

Abstract
The Qira black sheep and the Hetian sheep are two local breeds in the Northwest of China, which are characterized by high-fecundity and low-fecundity breed respectively. The elucidation of mRNA expression profiles in the ovaries among different sheep breeds representing fecundity extremes will helpful for identification and utilization of major prolificacy genes in sheep. In the present study, we performed RNA-seq technology to compare the difference in ovarian mRNA expression profiles between Qira black sheep and Hetian sheep. From the Qira black sheep and the Hetian sheep libraries, we obtained a total of 11,747,582 and 11,879,968 sequencing reads, respectively. After aligning to the reference sequences, the two libraries included 16,763 and 16,814 genes respectively. A total of 1,252 genes were significantly differentially expressed at Hetian sheep compared with Qira black sheep. Eight differentially expressed genes were randomly selected for validation by real-time RT-PCR. This study provides a basic data for future research of the sheep reproduction.
Introduction
Sheep are one of the important domestic animals in Xinjiang province in the northwest of China. High reproductive efficiency is one of the important goals in sheep breeding, because it plays an important role in efficiency of production. However, low-fecundity in the majority of domestic sheep is one of limiting factors for the development of sheep industry. Because of low heritability of the reproduction traits and sex-limited nature, traditional selection has been employed for improving litter size with only limited advancement [1]. So scientists pay more attention to search the candidate genes associated with ovulation rate and multiplets, which can lead to genetic improvement through the implementation of gene and/or marker assisted selection (MAS) [2].
With the development of molecular, sequencing and bioinformatics analysis technologies, high throughput RNA deep sequencing (RNA-seq) provides a platform for measuring large-scale gene expression patterns [3–5]. Recently, in order to identify the differentially expressed genes (DEGs) and novel transcript units, the RNA-seq has been widely used in domestic animals, including goats [6–7], pigs [8], cows [9], sheep [10–12], and others. Additionally, the efficacy of RNA-seq has also been demonstrated in mammalian reproductive tissues, for example in pig gonad [8, 13–14], bovine blastocyst [15–16], bovine granulosa cell [17], goat ovary [19], sheep oocyte and granulosa cell [18], and sheep ovary [20]. However, reports on the sheep DEGs and transcriptome information related to litter size or prolificacy trait that have applied RNA-seq technology are very limited. One previous study with the use of RNA-seq technology in ovary tissues reported that 1,924 and 1,501 DEGs were identified between the Surabaya fur sheep with the FecB (Booroola gene) genotype BB Han sheep and FecB genotype ++ Han sheep respectively [20].
Qira black sheep and Hetian sheep are two local breeds in Hetian region, Xinjiang province, China. The former is characterized by year-around estrous behavior and high-fecundity with an average lambing rate of 215.5% [21], while the latter is a low-fecundity breed with an average lambing rate of 102.5% [22]. The difference in fecundity between Qira black sheep and Hetian sheep may be associated with the difference of ovulation rate [23]. Significant fecundity differences between Qira black sheep and Hetian sheep have provided appropriate animal models for the identification and utilization of sheep major prolificacy genes [20]. In the present study, we detected the differential expression profiling in ovaries of the mRNAs in two groups (Qira black sheep and Hetian sheep) using RNA-seq technology. This work advances our understanding of the molecular mechanism of sheep fecundity, and provides basic data for future studies.
Materials and Methods
Ethics statement
The sheep experiments were performed in accordance with the “Guidelines for Experimental Animals” of the Ministry of Science and Technology (Beijing, China) and the “Regulations for the Experimental Animal Use Management” of Shihezi University. All ewes were housed in one group under the normal condition of natural lighting and free access to feed. All ewes were slaughtered humanely by anesthesia (Xylidinothiazoline, 1 mg/Kg; Feilong animal pharmaceutical factory, China), and performed by the professional workers at Qira Cattle and Sheep Abattoir.
Animal and ovary collection
According to the reproduction records, age and body size, Qira black ewes (high-fecundity individuals) and Hetian ewes (low-fecundity individuals) of similar age (3–4 years old) and body size were obtained from the Qira Sheep Breeding Farm located in Qira County, Hetian region, Xinjiang Province, China. A teaser ram was used to monitor the occurrence of estrus for all ewes twice a day. Four Qira black ewes and five Hetian ewes were killed between 12 and 24 h after spontaneous estrus was detected. Ovaries were dissected and all samples were collected with better ovulation points or preovulatory follicles on the surfaces of the ovaries. All samples were immediately frozen on liquid nitrogen for total RNA extraction.
