without siderosis (log-rank test: p = 0.0031).9 A small autopsy series of 6 CAA cases showed that multiple leptomeningeal arteries (and not parenchymal cortical vessels) can rupture into the subarachnoid space and the brain parenchyma, leading to large lobar hemorrhages.27,28 These observations might explain the relationship between disseminated CSS and CAA with ICH, as well as why patients with disseminated CSS have the highest risk of future ICH.9
Our study demonstrates that there might be APOE genotype-specific effects on the imaging and clinical expression of CAA-related disease. While APOE ε4 seems to be more associated with CAA without ICH, APOE ε2 is linked more strongly with bleeding and CSS. The APOE ε2 allele is reported to be associated with CAA-related lobar ICH, possibly due to the disease-related vasculopathic changes seen with this allele,13,29 and recently with CSS in a cohort of CAA defined by clinicoradiographic criteria.30 APOE ε4 (presence and number) is also associated with sporadic CAA and ICH risk.31–33 Our study raised the interesting possibility that APOE ε2 influences pathways causing both CSS and ICH. After accounting for the presence of vasculopathic changes, the apparent relation of APOE ε2 to CSS was not reduced, suggesting that vascular amyloid load and how extensively the vessel wall architecture is disrupted is not the sole driving force underlying these pathways and hence, clinical phenotypes. However, any mechanistic links are clearly complex and need to be treated with caution, as APOE genotype alone might not be necessary or sufficient to drive these effects. It is important to note that links may differ according to the presence or absence of AD pathology, particularly for APOE ε2, which has been associated with a reduced risk of late-onset Alzheimer dementia.31 This observation is in line with the differential associations between neuritic plaques and NFTs in our patients with CAA with and without ICH. There is significant variation in the pathologic appearance of CAA and its influence on AD pathology,34,35 further highlighting the concept of different phenotypes of CAA. It is important to acknowledge though that the overrepresentation of NFTs among the patients with CAA without ICH might also reflect a bias issue, since by definition these cases presented with cognitive impairment, partly driven by NFT pathology.
Although the natural history of patients with symptomatic CAA mainly presenting with cognitive impairment (i.e., without ICH at baseline) has not been fully investigated, our data suggest that this CAA phenotype may have a lower risk of developing ICH compared to CAA cases with a history of ICH. It is important to note that patients with CAA without major ICH (microbleeds or CSS-only CAA) presenting to stroke services with symptoms other than cognitive impairment (e.g., transient focal neurologic episodes) might still be at significant risk of future bleeding.9,36,37 This topic requires further investigation in prospective studies.
Major strengths of the study include the large sample of patients with histopathologically confirmed CAA and available MRI sequences, the systematic evaluation of MRI scans for a range of neuroimaging biomarkers of small vessel disease, and the testing of a prespecified hypothesis. The main limitations, inherent to any clinical-pathology series in CAA, include (1) the difference in pathology sampling between autopsied brains and biopsies/hematomas; (2) the biases regarding which patients get biopsied or autopsied, which might be skewed towards end stage or advanced disease; and (3) potential selection bias due to the requirement for both pathology and MRI and the unavailability of APOE genotype data in all patients. The differences in Alzheimer pathology between groups could potentially be explained by a longer duration of disease (taking time from MRI to pathology as a surrogate for disease duration) in patients with CAA without ICH or the different characteristics between the 2 groups (including APOE ε4 prevalence). Molecular imaging studies assessing amyloid and tau burden in vivo38 may help resolve this question. Furthermore, the degree and evolution of cognitive impairment in patients with CAA without ICH (data limited in our study) could influence pathologic findings, especially NFTs and amyloid plaques presence. Finally, the cross-sectional design of the current study did not allow us to assess potential causality of the reported associations.
Results from this neuropathologic-defined cohort of patients with CAA with and without ICH provide evidence for distinct disease phenotypes, and suggest that CSS and APOE ε2 are related to the hemorrhagic expression of the disease. Our study emphasizes the widening spectrum of CAA, with clinical phenotypes reflecting different neuroimaging and genetic features, and suggests divergent pathophysiologic mechanisms. Although these findings require external validation in larger CAA cohorts, they may be relevant for future biomarker studies and disease-modifying CAA trials.
ACKNOWLEDGMENT
The authors thank Dr. David J. Werring, Prof. Martin Brown, and the Bogue Research Fellowships (UCL) for supporting Dr. Andreas Charidimou's visiting fellowship at the Hemorrhagic Stroke Research Program, Harvard Medical School, Boston, MA.
GLOSSARY
- AD
Alzheimer disease
- BG
basal ganglia
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- CMB
cerebral microbleed
- CSO
centrum semiovale
- CSS
cortical superficial siderosis
- EPVS
enlarged perivascular spaces
- FLAIR
fluid-attenuated inversion recovery
- ICH
intracerebral hemorrhage
- IQR
interquartile range
- MGH
Massachusetts General Hospital
- NFT
neurofibrillary tangle
- OR
odds ratio
- PVS
perivascular spaces
- STRIVE
Standards for Reporting Vascular Changes on Neuroimaging
- SWI
susceptibility-weighted imaging
- T2*-GRE
T2*-weighted gradient-recalled echo
- WMH
white matter hyperintensities
Footnotes
Editorial, page 1190
AUTHOR CONTRIBUTIONS
A. Charidimou: project concept and design, imaging and data analysis, write up. S. Martinez-Ramirez: data collection, imaging analysis, critical revisions. A. Shoamanesh: critical revisions. J. Oliveira-Filho: data collection, critical revisions. M. Frosch: data collection, critical revisions. Anastasia Vashkevich: data collection and management. Alison Ayres: data collection and management. J. Rosand: funding, data collection, critical revisions. Mahmut Edip Gurol: data collection. Steven M. Greenberg: funding, data collection, critical revisions. A. Viswanathan: project concept and design, write up, critical revisions.
