

Abstract
Objectives:
To determine whether an MRI-based Alzheimer disease (AD) signature biomarker can detect tau-related neurodegeneration in preclinical AD, and to assess whether AD signature cortical thinning is associated with cognitive changes in cognitively normal (CN) older individuals.
Methods:
In a large cohort of CN individuals (n = 188), we measured the hippocampal volume and cortical thickness within independently defined AD signature regions. We cross-sectionally assessed the associations between AD signature cortical thinning or hippocampal atrophy with CSF biomarkers of tau (increased tau) and β-amyloid (Aβ) (decreased Aβ42). We also examined the impact of AD signature cortical thinning or other biomarker changes (i.e., hippocampal atrophy, reduced CSF Aβ42, or increased CSF tau) on cognitive performance in CN individuals.
Results:
Elevated CSF tau was associated with AD signature cortical thinning but not hippocampal atrophy. In contrast, decreased CSF Aβ42 was associated with hippocampal loss but not AD signature cortical thinning. In addition, AD signature cortical thinning was associated with lower visuospatial performance. Reduced CSF Aβ42 was related to poorer performance on episodic memory.
Conclusions:
Spatially distinct neurodegeneration is associated with Aβ and tau pathology in preclinical AD. Aβ deposition and AD signature cortical atrophy independently affect cognition in CN older individuals.
Isolated amyloidosis is the earliest stage of preclinical Alzheimer disease (AD), followed by neurodegeneration and then subtle cognitive decline.1 However, recent work has observed that some cognitively normal (CN) individuals have neurodegeneration (e.g., AD-like brain atrophy on structural MRI) independent of amyloidosis.2–4 These observations motivate the investigation of tau-related neurodegenerative process in preclinical AD. In addition, discrepant data exist regarding the relationships between amyloidosis or neurodegeneration and cognitive decline in preclinical AD.3,5
Neurodegeneration can be measured using the hippocampal volume or cortical thickness within the topography affected by AD (i.e., “AD signature”).6,7 The AD signature has been shown to predict prognosis in individuals without dementia,8 although an important gap remains. Specifically, the AD topography has been derived primarily as a function of clinical assessment and not biomarkers. Many supposed CN older persons may have biomarker evidence of AD pathology.9 Some individuals with clinically defined AD do not have biomarker evidence of AD pathology.10
Here, we refined the AD signature using a cohort of clinical and biomarker-confirmed CN and symptomatic AD individuals. In a separate group of CN individuals (n = 188), we investigated whether abnormal CSF biomarker of tau pathology was associated with neurodegenerative changes (AD signature cortical thinning or hippocampal atrophy) in preclinical AD. We also examined whether amyloidosis (reduced CSF β-amyloid [Aβ]42) mediated the effects of tau on neurodegeneration. We assessed the impact of neurodegeneration or amyloidosis on cognition in CN individuals.
METHODS
Participants.
Participants were research volunteers enrolled in longitudinal studies of memory and aging at the Knight Alzheimer's Disease Research Center at Washington University in Saint Louis. Recruitment procedures have been previously published.11 Inclusion criteria were as follows: (1) age 65 years or older, (2) brain MRI and CSF collection completed within 12 months of clinical assessment, and (3) normal cognition or mild dementia due to AD, indicated by a Clinical Dementia Rating (CDR) of 0 and 1, respectively,12 assessed at the time closest to MRI scanning and CSF collection. Participants (n = 308) were divided into 2 cohorts. Cohort 1 was used for definition of the topography of AD signature while cohort 2 was used for analyzing the relationships among MRI-based biomarkers (cortical thickness within AD signature and hippocampal volume), CSF biomarkers (Aβ42 and tau), and cognitive measures.
Standard protocol approvals, registrations, and patient consents.
The Human Research Protection Office at Washington University in Saint Louis School of Medicine approved this study. Written informed consent was obtained from each participant. The guidelines of the Strengthening the Reporting of Observational Studies in Epidemiology13 were followed if applicable.
Clinical and neuropsychological assessments.
The participant and a collateral source underwent separate semistructured interviews conducted by experienced clinicians. The presence or absence of dementia was determined by clinicians according to the principle of intraindividual decline relative to prior functional level.14 A diagnosis of dementia due to AD was made according to standard criteria.14
All participants completed a neuropsychological battery that assessed the following domains: episodic memory, executive function, visuospatial ability, and semantic. For each domain, a composite score was formed by averaging z scores of individual tests (appendix e-1 on the Neurology® Web site at Neurology.org). This assessment was performed within 2 months of clinical evaluation.