RNA isolation and RNA-seq
Trizol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract the total RNA from ovaries in the two groups following the manufacturer’s instructions, and then the DNase I was used to degrade any possible DNA contamination. According to the manufacturer’s manual, sequencing libraries were performed at Beijing Genomics Institute (BGI, Shenzhen, China) using the Illumina Truseq RNA Sample Preparation Kit (Illumina, San Diego, USA). Briefly, using the oligo (dT) magnetic beads, the mRNA was enriched from the total RNA and then the purified mRNA was interrupted into short fragments (about 200 bp) by the fragmentation buffer. Random hexamer-primer was added to synthesize the first strand cDNA. Adding buffer, dNTPs, DNA polymerase I (New England Biolabs) and RNase H (Invitrogen), the second strand cDNA was synthesized, and subsequently the double stranded cDNA was purified using the Qiaquick PCR extraction kit, washed with EB buffer for end repaired and Poly (A) tail addition. Finally, after ligating the sequencing adaptors (Illumina PE adaptors), the suitable fragments were selected for PCR amplification according to the results of agarose gel electrophoresis. The cDNA libraries were used for sequencing on an Illumina HiSeq 2000 instrument at BGI—Shenzhen, China.
Bioinformatics analysis
All clean reads were obtained by rejecting low quality sequence (more than half of the base qualities less than 5) or reads with more than 10% unknown nucleotides (N), and sequencing adapters. The clean reads were aligned to the gene sequences (downloaded from NCBI database) and sheep genome (oar3.1) through SOAP2 [24], with allowing less than 2 mismatches in the alignment. Unmapped or multi-position matched reads were excluded from further analyses. In addition, sequence saturation analyses of the two libraries were executed to provide an overview of the project.
Expression profiling
The number of mapped reads for each gene was normalized and calculated by using the RPKM (reads per kb per million reads) method, which is an effective method for eliminating the influence of sequencing discrepancy and gene length [25]. Based on the method described by Audic and Claverie [26], we determined the statistical significance of differential expression profile for each gene. The threshold of P-value was determined according to the false discovery rate (FDR) [27]. In this study, to judge the significance of gene expression difference, the absolute value of log2 Ratio ≥ 1 and FDR ≤ 0.001 were used as the criteria [28].
Gene Ontology (GO) and KEGG pathway enrichment analysis
As an international standardized gene functional classification system, GO offers a dynamic-updated controlled vocabulary and a strictly defined concept to comprehensively describe properties of genes and their products [20]. In order to identify the significantly enriched GO terms, all DEGs were mapped to GO terms in GO database (http://www.geneontology.org/) by hypergeometric test [29]. And the Bonferroni-corrected P-value ≤ 0.05 was used as the threshold. GO covers three domains: cellular component, molecular function and biological process [29]. The GO annotation analysis and the plotting of GO annotations were performed using WEGO software [30]. Moreover, according to the KEGG database, the significantly enriched biological pathways were identified [31]. Pathways with a Q-value ≤ 0.05 were considered significant.
Quantitative real-time PCR (qRT-PCR) confirmation
In order to verify the RNA-seq results, eight candidate genes were selected and detected using qRT-PCR. Three genes (follistatin-related protein 3 precursor, latent-transforming growth factor beta-binding protein 4, and transforming growth factor beta-1-induced transcript 1 protein) were associated with TGFβ; two genes (cytochrome P450 19A1 and steroidogenic acute regulatory protein) were associated with hormone regulation; and three genes (estrogen receptor beta, prolactin receptor, and scavenger receptor class B member 1) were grouped as receptor. β-actin was used as a reference control. Total RNA (1.0 mg) was used to synthesize first-strand cDNA using EasyScript RT reagent Kit (Transgen, Beijing, China) and the qRT-PCR was performed using qPCR SuperMix Kit (Transgen, Beijing, China). The amplification conditions were 95°C for 2 min of initial stage, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s and performed on Mx3000P System. The reactions of all genes were run on one plate in triplicate for each biological replicate. Values of real-time RT-PCR were calculated by the 2−ΔΔCt method [20, 32]. The relative expression level of each gene (ΔCt) was calculated as follows: Ct target gene—Ct β-actin, and the relative expression level of each gene in two sheep (ΔΔCt) was calculated as follows: Ct Hetian sheep—Ct Qira black sheep [20, 32].
