

Abstract
Insulin has been shown to act on pancreatic β cells to regulate its own secretion. Currently the mechanism underlying this effect is unclear. INS-2, a novel inositol glycan pseudo-disaccharide containing D-chiro-inositol and galactosamine, has been shown to function as an insulin mimetic and a putative insulin mediator. In the present study we found that INS-2 stimulates insulin secretion in MIN6 β cells and potentiates glucose stimulated insulin secretion in isolated mouse islets. Importantly, INS-2 failed to potentiate insulin secretion induced by tolbutamide, which stimulates insulin release by closing ATP sensitive potassium channels (KATP). Electrophysiological studies showed that INS-2 inhibited sulfonylurea-sensitive KATP conductance. The effect of INS-2 on inhibiting KATP channel is mediated by protein phosphatase 2C (PP2C), as knocking down PP2C expression in MIN6 cells by PP2C small hairpin RNA completely abolished the effect of INS-2 on KATP and consequently attenuated INS-2 induced insulin secretion. In conclusion, the present study identifies a novel mechanism involving PP2C in regulating KATP channel activity and consequently insulin secretion.
Keywords: INS-2, insulin secretion, KATP, PP2C, islet, MIN6
1. Introduction
Insulin from pancreatic β cells plays a pivotal role in regulating glucose homeostasis. Thus, it is not surprising that insulin secretion is tightly regulated by multiple factors and signals [1,2]. The release is predominantly controlled by glucose, as glucose alone can effectively stimulate insulin secretion [3]. In addition to glucose, a number of secretagogues including glucagon-like peptide 1 (GLP-1) and gastric inhibitory peptide (GIP) stimulate insulin release in a glucose dependent manner [4,5].
Accumulating evidence has shown that intra-islet communications via both autocrine and paracrine interactions also play a critical role in insulin secretion and ultimately glucose homeostasis. For example, glucagon secreted from pancreatic β cells potentiates insulin secretion, while somatostatin from δ cells inhibits both insulin and glucagon release [6]. In addition to these paracrine relating hormones, a number of factors including amylin [7] and urocortin 3 [8-10] are expressed in pancreatic β cells and functionally serve as local autocrine factors in modulating insulin secretion.
In addition to β cell released local factors, several lines of evidence support the notion that insulin itself also modulates its own secretion. Functional insulin receptors have been identified on pancreatic β cells [11]. Pharmacological studies have shown that the insulin receptor is involved in insulin secretion [11-13]. Further, mice with insulin receptor deleted in pancreatic β cells exhibited defective insulin secretion [14,15]. Currently the underlying mechanism by which insulin regulates insulin secretion remains elusive.
Early studies indicated the existence of second messengers containing inositols and amino hexoses in insulin target tissues to mediate some of insulin's actions [16,17]. 4-O (2-amino-2-deoxy- β -D-galactopyranosyl)-3-O-methyl-D-chiro-inositol, abbreviated as INS-2, was first isolated in insulin-stimulated liver as an inositol glycan with structure determined and later synthesized as β-1,4-galactosamine pinitol, where pinitol is the 3-O-methyl ether of D-chiro-inositol [18]. Functional studies demonstrated that INS-2 is insulin mimetic and insulin sensitizing [18]. Similar to insulin, INS-2 can lower blood glucose dose dependently in type 2 diabetic rats [18] and can sensitize low dose insulin in lowering hyperglycemia in type 1 diabetic rats [19]. Further, INS-2 stimulates glycogen synthesis dose dependently in hepatoma cells [18], and testosterone production in ovarian theca cells [20]. Most importantly, an INS-2 polyclonal antibody blocks both INS-2 action and insulin action in stimulating testosterone synthesis in ovarian theca cells [20]. This result thus suggests that some cellular effects of insulin are mediated by an extracellularly generated INS-2 like messenger, transported intracellularly for its action [17]. Both extracellular generation of inositol glycans [21] as well as the presence of an ATP dependent inositol glycan transporter have been reported [22].
In the present study, we show that INS-2 stimulates insulin secretion in β cells and potentiates glucose-induced insulin secretion. We then explored the possible mechanism underlying the insulin releasing effect of INS-2. We found that INS-2 regulates insulin secretion by closing of ATP sensitive potassium channels (KATP) and that this effect of INS-2 requires protein phosphatase 2C (PP2C).
2. Research Design and Methods
2.1 Reagents
INS-2 and C-INS-2 compounds was synthesized as described previously [18,23]. Chemicals and drugs were purchased from Sigma (St. Louis, MO). Cell culture reagents were purchased from Life Technologies (Grand Island, NY).
2.2 Cell culture
MIN6 cells, a mouse clonal β cell line, were cultured in high glucose Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1mM sodium pyruvate, 1% penicillin/streptomycin and 86 M β-mercaptoethanol. MIN6 cells were transduced with lentiviral particles and cultured for 72 hours prior to use in experiments. All cell culture reagents were purchased from Invitrogen.
2.3 Islet Isolation
Islets were isolated from adult male C57BL/6J mice (aged between 15-18 weeks) purchased from Jackson Laboratory (Bar Harbor, ME) via intraductal collagenase digestion and density centrifugation as described previously [24]. After isolation, islets were hand-picked, size-matched and maintained in RPMI media supplemented with 10% (v/v) FBS and 1% penicillin/streptomycin. Animal procedures were approved by the University of Virginia Animal Care and Use Committee.