CSF collection and APOE genotyping.
After an overnight fast, CSF (20–30 mL) was obtained and analyzed for Aβ42, tau, and phosphorylated tau181 by plate-based ELISA (INNOTEST; Innogenetics, Ghent, Belgium).15 Genotyping for APOE was conducted using procedures previously reported.16
Structural MRI acquisition and preprocessing.
Participants were scanned on either Siemens 3T Trio (n = 110 in cohort 1 or n = 155 in cohort 2) or 1.5T Vision (n = 10 in cohort 1 or n = 33 in cohort 2) scanner (Siemens Medical Systems, Erlangen, Germany). High-resolution structural scans were obtained with a T1-weighted magnetization-prepared rapid gradient echo sequence. Images were processed with FreeSurfer (version 5.10) (http://surfer.nmr.mgh.harvard.edu) (appendix e-2).
Definition of the topography of AD signature.
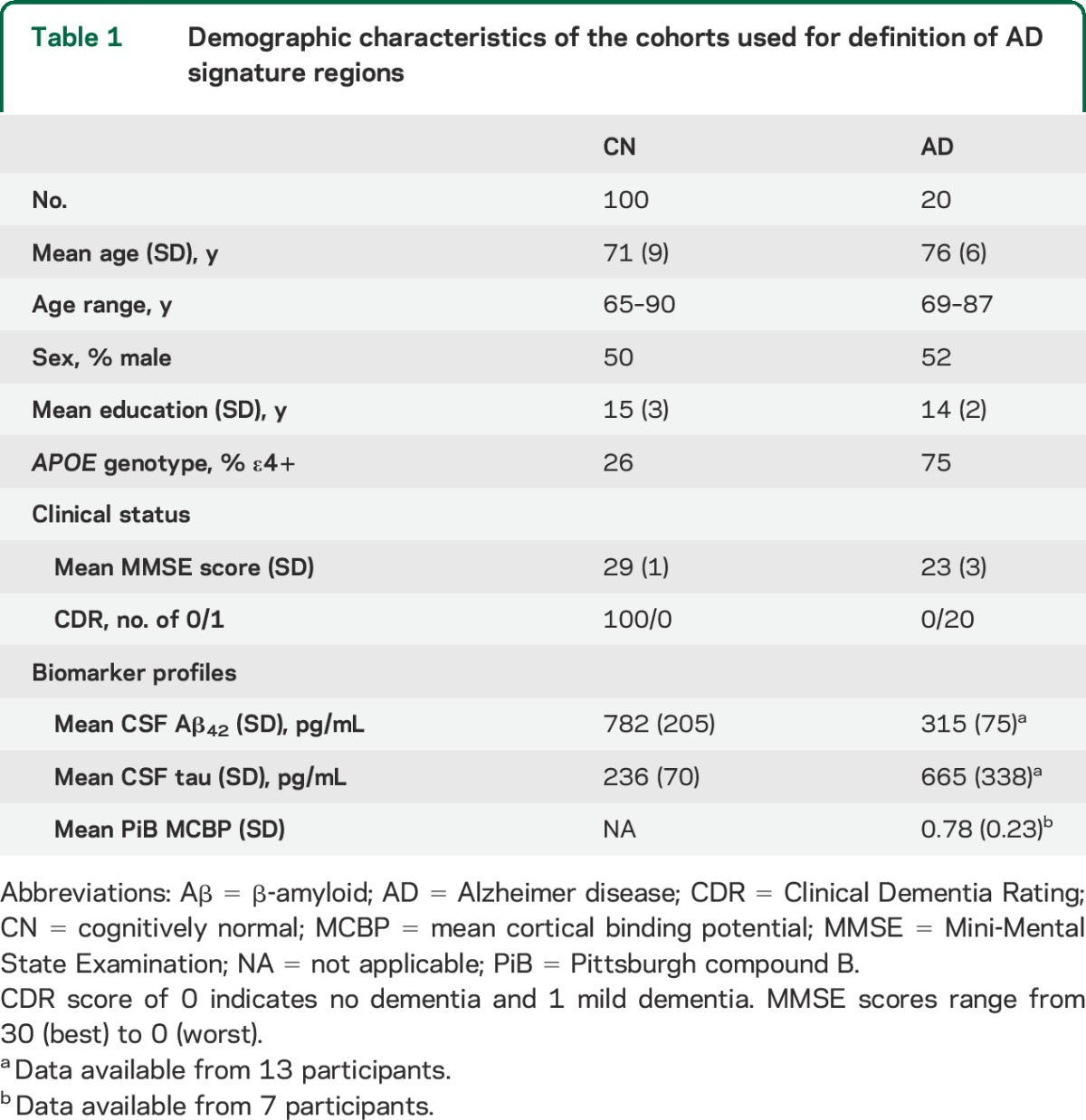
The topography of AD signature was identified in cohort 1 comprising participants with mild AD (CDR 1, n = 20) and a portion of CN participants (CDR 0, n = 100). The CN participants were randomly chosen from CN individuals who were negative for CSF Aβ42 (>500 pg/mL) and CSF tau (<500 pg/mL).17 All participants with mild AD had either reduced CSF Aβ42 (≤500 pg/mL) or elevated Pittsburgh compound B binding (mean cortical binding potential ≥0.18).17 Demographics and biomarker data for cohort 1 are provided in table 1.