Results
Sequencing data summary
In this study, we sequenced two cDNA libraries from the Qira black sheep and Hetian sheep, respectively, using a Illumina HiSeq 2000 sequencing platform at BGI-Shenzhen, China, and approximately 2.36 Gb reads (1.17 and 1.19 Gb for the Qira black sheep and Hetian sheep, respectively) were obtained. Next, according to the BGI bioinformatics protocols for RNA-seq, these data were preliminarily analyzed. The major characteristics of the two DGE libraries are described in Table 1. The result showed varying amount of sequencing reads for these samples. In both libraries, although 41.99% of the reads in Qira black sheep and 43.81% of the reads in Hetian sheep could be mapped to reference genes, approximately 82% of the reads mapped to sheep genome (Qira black sheep with 82.39%, Hetian sheep with 82.42%). For the unique match, a little more than one-third and two-thirds of the reads corresponded to reference genes and genome respectively. In addition, 33.76% of the reads in Qira black sheep and 34.65% of the reads in Hetian sheep could be perfectly matched to the reference genes, and approximately 65% of the reads perfectly matched to genome. The results of saturation analyses (Fig. 1) showed that when the number of sequenced reads reached 2 M or more, the number of detected genes almost ceased increasing, which validates the integrity of the library for use in further analysis.
Table 1. A summary of the sequencing reads alignment to the Ovis aries genome and reference genes.
| Sample | alignment to genome | alignment to reference genes | ||
|---|---|---|---|---|
| Qira black sheep | Hetian sheep | Qira black sheep | Hetian sheep | |
| Total Reads | 11,747,582 | 11,879,968 | 11,747,582 | 11,879,968 |
| Total Base-Pairs | 575,631,518 | 582,118,432 | 575,631,518 | 582,118,432 |
| Total Mapped Reads | 9,679,005(82.39%) | 9,790,981(82.42%) | 4,932,905(41.99%) | 5,204,429(43.81%) |
| Perfect Match | 7,605,875(64.74%) | 7,712,860(64.92%) | 3,965,493(33.76%) | 4,116,347(34.65%) |
| < = 2bp Mismatch | 2,073,130(17.65%) | 2,078,121(17.49%) | 967,412(8.23%) | 1,088,082(9.16%) |
| Unique Match | 8,052,464(68.55%) | 8,252,218(69.46%) | 4,061,762(34.58%) | 4,435,953(37.34%) |
| Multi-position Match | 1,626,541(13.85%) | 1,538,763(12.95%) | 871,143(7.42%) | 768,476(6.47%) |
| Total Unmapped Reads | 2,068,577(17.61%) | 2,088,987(17.58%) | 6,814,677(58.01%) | 6,675,539(56.19%) |
Fig 1. Saturation analyses of Qira black sheep (A) and Hetian sheep (B).