2.4 Lentiviral Vector Production
A sequence to knockdown mouse PP2C expression was designed based on earlier reports [25,26]. A lentiviral vector expressing small hairpin RNA (shRNA) against mouse PP2C was generated as previously described [27]. Briefly, a PP2C shRNA expressing cassette driven by a U6 promoter was constructed and cloned into a lentiviral packaging vector (p156RRLsinPPtCMV-GFP-PREU3Nhe, kindly provided by Dr. Inder Verma, The Salk Institute). The viral vector expresses green fluorescent protein (GFP) under the control of a CMV promoter, allowing visual identification of cells infected by the viral vector. The shRNA PP2C vector and lentiviral packaging plasmids were co-transfected into HEK239FT cells. After transfection, the supernatant containing viral particles was harvested, clarified, and concentrated by centrifugation. In addition to the shRNA PP2C lentiviral vectors, a control vector expressing a non-coding scrambled sequence was also generated using the same method.
2.5 Insulin Secretion
Ten islets were incubated in 250 L of Kreb-Ringer bicarbonate HEPES buffer (KRB) consisting of the following in mM: NaCl (129), KCl (4.8), CaCl2 (2.5), MgCl2 (1.2), NaH2PO4 (1.2), NaHCO3 (5), HEPES (10) and 0.1 % BSA per well in a 48 well plate or 80,000 MIN6 cells were seeded per well in a 48 well plate. Both the cells and islets were equilibrated for 15 minutes at 37°C in KRB buffer. The preincubation was followed immediately by a 30 minute pretreatment with testing agents then stimulated with high glucose (16.8mM) for 60 minutes. Insulin concentrations in KRB buffer were determined with an insulin ELISA kit (Millipore). Insulin secretion assays were performed in triplicate and insulin ELISAs were averaged in duplicate.
2.6 Electrophysiology
Whole cell voltage clamp recordings were obtained from MIN6 cells on a fluorescence microscope (Zeiss Axioskop FS) using pCLAMP software interfaced with an Axopatch 200B amplifier via a Digidata 1322A digitizer (all from Molecular Devices, Foster City, CA). In some cases, cells were infected either with noncoding scramble or PP2C shRNA viral constructs and identified by GFP fluorescence. All recordings were performed at room temperature. Pipettes were pulled from borosilicate glass capillaries to a DC resistance ranging from 3 to 5 MΩ and coated with Sylgard 184 (Dow Corning, Midland, MA). The pipette solution contained (in mM): 120 KCH3SO3, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 10 HEPES Na, 10 EGTA (pH 7.2 adjusted by KOH). The bath solution contained (in mM): 140 NaCl, 3 KCl, 10 HEPES Na, 5 glucose, 2 CaCl2, and 2 MgCl2 (pH 7.3, adjusted by NaOH). ATP was omitted from the pipette solution and glucose was omitted from bath solution to induce activation of KATP channels. INS-2 and/or Glibenclamide were applied at 10 μM concentration where indicated. Series resistance was compensated (60-75%) and monitored throughout the recordings to ensure adequate compensation. Cells were held at -60 mV, and hyperpolarizing ramps (70 mV/sec, from -60 to -130 mV) were applied at 10 sec intervals. For analysis, slope conductance (G) was evaluated by linear fits to currents over the membrane potential range from -70 to -100 mV.
2.7 Statistics
Results are expressed as the mean ± SEM. Statistical analyses were performed using repeated measures, two-way ANOVA with post hoc tests or student's t-tests when appropriate using Prism 5 software (GraphPad).
3. Results
3.1 INS-2 stimulates insulin secretion in β cells
To investigate the effect of INS-2 on insulin secretion, MIN6 cells were treated with various concentrations of INS-2 and insulin levels in the media were determined by ELISA. As shown in Fig. 1A, INS-2 stimulates insulin secretion in MIN6 cells in a dose dependent manner under low glucose conditions. Importantly, INS-2 potentiates glucose-induced insulin secretion (GSIS; Fig. 1B). The effect of INS-2 on insulin secretion was also determined using isolated mouse islets. Similar to MIN6 cells, INS-2 potentiated GSIS in isolated mouse islets (Fig. 1C); however, in contrast to MIN6 cells, INS-2 had a mild effect on insulin release in 2.8 mM glucose conditions (Fig. 1C).
Fig. 1. INS-2 stimulates insulin release from pancreatic β cells.