Table 1.
Demographic characteristics of the cohorts used for definition of AD signature regions
For each participant in cohort 1, following the preprocessing of structural MRI data, the cortical surface representing the gray-white boundary was inflated into a sphere.18 The inflated spheres were registered to a common spherical coordinate system that aligned cortical folding patterns across participants.19 The surface maps of cortical thickness were compared between the mild AD and CN participants using a general linear model with adjustment for age, sex, the presence of APOE ε4 allele, and scanner type (3T Trio vs 1.5T Vision). The group difference map was thresholded at a vertex-wise p < 0.001 and cluster size ≥100 mm2. The participants with mild AD exhibited cortical thinning in the entorhinal cortex, fusiform gyrus, inferior, middle and superior temporal gyri, superior and inferior parietal lobules, posterior cingulate gyrus, and precuneus (figure 1). These regions hereafter are collectively referred to as the AD signature.
Figure 1. Topography of AD signature.
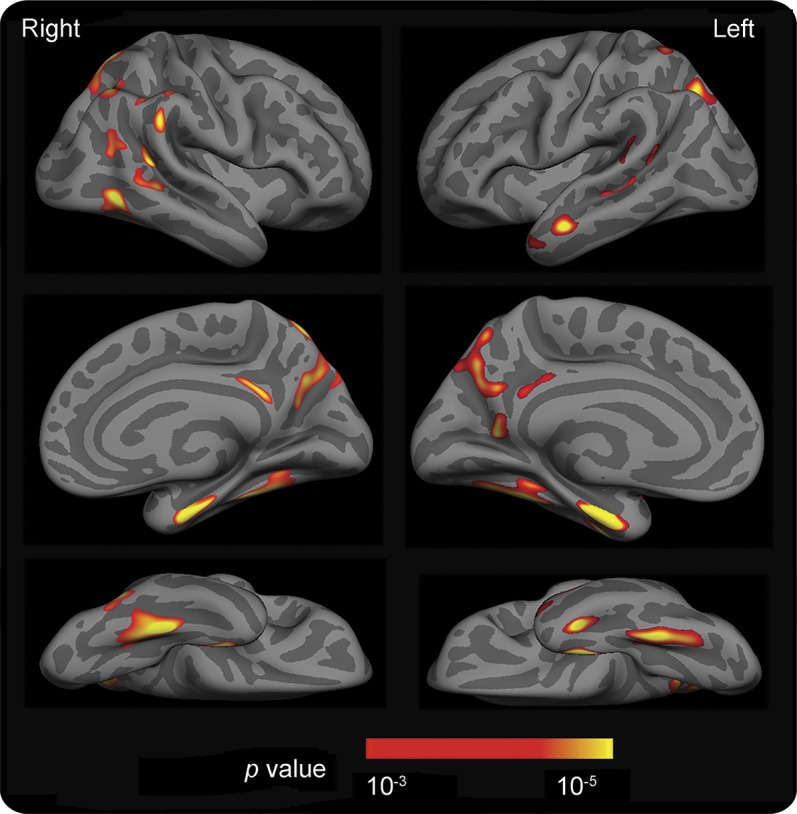
Surface maps of cortical thickness were compared between cognitively normal (CN) individuals (n = 100) who were negative for CSF Aβ42 (>500 pg/mL) and CSF tau (<500 pg/mL) and individuals with Alzheimer disease (AD) (n = 20) who were positive for either CSF Aβ42 (≤500 pg/mL) or amyloid imaging with Pittsburgh compound B (mean cortical binding potential ≥0.18) using a general linear model. A group difference map (AD < CN) was thresholded at a vertex-level p < 0.001 and cluster size >100 mm2 after adjustment for age, sex, presence of APOE ε4 allele, and scanner type (3T Trio vs 1.5T Vision). The map is displayed on the semi-inflated cortical surface of the FreeSurfer average brain with light gray regions representing gyri and dark gray regions representing sulci.