STUDY FUNDING
Supported by NIH grants K23AG028726-05, P50AG005134-30, 2R01AG26484, 5R01AG026484-10, and 5K23AG028726-05.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012;83:124–137. [DOI] [PubMed] [Google Scholar]
- 2.Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011;70:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinters HV. Cerebral amyloid angiopathy: a critical review. Stroke 1987;18:311–324. [DOI] [PubMed] [Google Scholar]
- 4.Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993;43:2073–2079. [DOI] [PubMed] [Google Scholar]
- 5.Ellis RJ, Olichney JM, Thal LJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, part XV. Neurology 1996;46:1592–1596. [DOI] [PubMed] [Google Scholar]
- 6.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009;8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linn J, Wollenweber FA, Lummel N, et al. Superficial siderosis is a warning sign for future intracranial hemorrhage. J Neurol 2013;260:176–181. [DOI] [PubMed] [Google Scholar]
- 9.Charidimou A, Peeters AP, Jager R, et al. Cortical superficial siderosis and intracerebral hemorrhage risk in cerebral amyloid angiopathy. Neurology 2013;81:1666–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 1995;38:254–259. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg SM, Briggs ME, Hyman BT, et al. Apolipoprotein E epsilon 4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke 1996;27:1333–1337. [DOI] [PubMed] [Google Scholar]
- 12.Rannikmae K, Samarasekera N, Martinez-Gonzalez NA, Al-Shahi Salman R, Sudlow CL. Genetics of cerebral amyloid angiopathy: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2013;84:901–908. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 1998;50:961–965. [DOI] [PubMed] [Google Scholar]
- 14.Charidimou A, Jager HR. Developing biomarkers for cerebral amyloid angiopathy trials: do potential disease phenotypes hold promise? Lancet Neurol 2014;13:538–540. [DOI] [PubMed] [Google Scholar]
- 15.Brouwers HB, Biffi A, McNamara KA, et al. Apolipoprotein E genotype is associated with CT angiography spot sign in lobar intracerebral hemorrhage. Stroke 2012;43:2120–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP., Jr Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991;30:637–649. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg SM, Vonsattel JP. Diagnosis of cerebral amyloid angiopathy: sensitivity and specificity of cortical biopsy. Stroke 1997;28:1418–1422. [DOI] [PubMed] [Google Scholar]
- 18.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kidwell CS, Greenberg SM. Red meets white: do microbleeds link hemorrhagic and ischemic cerebrovascular disease? Neurology 2009;73:1614–1615. [DOI] [PubMed] [Google Scholar]
- 20.Charidimou A, Jager RH, Fox Z, et al. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013;81:626–632. [DOI] [PubMed] [Google Scholar]
- 21.Charidimou A, Jaunmuktane Z, Baron JC, et al. White matter perivascular spaces: an MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology 2014;82:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 2010;41:450–454. [DOI] [PubMed] [Google Scholar]
- 23.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. AJR Am J Roentgenol 1987;149:351–356. [DOI] [PubMed] [Google Scholar]
- 24.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 2007;370:1453–1457. [DOI] [PubMed] [Google Scholar]
- 25.Zonneveld HI, Goos JD, Wattjes MP, et al. Prevalence of cortical superficial siderosis in a memory clinic population. Neurology 2014;82:698–704. [DOI] [PubMed] [Google Scholar]
- 26.Linn J, Herms J, Dichgans M, et al. Subarachnoid hemosiderosis and superficial cortical hemosiderosis in cerebral amyloid angiopathy. AJNR Am J Neuroradiol 2008;29:184–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda S, Yamazaki K, Miyakawa T, et al. Subcortical hematoma caused by cerebral amyloid angiopathy: does the first evidence of hemorrhage occur in the subarachnoid space? Neuropathology 2003;23:254–261. [DOI] [PubMed] [Google Scholar]
- 28.Takeda S, Hinokuma K, Yamazaki K, et al. The hemorrhage caused by sporadic-type cerebral amyloid angiopathy occurs primarily in the cerebral sulci. Neuropathology 2012;32:38–43. [DOI] [PubMed] [Google Scholar]
- 29.Nicoll JA, Burnett C, Love S, et al. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol 1997;41:716–721. [DOI] [PubMed] [Google Scholar]
- 30.Shoamanesh A, Martinez-Ramirez S, Oliveira-Filho J, et al. Interrelationship of superficial siderosis and microbleeds in cerebral amyloid angiopathy. Neurology 2014;83:1838–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol 2011;10:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 1998;57:353–359. [DOI] [PubMed] [Google Scholar]
- 33.Rannikmae K, Kalaria RN, Greenberg SM, et al. APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J Neurol Neurosurg Psychiatry 2014;85:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen N, Robinson AC, Snowden J, Davidson YS, Mann DM. Patterns of cerebral amyloid angiopathy define histopathological phenotypes in Alzheimer's disease. Neuropathol Appl Neurobiol 2014;40:136–148. [DOI] [PubMed] [Google Scholar]
- 35.Attems J, Jellinger KA, Lintner F. Alzheimer's disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol 2005;110:222–231. [DOI] [PubMed] [Google Scholar]
- 36.Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012;43:2324–2330. [DOI] [PubMed] [Google Scholar]
- 37.van Etten ES, Auriel E, Haley KE, et al. Incidence of symptomatic hemorrhage in patients with lobar microbleeds. Stroke 2014;45:2280–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement 2013;9:666–676. [DOI] [PubMed] [Google Scholar]