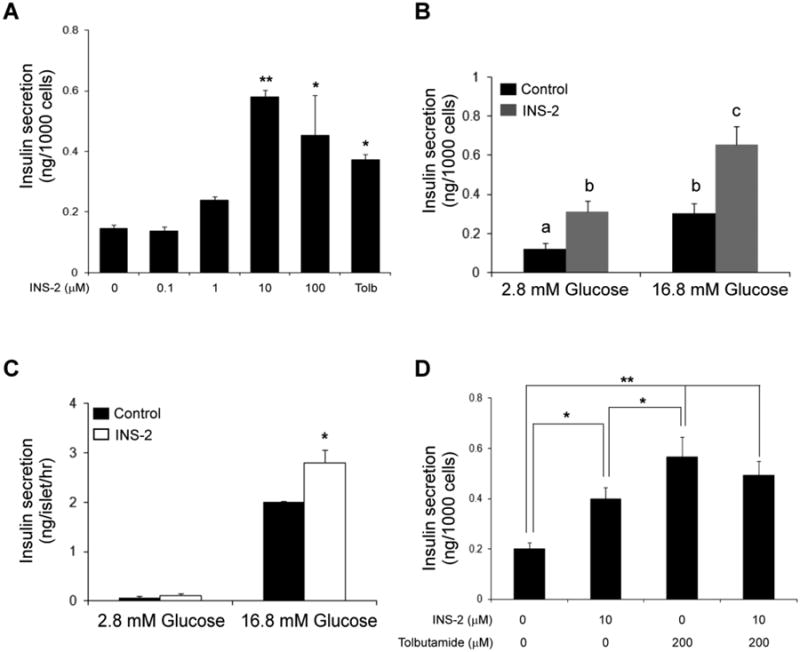
A: MIN6 cells were incubated with varying concentrations of INS-2 (0.1 – 100 μM) or tolbutamide (Tolb; 200 μM) in 2.8 mM glucose for 90 min and insulin concentrations in media determined by ELISA. B: Insulin secretion in MIN6 cells pre-treated with INS-2 (10 μM) for 15 min before stimulated with basal glucose (2.8 mM) or high glucose (16.8 mM) for 90 min. Groups with different letters are significantly different (P<0.05). C: Insulin secretion in islets (10) treated with INS-2 (10 μM) for 15 min before stimulation with either 2.8 or 16.8 mM glucose for 60 min. D: Insulin secretion in MIN6 cells treated with INS-2 (10 μM) alone, tolbutamide (Tolb; 200 μM) alone and a combination of the two compounds in the presence of low glucose (2.8 mM). **P<0.01, *P<0.05 vs. control.
3.2 Effect of sulfonylurea receptor agonist on INS-2-induced insulin secretion
To investigate the possible mechanism underlying the effect of INS-2 stimulated insulin release, we treated MIN6 cells with tolbutamide, a sulfonylurea receptor (SUR) agonist, with or without INS-2. As shown in Fig. 4, INS-2 and tolbutamide alone greatly stimulated insulin secretion. Interestingly, pre-treating cells with INS-2 failed to further potentiate tolbutamide-induced insulin secretion (Fig. 1D). The mechanism by which tolbutamide stimulates insulin secretion is that tolbutamide binds and closes ATP-sensitive potassium channels (KATP) [28,29]. The lack of additional insulin releasing effect of INS-2 in the presence of tolbutamide suggests either that the insulin release mechanism was saturated or that INS-2 may regulate insulin secretion via a mechanism involving KATP channels.
Fig. 4. Abrogating PP2C in β cells attenuated INS-2 induced insulin secretion.
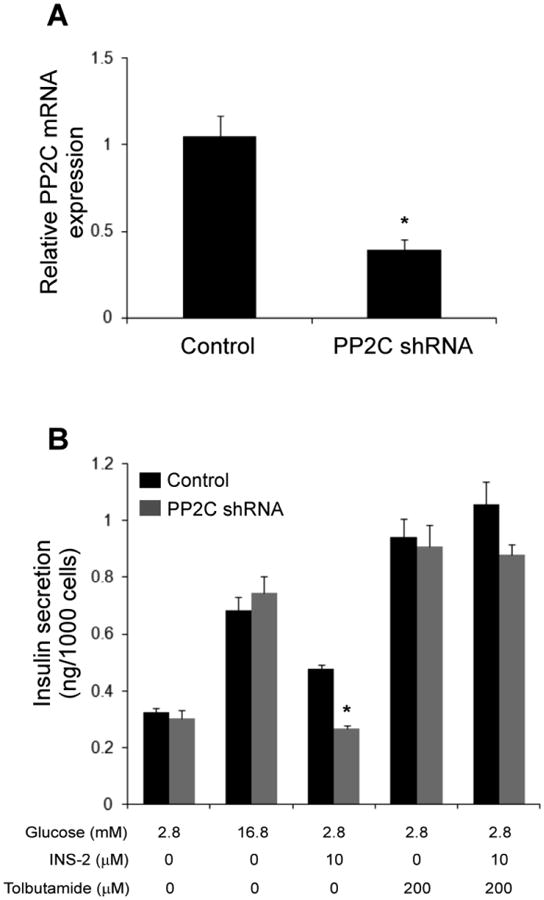
A: PP2C mRNA levels in MIN6 cells infected with lentiviral vector that expresses PP2C shRNA or noncoding scramble sequence (Control). B: Insulin secretion in MIN6 cells infected with lentiviral vector expression scrambled control or PP2C shRNA before stimulation with testing agents. *: p<0.05.
3.3 INS-2 inhibits KATP channels in pancreatic β cells
In order to test directly if INS-2 acts via inhibition of KATP channels, we recorded from MIN6 cells using whole cell voltage clamps (Fig. 2). After establishing the whole-cell configuration at -60 mV with an ATP-free pipette solution, we always observed a gradual increase in outward holding current; this time-dependent current was attributed to activation of a KATP conductance since it was abrogated in the presence of glibenclamide (Fig. 2). The glibenclamide-sensitive outward current typically saturated at the maximum level within 6 min. Once a stable peak current was attained, we examined the effects of INS2 and glibenclamide in either order of application. When applied first, both INS2 (Fig. 2A) and glibenclamide (Fig. 2C) inhibited conductance rapidly (in < 30 s) and strongly (72.1 ± 3.6 % and 63 ± 4.7 %, respectively). After INS2-induced block, glibenclamide had only modest effects on the residual current and, correspondingly, after glibenclamide-induced block, INS-2 had little further effect. In addition, the I-V characteristics of INS-2- and glibenclamide-sensitive currents were essentially identical (Fig. 2B, 2D and Insets). These data indicate that INS-2 inhibits glibenclamide-sensitive KATP channels in MIN6 cells.