Measurements of AD signature cortical thickness and hippocampal volume in CN individuals.
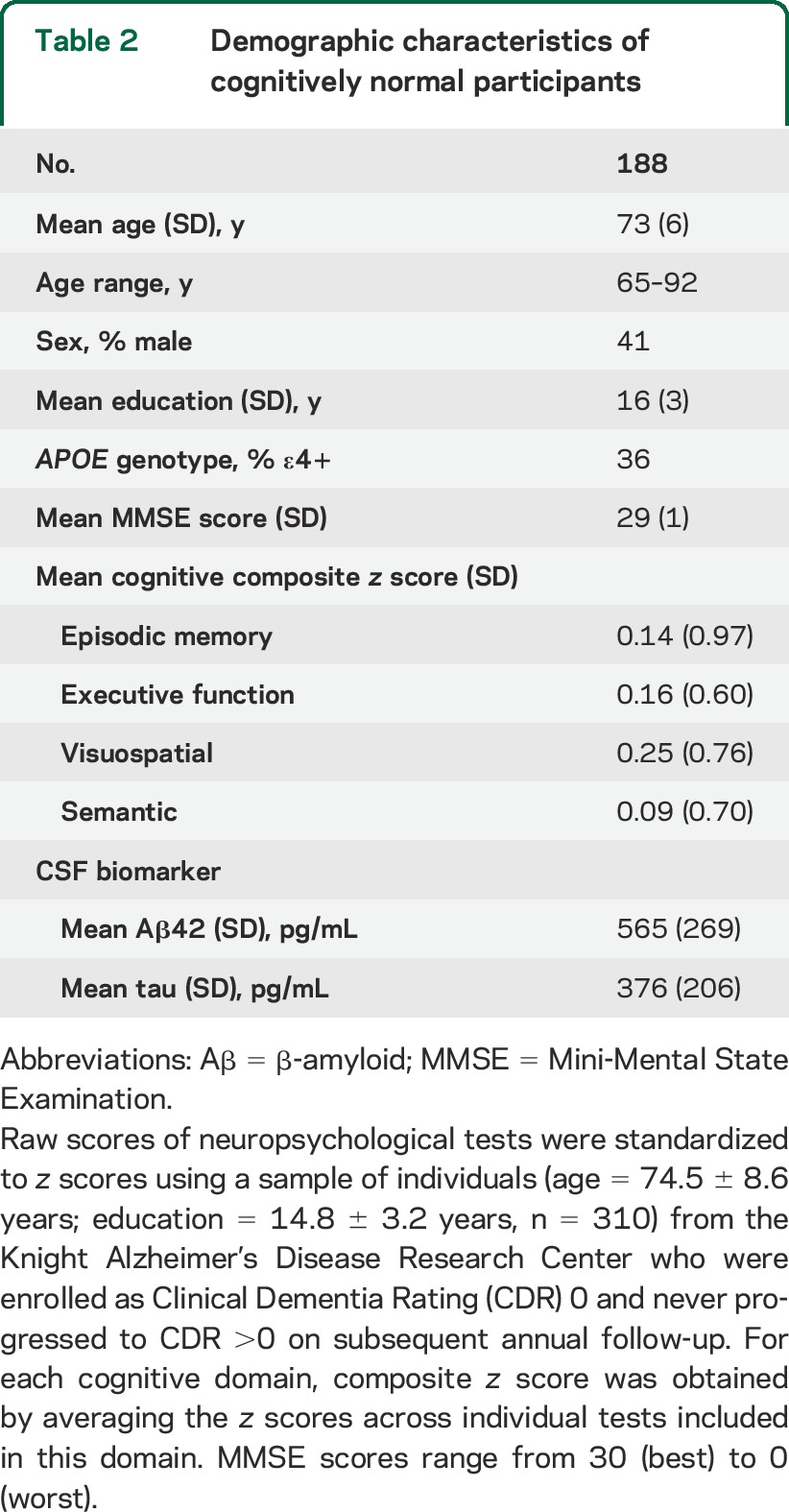
Demographics and biomarker data for cohort 2 (n = 188) are provided in table 2. For each participant included in cohort 2, AD signature regions, as identified above, were mapped back to this participant's individual space. Thickness values were obtained from each region and averaged across all AD signature regions to form a composite score. In addition, hippocampal volumes were measured using FreeSurfer's subcortical stream.20 The details of FreeSurfer's subcortical stream have been previously documented.20 Briefly, the hippocampus was segmented using automated procedures that examined variations in voxel intensities and spatial relationships to classify subcortical regions.20 Hippocampal volume was calculated for each participant as the product of the voxel volume and the number of voxels. Hippocampal volumes were then averaged across hemispheres and adjusted for intracranial volume. For each of the samples scanned with 3T Trio (n = 155) or 1.5T Vision (n = 33) scanner, individual composite scores of AD signature cortical thickness and adjusted hippocampal volumes were transformed to z scores using corresponding mean and SD derived from this sample.
Table 2.
Demographic characteristics of cognitively normal participants
Statistical analysis.
The present statistical approach was informed by existing studies, which have found that (1) the relationship between CSF Aβ42 and brain atrophy differs in Aβ42-positive vs Aβ42-negative individuals (i.e., an interaction between the continuous and categorical measures of CSF Aβ42),21,22 (2) the interaction between CSF Aβ42 and CSF phosphorylated tau181 is related to brain atrophy,23 and (3) Aβ mediates the effect of tau on neurodegeneration (autopsy data).24 Thus, we analyzed the associations of AD-associated atrophy with CSF biomarkers using equation 1, which includes the interactions between the continuous and categorical measures of CSF Aβ42, between the continuous and categorical measures of CSF tau, between CSF Aβ42 (categorical) and CSF tau (categorical), and between CSF Aβ42 (categorical) and CSF tau (continuous):
 |
Here, AD-associated atrophy denotes the z scores of AD signature cortical thickness or hippocampal volume. CSF_Aβ42 and CSF_tau denote the continuous measures of CSF Aβ42 and CSF tau, respectively. Aβ_status and tau_status were categorical-defined as positive if CSF Aβ42 ≤500 pg/mL and CSF tau ≥500 pg/mL, respectively, and negative if CSF Aβ42 >500 pg/mL and CSF tau <500 pg/mL, respectively.17 Age, sex, and APOE genotype (the presence vs absence of ε4 allele) were used as covariates. The interactions were first tested and reported if confirmed. After any detected interaction involving Aβ_status or tau_status, relationships between AD-associated atrophy and CSF biomarker were assessed separately within individuals who were positive or negative for the involved biomarker using equation 2:
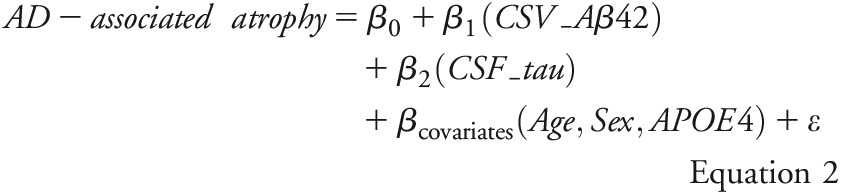 |
Multiple regression models were used to assess whether cognitive measures were associated with AD signature cortical thickness, hippocampal volume, CSF Aβ42, and CSF tau in CN individuals. Specifically, composite scores of each cognitive measure (i.e., episodic memory, executive function, visuospatial, and semantic) were used as dependent variables separately. For each model, AD signature cortical thickness, hippocampal volume, CSF Aβ42, and CSF tau were analyzed individually to determine which of them explained a significant portion of the variance (i.e., R2). Furthermore, the 4 independent variables were examined recursively to determine which one explained a significant additional portion of the variance (i.e., increase in R2 or ΔR2) after accounting for the other 3. All models included age, sex, education, and APOE genotype (the presence vs absence of ε4 allele) as covariates. Analyses were implemented using SPSS (version 21.0; IBM Corp., Armonk, NY) with a statistical threshold for significance of p < 0.05, corrected for multiple comparisons for equation 1.
RESULTS
Associations of AD signature cortical thickness with CSF Aβ42 and CSF tau in CN individuals.