Fig. 2. INS-2 inhibits outward current through ATP-sensitive potassium channels (KATP) in pancreatic β cells.
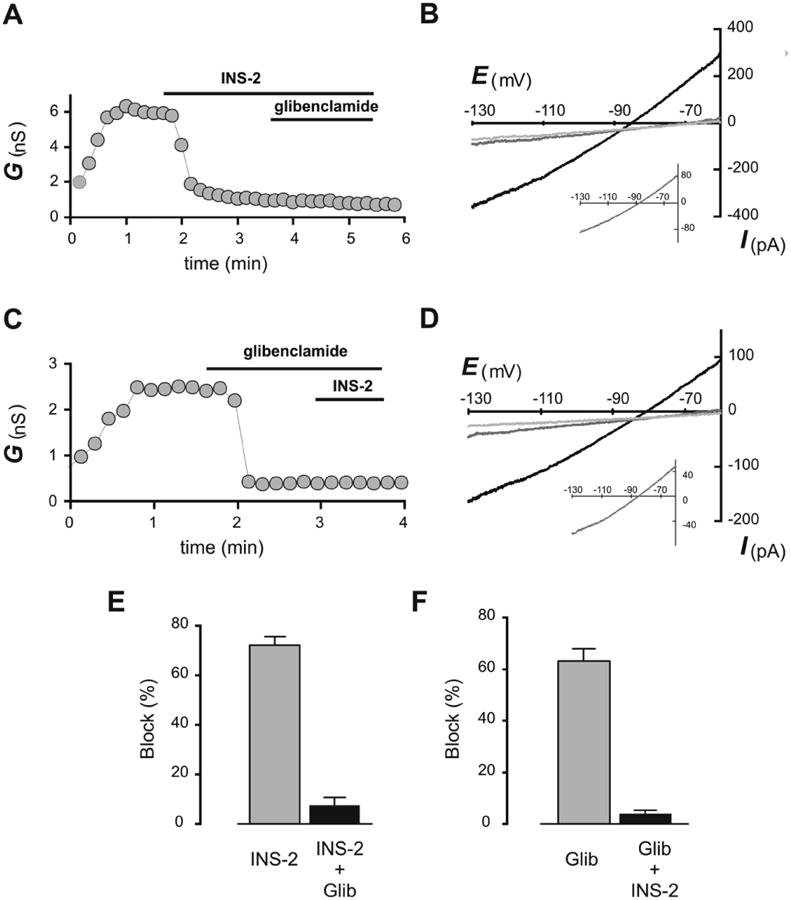
A, C. Representative time-series of conductance derived from slopes of the corresponding I-V curves (in B & D, between -70 and -110 mV); glibenclamide and INS-2 were applied as indicated. B, D. Representative I-V relationships of currents recorded in response to hyperpolarizing ramps before drug exposure (black), during exposure to the first drug (INS- 2 in B, glibenclamide in D, dark grey) and after exposure to both drugs (glibenclamide + INS-2, light grey). Insets show averaged I-V-relationship of INS-2-sensitive current (in B) and glibenclamide-sensitive current (in D, n = 6 for each). E, F. Averaged inhibition of conductance by initial applications of INS-2 (72.1 ± 3.6%, n = 6) and glibenclamide (Glib; 63 ± 4.7%, n = 6). Note that INS-2 and glibenclamide had similar inhibitory effects on KATP currents.
3.4 Phosphoprotein phosphatase receptor is involved in INS-2 stimulated insulin secretion
It has been determined that INS-2 interacts with at least two prominent protein targets: protein phosphatase 2C (PP2C) and pyruvate dehydrogenase phosphatase-1 (PDHP-1) [18,19]. C-INS-2, a carbon bridge analog of INS-2, has been shown to retain activity on PDHP-1, but was inactive on PP2C [23]. To investigate the possible downstream target of INS-2 in insulin secretion, we treated MIN6 cells with C-INS-2. As shown in Fig. 3, in contrast to INS-2 (100 μM) which again stimulated insulin secretion, C-INS-2 at the same concentration was completely inactive. The above results suggest that INS-2 stimulation of insulin secretion is not due to effects on PDHP-1, and that PP2C may be the relevant target for INS-2 action.
Fig. 3.
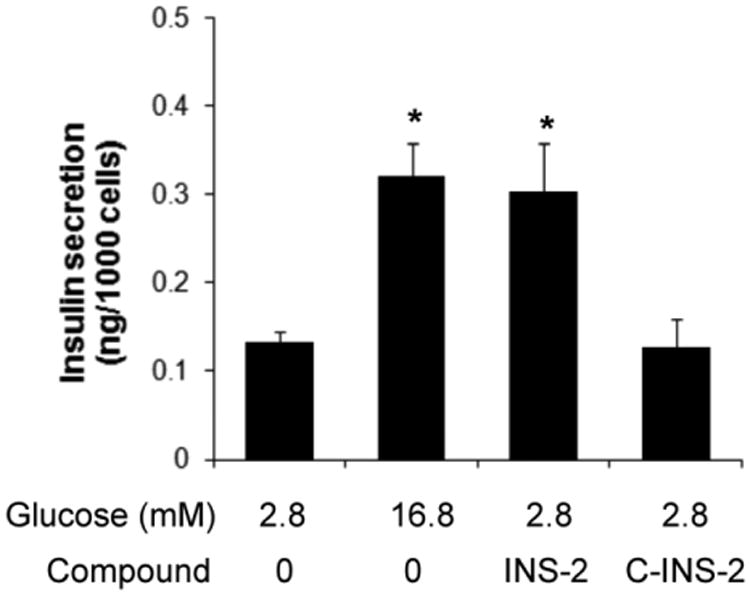
Insulin secretion in MIN6 cells treated with basal, 16.8 mM glucose, INS-2 (100 μM) or C-INS-2 (100 μM) for 90 min. *: p<0.05 vs. 2.8 mM G.