Analyses of cohort 2 using equation 1 found interactions between CSF Aβ42 (continuous) and Aβ status (categorical) (p = 0.02), and between CSF tau (continuous) and tau status (categorical) (p = 0.01), but not between Aβ status and tau status (p = 0.84), and not between CSF tau and Aβ status (p = 0.53) regarding AD signature cortical thickness (figure e-1A). Since observed interactions suggested that the relationship between AD signature and CSF Aβ42 (continuous) was different regarding Aβ status and that the relationship between AD signature and CSF tau (continuous) was distinct regarding tau status, AD signature–CSF biomarker relationships were analyzed separately within biomarker-negative or -positive group using equation 2. Within the CSF Aβ42-positive (≤500 pg/mL, n = 104) and CSF Aβ42-negative (>500 pg/mL, n = 84) groups, the relationship between CSF Aβ42 and AD signature cortical thickness was not found (both p ≥ 0.13) after adjusting for age, sex, APOE ε4, and CSF tau. An inverse relationship was observed between CSF tau and AD signature cortical thickness in the CSF tau-positive group (≥500 pg/mL, n = 46) (partial η2 = 0.15, p = 0.01), but no relationship was seen in the CSF tau-negative group (<500 pg/mL, n = 142) (p = 0.84) after adjusting for age, sex, APOE ε4, and CSF Aβ42 (table e-1).
Associations of hippocampal volume with CSF Aβ42 and CSF tau in CN individuals.
Analyses of cohort 2 using equation 1 revealed an interaction between CSF Aβ42 and Aβ status (p = 0.01), but not between Aβ status and tau status and not between Aβ status and CSF tau (both p ≥ 0.71) regarding the hippocampal volume. Neither main effects of CSF tau or tau status nor the interaction between them was observed (all p ≥ 0.30) for the hippocampal volume (figure e-1B). Since hippocampal volume was differentially associated with CSF Aβ42 (continuous) according to Aβ status, as indicated by the presence of CSF Aβ42–Aβ status interaction, we analyzed the hippocampal–CSF Aβ42 association separately within the CSF Aβ42-negative or -positive group using equation 2. A positive relationship was seen between CSF Aβ42 and hippocampal volume in the CSF Aβ42-positive group (≤500 pg/mL) (partial η2 = 0.07, p = 0.009), but not in the CSF Aβ42-negative group (>500 pg/mL) (p = 0.79) after adjusting for age, sex, APOE ε4, and CSF tau (table e-2). Results are presented in appendix e-3 regarding evaluation of the choice of CSF biomarker cutoffs for studied relationships.
Associations of cognitive performance with AD signature cortical thickness, hippocampal volume, CSF Aβ42, and CSF tau in CN individuals.
AD signature cortical thickness, hippocampal volume, CSF Aβ42, and CSF tau were first analyzed individually regarding each cognitive composite (episodic memory, executive function, visuospatial, and semantic) after controlling for age, sex, education, and APOE ε4. Hippocampal volume or CSF Aβ42 explained a portion of the variance in episodic memory (R2 = 0.02, p = 0.03, and R2 = 0.03, p = 0.02, respectively). After accounting for each other, AD signature cortical thickness, and CSF tau, CSF Aβ42 explained an additional portion of the variance in episodic memory (ΔR2 = 0.02, p = 0.03) while hippocampal volume was not noteworthy (ΔR2 = 0.02, p = 0.07). Episodic memory had no relationship with AD signature cortical thickness or CSF tau (both p ≥ 0.58). AD signature cortical thickness explained some of the variance in visuospatial performance (R2 = 0.02, p = 0.03) and continued to account for the same amount of variance after controlling for hippocampal volume, CSF Aβ42, and CSF tau. Visuospatial performance had no association with hippocampal volume, CSF Aβ42, or CSF tau (all p ≥ 0.25). Neither executive nor semantic composite scores had any notable relationship with AD signature cortical thickness, hippocampal volume, CSF Aβ42, or CSF tau (all p ≥ 0.10).
DISCUSSION
Our work demonstrates that increased CSF tau was related to AD signature cortical thinning but not reduced hippocampal volume in CN individuals with higher CSF tau levels. In contrast, decreased CSF Aβ42 was associated with reduced hippocampal volume but not AD signature cortical thinning in CN individuals with lower CSF Aβ42 levels. In particular, the effect of increased CSF tau or decreased CSF Aβ42 was not attributed to age, sex, and APOE genotype. In addition, AD signature cortical thinning was associated with lower visuospatial performance. Reduced CSF Aβ42 was related to poorer performance on episodic memory.
AD-like brain atrophy parallels amyloidosis in preclinical AD.2–4 However, the pathophysiologic correlates of Aβ-independent atrophy remain unclear. Our work shows that AD-like neurodegeneration (i.e., AD signature cortical thinning) has a direct relationship with abnormal biomarker of tau pathology. Moreover, the topography of AD signature largely parallels the initiation (entorhinal) and early progression (posterior cingulate and temporal neocortex) of neurofibrillary tangles.25 This topographic correspondence suggests that the observed CSF tau–cortical atrophy association may reflect tangle-related neurodegeneration in the brain. This finding also indicates that the refined AD signature can detect early neurodegeneration in preclinical AD.