To establish more directly that PP2C is a target of INS-2 in insulin secretion, a lentiviral vector expressing small hairpin (shRNA) against mouse PP2C was constructed to knock down expression of PP2C in MIN6 cells (Fig. 4). We found that INS-2 failed to induce insulin secretion in MIN6 cells infected with PP2C shRNA lentiviral vector compared to cells infected with control noncoding viral vector (Fig. 4). Thus, PP2C is established as a target of INS-2 action to stimulate insulin secretion in MIN6 cells. Notably, tolbutamide retained the ability to induce insulin secretion in PP2C-depleted cells, suggesting that loss of PP2C did not directly affect KATP channel function and that PPC2 is an intermediary in the signaling pathway from INS-2 to KATP channels.
3.5 INS-2 induced block of KATP channel requires PP2C
To further test this proposed role for PP2C in INS-2-mediated KATP channel inhibition, we examined KATP channel activity in MIN6 cells treated with PP2C shRNA viral vector. As shown in Fig. 5, and consistent with our earlier results in uninfected cells, INS-2 potently blocked ∼80% of KATP current in cells infected with control viral vector (Fig. 5A, 5C). However, in cells infected with PP2C shRNA virus (Fig. 5B, 5D), INS2 was unable to inhibit KATP current (decreased by only ∼4%) which could subsequently be nearly completely blocked by glibenclamide. Thus, INS-2-induced block of KATP channels, similar to INS-2-evoked insulin secretion, requires PP2C.
Fig. 5. INS-2 block of KATP channel requires PP2C.
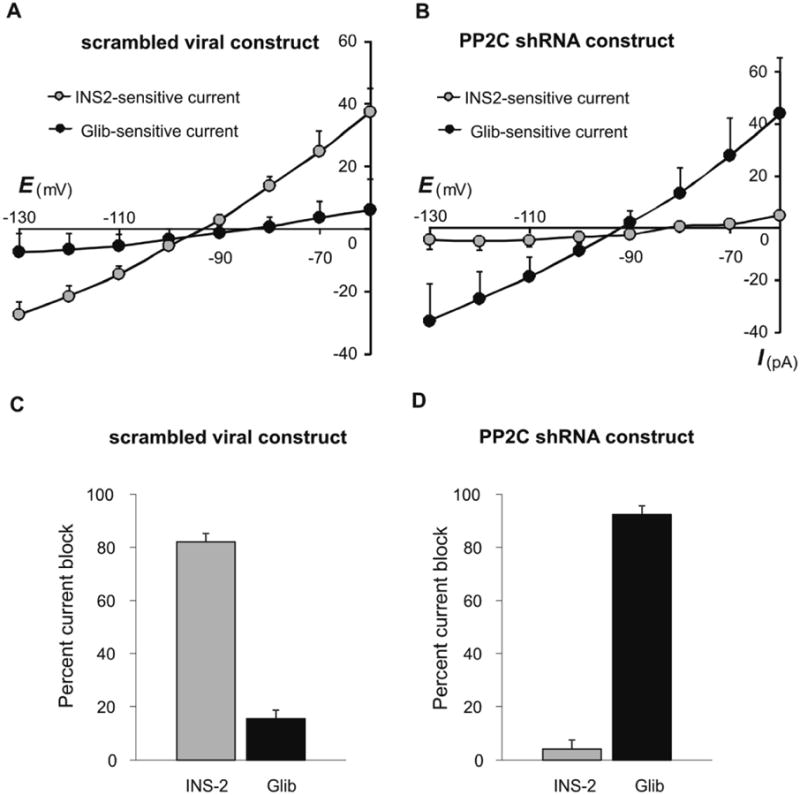
A, B. Averaged I-V curves of INS-2- and glibenclamide (Glib)-sensitive currents recorded in MIN6 cells infected with control viral construct (A, n = 7) or infected with a PP2C shRNA construct (B, n = 7). C. In cells infected with control virus, INS-2 blocked nearly all of the KATP current (82.0 ± 3.2%); after INS-2, glibenclamide had little further effect (15.3 ± 3.3%, n = 7). D. In cells infected with PP2C shRNA, INS-2 was ineffective at inhibiting KATP current (4.0 ± 4.2%) which could be subsequently blocked by glibenclamide (92.4 ± 4.8%, n = 7).
4. Discussion
The ability of insulin to regulate its own secretion has been proposed [14,30,31] but a mechanism has not been established. The present study demonstrates that INS-2, an insulin mimetic and putative second messenger, stimulates insulin secretion in both MIN6 cells and isolated mouse islets. Further, we found INS-2 failed to potentiate insulin secretion induced by Tolbutamide, which stimulates insulin release from β cells by closing KATP channels [28]. This result argues that INS-2 and tolbutamide share a similar mechanism in regulating insulin secretion. Consistent with this notion, our electrophysiological data showed that INS-2 inhibits KATP channels. Importantly, glibenclamide, a second generation sulfonylurea, had no effect on current when cells were first treated with INS-2 and vice versa. These results further support our hypothesis that INS-2 stimulates insulin secretion, at least in part, by regulating KATP channel activity.
A key question is then how INS-2 regulates KATP channel activity. Earlier studies have identified at least two targets of INS-2: PP2C and PDHP-1 [18,19]. They both contain potential allosteric binding sites for INS-2 adjacent to the catalytic sites using in-silico studies based on X-ray crystal structures of the enzymes [19,23]. We have further shown that an acidic amino acid is required for allosteric binding of INS-2 in each enzyme; aspartic acid at position 243 [19] in PP2Cα and glutamic acid at position 351in PDHP-1 [23]. The current study showed that C-INS-2, a modified carbon bridge analog of INS-2 [23], had no effect in insulin secretion. As C-INS-2 retains activity on PDHP-1, but is inactive on PP2C [23], these results argues against a role for PDHP-1 and suggests that INS-2 likely targets PP2C in β cells in regulating insulin secretion. Moreover, Yoshizaki and co-workers have determined that PP2C is involved in mediating the effect of insulin in fat cells [32]. They found that PP2C promotes insulin action in adipocytes by dephosphorylating the p85 regulatory subunit of PI3 kinase (PI3K), thus facilitating the dissociation of the regulatory subunit from p110 catalytic subunit [32]. Taken together we postulate that INS-2 regulates KATP activity through PP2C.
Consistent with our hypothesis, suppressing PP2C expression in MIN6 cells by shRNA against PP2C effectively abrogated the effect of INS-2 in insulin secretion, confirming that PP2C is involved in INS-2 stimulated insulin release. Our electrophysiological study further indicates that in PP2C knockdown MIN6 cells, INS-2 failed to modify KATP channel activity while, interestingly, glibenclamide remained effective in closing KATP. This result suggests that: (1) INS-2 must interact at a site distinct from the drug binding site on the SUR subunit of the KATP channel complex, and (2) PP2C is not required for glibenclamide binding to SUR. Our study also indicates that INS-2 regulates KATP channel activity through PP2C but not SUR, or at least not on sulfonylurea binding sites [28,29]. Several protein phosphatases including PP2A and PP2B have been found in β cells [33]. On the other hand, there are no reports of detection of PP2C in β cells or islets. The present study shows for the first time that PP2C is expressed in MIN6 cells and is involved in regulating insulin secretion. Thus, the present study identifies a novel mechanism involving PP2C in regulating KATP channel activity and consequently insulin secretion.
Currently it remains to be determined as to how PP2C regulates KATP channel activity. PP2C has been shown to directly bind and regulate Ca2+ channels in neurons [34]. Flajolet et al. has also shown that PP2C binds and dephosphorylates metabotropic glutamate receptors [35]. Thus, it is conceivable that PP2C interacts directly with the KATP channel to modulate the channel activity by dephosphorylation of key residues on the channel. Both the ATP inhibited pore-forming K+ channel as well as the SUR have been reported to have kinase phosphorylation sites [36,37]. Of interest, both serine as well as threonine sites have been identified. Threonine is known as the preferred substrate for PP2C [38]. Further experiments are needed to identify the site(s) dephosphorylated by PP2C via activation by INS-2 and the mechanism of increased insulin secretion. Clearly this work defines a novel mechanism of inositol glycan stimulated insulin secretion.
In the present study, we show that INS-2 stimulates insulin secretion in MIN6 cells under basal glucose conditions. On the other hand, in isolated mouse islets the compound potentiates GSIS without significant effect on insulin release under basal conditions. The discrepancy may be due to a number of issues including differences in levels of PP2C or KATP between MIN6 cells and mouse islets and may be of interest to explore. It is also noteworthy that INS-2 potentiates GSIS in both MIN6 cells and isolated mouse islets, suggesting the mechanism underlying INS-2 action is still operational under high glucose conditions. Glucose induces insulin secretion by elevating intracellular ATP levels, which then in turn closes KATP channels [39] to open voltage dependent calcium channels [40,41] and stimulates extracellular purinergic receptors to promote insulin secretion [42,43]. It is not immediately clear as to how INS-2 remains effective in facilitating insulin release in the face of elevated ATP levels. It is possible that elevated ATP in cells does not bind and close all the KATP channels in β cells, thus allowing INS-2 to participate in closing additional channels. It is also possible that in addition to KATP, INS-2 and/or PP2C acts on as yet unidentified targets to regulate insulin secretion. More studies are needed to further resolve this issue.
In conclusion, we have determined that INS-2, an insulin mimetic and possible second messenger, stimulates insulin section and potentiates glucose induced insulin secretion. The mechanism by which inositol glycans stimulates insulin secretion is due, at least in part, to the closure of KATP channels in a PP2C dependent manner.
Highlights.
INS-2 is a D-chirl-inositol with insulin mimicking effect.
INS-2 stimulates insulin secretion from pancreatic cells
INS-2 stimulates insulin release by inhibiting ATP-sensitive K+ channels (KATP).
The inhibitory effect of INS-2 on KATP is mediated by protein phosphatase 2C.
Acknowledgments
We thank David Lin and Michael Digruccio for providing technical assistance in insulin ELISA. We also thank Drs. Michael Thorner, David Brautigan and Peilin Chen for providing comments on the manuscript.
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK-078049 (C.L.) and R01 NS33583 (D.A.B.).