Studies of autopsy24 and transgenic mice26 postulate that Aβ mediates the effect of tau on neurodegeneration, with tau-related neurodegeneration more prevalent in individuals with higher Aβ burden than in those with lower Aβ burden. We assessed this theory within the entire cohort but found no significant difference in the association of CSF tau and AD signature cortical thickness with respect to Aβ status (i.e., CSF Aβ42-positive vs CSF Aβ42-negative). A secondary analysis within the CSF tau-positive group showed that a significant association of CSF tau with AD signature cortical thickness was seen in CSF Aβ42-positive but not CSF Aβ42-negative individuals (data not shown). However, the difference in the CSF tau–AD signature association remained nonsignificant regarding Aβ status within the CSF tau-positive group. The present observation may provide some preliminary evidence in support of the view that cortical neurodegeneration results from the synergistic effect of Aβ and tau.23,24,27 Further work assessing this view with more sensitive and specific measure of tau pathology (e.g., tau imaging tracer) is needed.
Longitudinal studies have observed an accelerating decline in visuospatial ability in CN individuals who subsequently developed AD dementia compared with those who remained cognitively unimpaired.28 The present result suggests a pathophysiologic correlate for the previous observation by showing that visuospatial decline is explained by subtle cortical neurodegeneration that at least is partly linked to tau pathology. In addition, we observed that lower performance on episodic memory is related to Aβ deposition or hippocampal atrophy, the former consistent with several recent reports29,30 but not others.3 Notably, after accounting for each other, the effect of Aβ on memory remained significant while the relationship with hippocampal atrophy was marginal. This result suggests that the impact of Aβ on memory may be independent of its effects on neurodegeneration in CN individuals. The mechanisms underlying observed Aβ-related lower memory performance remain to be explored.
Overall, our data suggest that Aβ and tau are preferentially associated with spatially distinct neurodegeneration during the preclinical phase of AD. Specifically, Aβ deposition alone may be sufficient for the atrophy in the hippocampus but not AD signature regions, which instead is related to tau pathology, or likely the mediation of Aβ on tau. These results not only agree with the emerging view of existence of “Aβ-dependent” and “Aβ-independent” pathways to preclinical AD,2–4,31–33 but also stress that the spatial distribution of the different pathways needs to be considered. In addition, our work demonstrates that memory and nonmemory domains may be differentially affected by Aβ and neurodegeneration. This result explains several recent observations27,34 that the joint presence of amyloidosis and neurodegeneration may be required for the emergence of clinical symptoms of AD.
Our work has limitations. We observed the association between higher CSF tau and AD signature cortical thinning after adjusting for age (and other confounders). However, age adjustment may be unable to completely remove the aging effect that is associated with tau pathology and cortical atrophy. Longitudinal studies are needed to estimate the extent to which observed CSF tau-related AD signature atrophy is specific to AD.
ACKNOWLEDGMENT
The authors thank the Knight ADRC's Clinical Core for participant assessments, Imaging Core for MRI data acquisition and preprocessing, Biomarker Core for CSF collection and assays, and Genetics Core for APOE genotyping. The authors especially thank all of the research participants.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- CDR
Clinical Dementia Rating
- CN
cognitively normal
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
L.W.: study concept and design, analysis and interpretation, critical revision of manuscript. T.L.B.: analysis and interpretation, collection of data, study supervision. J.H.: analysis and interpretation, critical revision of manuscript. T.B.: analysis and interpretation. C.O.: analysis and interpretation. J.L.: analysis and interpretation. A.M.F.: analysis and interpretation, collection of data, critical revision of manuscript. J.C.M.: critical revision of manuscript, study supervision. B.M.A.: analysis and interpretation, collection of data, critical revision of manuscript, study supervision.
STUDY FUNDING
This work was supported by Knight Alzheimer's Disease Research Center (ADRC) pilot grant (3255 ADRC 26) (B.M.A.), National Institute of Mental Health (R21MH099979) (B.M.A.), National Institute of Nursing Research (NINR) (R01NR014449, R01NR012657, R01NR012907) (B.M.A.), Alzheimer's Association (B.M.A.), NIA P01AG026276, P01AG03991, and P50 AG05681 (J.C.M.), and the generous support of Fred Simmons and Olga Mohan and the Paula and Rodger O. Riney Fund. Research reported in this article was also supported by the Washington University Institute of Clinical and Translational Sciences grant UL1 TR000448 from the National Center for Advancing Translational Sciences (NCATS) of the NIH. The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
DISCLOSURE
L. Wang reports no disclosures relevant to the manuscript. T. Benzinger receives research support from Avid Radiopharmaceuticals. J. Hassenstab, T. Blazey, C. Owen, and J. Liu report no disclosures relevant to the manuscript. A. Fagan consults for IBL International. Dr. Benzinger has participated or is currently participating in clinical trials of antidementia drugs sponsored by Eli Lilly and Roche. J. Morris has participated or is currently participating in clinical trials of antidementia drugs sponsored by Janssen Immunotherapy and Pfizer. Dr. Morris has served as a consultant for Lilly USA. He receives research support from Eli Lilly/Avid Radiopharmaceuticals. Neither Dr. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. B. Ances reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Brain injury biomarkers are not dependent on beta-amyloid in normal elderly. Ann Neurol 2013;73:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM, Jagust WJ. Alzheimer's disease neurodegenerative biomarkers are associated with decreased cognitive function but not beta-amyloid in cognitively normal older individuals. J Neurosci 2013;33:5553–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jack CR, Jr, Wiste HJ, Weigand SD, et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology 2013;81:1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling RA, Johnson KA, Doraiswamy PM, et al. Amyloid deposition detected with florbetapir F 18 ((18)F-AV-45) is related to lower episodic memory performance in clinically normal older individuals. Neurobiol Aging 2013;34:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vemuri P, Whitwell JL, Kantarci K, et al. Antemortem MRI based Structural Abnormality Index (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage 2008;42:559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009;19:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickerson BC, Wolk DA; Alzheimer's Disease Neuroimaging Initiative. Biomarker-based prediction of progression in MCI: comparison of AD signature and hippocampal volume with spinal fluid amyloid-beta and tau. Front Aging Neurosci 2013;5:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006;67:446–452. [DOI] [PubMed] [Google Scholar]
- 10.Serrano-Pozo A, Qian J, Monsell SE, et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann Neurol 2014;75:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berg L, McKeel DW, Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 1998;55:326–335. [DOI] [PubMed] [Google Scholar]
- 12.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 13.Vandenbroucke JP, von Elm E, Altman DG, et al. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): explanation and elaboration. PLoS Med 2007;4:e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 2006;20:210–216. [DOI] [PubMed] [Google Scholar]
- 15.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 2006;59:512–519. [DOI] [PubMed] [Google Scholar]
- 16.Pastor P, Roe CM, Villegas A, et al. Apolipoprotein Eepsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol 2003;54:163–169. [DOI] [PubMed] [Google Scholar]
- 17.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis: II: inflation, flattening, and a surface-based coordinate system. Neuroimage 1999;9:195–207. [DOI] [PubMed] [Google Scholar]
- 19.Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp 1999;8:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 21.Fjell AM, Walhovd KB, Fennema-Notestine C, et al. Brain atrophy in healthy aging is related to CSF levels of Abeta1-42. Cereb Cortex 2010;20:2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schott JM, Bartlett JW, Fox NC, Barnes J; Alzheimer's Disease Neuroimaging Initiative I. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Abeta1-42. Ann Neurol 2010;68:825–834. [DOI] [PubMed] [Google Scholar]
- 23.Desikan RS, McEvoy LK, Thompson WK, et al. Amyloid-beta associated volume loss occurs only in the presence of phospho-tau. Ann Neurol 2011;70:657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 1999;45:358–368. [DOI] [PubMed] [Google Scholar]
- 25.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 26.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 2007;316:750–754. [DOI] [PubMed] [Google Scholar]
- 27.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with beta-amyloidosis. JAMA Neurol 2013;70:1030–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol 2009;66:1254–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hedden T, Mormino EC, Amariglio RE, et al. Cognitive profile of amyloid burden and white matter hyperintensities in cognitively normal older adults. J Neurosci 2012;32:16233–16242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol 2013;12:357–367. [DOI] [PubMed] [Google Scholar]
- 31.Storandt M, Head D, Fagan AM, Holtzman DM, Morris JC. Toward a multifactorial model of Alzheimer disease. Neurobiol Aging 2012;33:2262–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chetelat G. Alzheimer disease: Abeta-independent processes: rethinking preclinical AD. Nat Rev Neurol 2013;9:123–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyman BT. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol 2011;68:1062–1064. [DOI] [PubMed] [Google Scholar]
- 34.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 2007;64:343–349. [DOI] [PubMed] [Google Scholar]