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Malaisse WJ. Regulation of insulin release by the intracellular mediators cyclic AMP, Ca2+, Inositol 1,4,5-trisphosphate, and diacylglycerol. In: Cuatrecasas P, Jacobs SJ, editors. Insulin. Springer-Verlag; Berlin: 1990. pp. 113–124. [Google Scholar]
- 2.Taborsky GJ., Jr . Insulin and glucagon secretion in vivo and its neural control. In: Jefferson LS, Cherrington AD, editors. Handbook of Physiology. Oxford University Press; New York: 2001. pp. 153–176. [Google Scholar]
- 3.Newgard CB, Matschinsky FM. Substrate control of insulin release. In: Jefferson LS, Cherrington AD, editors. Handbook of Physiology. Oxford University Press; New York: 2001. pp. 125–152. [Google Scholar]
- 4.Holst JJ, Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab. 2004;287:E199–206. doi: 10.1152/ajpendo.00545.2003. [DOI] [PubMed] [Google Scholar]
- 5.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–57. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 6.Samols E, Bonner-Weir S, Weir GC. Intra-islet insulin-glucagon-somatostatin relationships. Clin Endocrinol Metab. 1986;15:33–58. doi: 10.1016/s0300-595x(86)80041-x. [DOI] [PubMed] [Google Scholar]
- 7.Cooper GJ. Amylin and related proteins: physiology and pathphysiology. In: Jefferson LS, C AD, editors. Handbook of Physiology. Oxford University Press; New York: 2001. pp. 303–396. [Google Scholar]
- 8.Li C. Urocortin III Is Expressed in Pancreatic -Cells and Stimulates Insulin and Glucagon Secretion. Endocrinology. 2003;144:3216–3224. doi: 10.1210/en.2002-0087. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Chen P, Vaughan J, Lee KF, Vale W. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc Natl Acad Sci U S A. 2007;104:4206–11. doi: 10.1073/pnas.0611641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30:261–4. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leibiger IB, Leibiger B, Moede T, Berggren PO. Exocytosis of insulin promotes insulin gene transcription via the insulin receptor/PI-3 kinase/p70 s6 kinase and CaM kinase pathways. Mol Cell. 1998;1:933–8. doi: 10.1016/s1097-2765(00)80093-3. [DOI] [PubMed] [Google Scholar]
- 12.Aspinwall CA, Lakey JR, Kennedy RT. Insulin-stimulated insulin secretion in single pancreatic beta cells. J Biol Chem. 1999;274:6360–5. doi: 10.1074/jbc.274.10.6360. [DOI] [PubMed] [Google Scholar]
- 13.Argoud GM, Schade DS, Eaton RP. Insulin suppresses its own secretion in vivo. Diabetes. 1987;36:959–62. doi: 10.2337/diab.36.8.959. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-Specific Knockout of the Insulin Receptor in Pancreatic [beta] Cells Creates an Insulin Secretory Defect Similar to that in Type 2 Diabetes. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 15.Okada T, Liew CW, Hu J, Hinault C, Michael MD, Krtzfeldt J, Yin C, Holzenberger M, Stoffel M, Kulkarni RN. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A. 2007;104:8977–82. doi: 10.1073/pnas.0608703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larner J, Galasko G, Cheng K, DePaoli-Roach AA, Huang L, Daggy P, Kellogg J. Generation by insulin of a chemical mediator that controls protein phosphorylation and dephosphorylation. Science. 1979;206:1408–10. doi: 10.1126/science.228395. [DOI] [PubMed] [Google Scholar]
- 17.Larner J, Brautigan DL, Thorner MO. D-chiro-inositol glycans in insulin signaling and insulin resistance. Mol Med. 2010;16:543–52. doi: 10.2119/molmed.2010.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larner J, Price JD, Heimark D, Smith L, Rule G, Piccariello T, Fonteles MC, Pontes C, Vale D, Huang L. Isolation, structure, synthesis, and bioactivity of a novel putative insulin mediator. A galactosamine chiro-inositol pseudo-disaccharide Mn2+ chelate with insulin-like activity. J Med Chem. 2003;46:3283–91. doi: 10.1021/jm030071j. [DOI] [PubMed] [Google Scholar]
- 19.Brautigan DL, Brown M, Grindrod S, Chinigo G, Kruszewski A, Lukasik SM, Bushweller JH, Horal M, Keller S, Tamura S, Heimark DB, Price J, Larner AN, Larner J. Allosteric activation of protein phosphatase 2C by D-chiro-inositol-galactosamine, a putative mediator mimetic of insulin action. Biochemistry. 2005;44:11067–73. doi: 10.1021/bi0508845. [DOI] [PubMed] [Google Scholar]
- 20.Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83:2001–5. doi: 10.1210/jcem.83.6.4886. [DOI] [PubMed] [Google Scholar]
- 21.Alvarez JF, Varela I, Ruiz-Albusac JM, Mato JM. Localisation of the insulin-sensitive phosphatidylinositol glycan at the outer surface of the cell membrane. Biochem Biophys Res Commun. 1988;152:1455–62. doi: 10.1016/s0006-291x(88)80449-2. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez JF, Sanchez-Arias JA, Guadano A, Estevez F, Varela I, Feliu JE, Mato JM. Transport in isolated rat hepatocytes of the phospho-oligosaccharide that mimics insulin action. Effects of adrenalectomy and glucocorticoid treatment. Biochem J. 1991;274(Pt 2):369–74. doi: 10.1042/bj2740369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hans SK, Camara F, Altiti A, Martin-Montalvo A, Brautigan DL, Heimark D, Larner J, Grindrod S, Brown ML, Mootoo DR. Synthesis of C-glycoside analogues of beta-galactosamine-(1→4)-3-O-methyl-D-chiro-inositol and assay as activator of protein phosphatases PDHP and PP2Calpha. Bioorg Med Chem. 2010;18:1103–10. doi: 10.1016/j.bmc.2009.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter JD, Dula SB, Corbin KL, Wu R, Nunemaker CS. A practical guide to rodent islet isolation and assessment. Biol Proced Online. 2009;11:3–31. doi: 10.1007/s12575-009-9021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sathe MN, Woo K, Kresge C, Bugde A, Luby-Phelps K, Lewis MA, Feranchak AP. Regulation of purinergic signaling in biliary epithelial cells by exocytosis of SLC17A9-dependent ATP-enriched vesicles. J Biol Chem. 2011;286:25363–76. doi: 10.1074/jbc.M111.232868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y. Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci U S A. 2008;105:5683–6. doi: 10.1073/pnas.0800141105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chao H, Digruccio M, Chen P, Li C. Type 2 corticotropin-releasing factor receptor in the ventromedial nucleus of hypothalamus is critical in regulating feeding and lipid metabolism in white adipose tissue. Endocrinology. 2012;153:166–76. doi: 10.1210/en.2011-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev. 2003;55:105–31. doi: 10.1124/pr.55.1.7. [DOI] [PubMed] [Google Scholar]
- 29.Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–91. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- 30.Bouche C, Lopez X, Fleischman A, Cypess AM, O'Shea S, Stefanovski D, Bergman RN, Rogatsky E, Stein DT, Kahn CR, Kulkarni RN, Goldfine AB. Insulin enhances glucose-stimulated insulin secretion in healthy humans. Proc Natl Acad Sci U S A. 2010;107:4770–5. doi: 10.1073/pnas.1000002107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leibiger B, Leibiger IB, Moede T, Kemper S, Kulkarni RN, Kahn CR, de Vargas LM, Berggren PO. Selective Insulin Signaling through A and B Insulin Receptors Regulates Transcription of Insulin and Glucokinase Genes in Pancreatic [beta] Cells. Molecular Cell. 2001;7:559–570. doi: 10.1016/s1097-2765(01)00203-9. [DOI] [PubMed] [Google Scholar]
- 32.Yoshizaki T, Maegawa H, Egawa K, Ugi S, Nishio Y, Imamura T, Kobayashi T, Tamura S, Olefsky JM, Kashiwagi A. Protein phosphatase-2C alpha as a positive regulator of insulin sensitivity through direct activation of phosphatidylinositol 3-kinase in 3T3- L1 adipocytes. J Biol Chem. 2004;279:22715–26. doi: 10.1074/jbc.M313745200. [DOI] [PubMed] [Google Scholar]
- 33.Jones PM, Persaud SJ. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic beta-cells. Endocr Rev. 1998;19:429–61. doi: 10.1210/edrv.19.4.0339. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Wang F, Lai M, Chen Y, Zhang JF. A protein phosphatase 2calpha-Ca2+ channel complex for dephosphorylation of neuronal Ca2+ channels phosphorylated by protein kinase C. J Neurosci. 2005;25:1914–23. doi: 10.1523/JNEUROSCI.4790-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flajolet M, Rakhilin S, Wang H, Starkova N, Nuangchamnong N, Nairn AC, Greengard P. Protein phosphatase 2C binds selectively to and dephosphorylates metabotropic glutamate receptor 3. Proc Natl Acad Sci U S A. 2003;100:16006–11. doi: 10.1073/pnas.2136600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang TJ, Chen WP, Yang C, Lu PH, Liang YC, Su MJ, Lee SC, Chuang LM. Serine-385 phosphorylation of inwardly rectifying K+ channel subunit (Kir6.2) by AMP- dependent protein kinase plays a key role in rosiglitazone-induced closure of the K(ATP) channel and insulin secretion in rats. Diabetologia. 2009;52:1112–21. doi: 10.1007/s00125-009-1337-4. [DOI] [PubMed] [Google Scholar]
- 37.Lin YF, Jan YN, Jan LY. Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. Embo J. 2000;19:942–55. doi: 10.1093/emboj/19.5.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donella Deana A, Mac Gowan CH, Cohen P, Marchiori F, Meyer HE, Pinna LA. An investigation of the substrate specificity of protein phosphatase 2C using synthetic peptide substrates; comparison with protein phosphatase 2A. Biochim Biophys Acta. 1990;1051:199–202. doi: 10.1016/0167-4889(90)90194-i. [DOI] [PubMed] [Google Scholar]
- 39.Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311:271–3. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- 40.Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- 41.Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiol Rev. 1981;61:914–73. doi: 10.1152/physrev.1981.61.4.914. [DOI] [PubMed] [Google Scholar]
- 42.Petit P, Lajoix AD, Gross R. P2 purinergic signalling in the pancreatic beta-cell: control of insulin secretion and pharmacology. Eur J Pharm Sci. 2009;37:67–75. doi: 10.1016/j.ejps.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Geisler JC, Corbin KL, Li Q, Feranchak AP, Nunemaker CS, Li C. Vesicular nucleotide transporter-mediated ATP release regulates insulin secretion. Endocrinology. 2013;154:675–84. doi: 10.1210/en.2012-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]