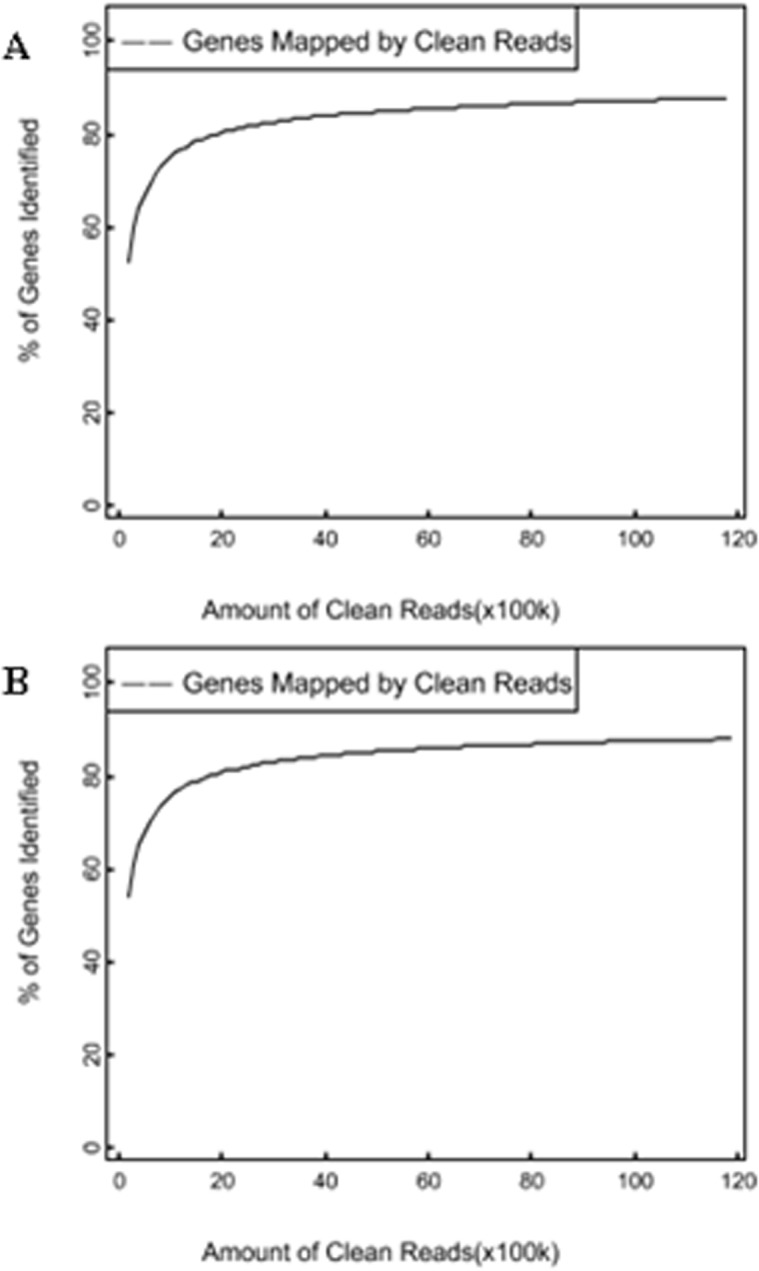
The number of detected genes continued increasing as the total number of sequencing reads increased. When the number of reads reaches a certain amount, the number of detected genes almost ceased increasing.
Identification and analysis of DEGs
The RPKM method was used to evaluate the gene expression levels. As a result, 16,763 and 16,814 reference genes were identified from Qira black sheep and Hetian sheep libraries, respectively, which shared 16,425 genes in common (S1 Table). As shown in Table 2, approximately 88% and a little more than 1.7% of the reference genes were expressed at less than 100 RPKM and more than 500 RPKM respectively. As shown in Fig. 2, 20% of the reference genes had 80–90% coverage, and 31% and 35% of the annotated genes had 90–100% coverage in Qira black sheep and Hetian sheep libraries respectively, suggesting that the read distributions are similar between the two libraries.
Table 2. Gene expression and annotation by RPKM.
| RPKM | Gene number of Hetian sheep (%) | Gene number of Qira black sheep (%) |
|---|---|---|
| 0–50 | 12,513 (74.42%) | 12,538 (74.80%) |
| 50–100 | 2233 (13.28%) | 2243 (13.38%) |
| 100–500 | 1779 (10.58%) | 1685 (10.05%) |
| >500 | 289 (1.72%) | 297 (1.77%) |
| Total | 16,814 | 16,763 |
Fig 2. Distribution of genes′ coverage in the two ovine libraries.
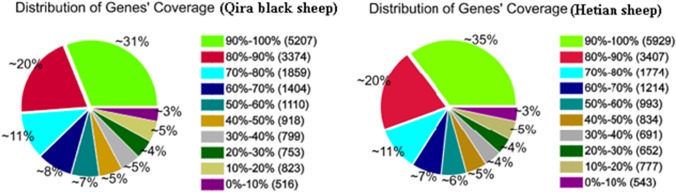
Gene coverage is the percentage of a gene covered by reads. This value is equal to the ratio of total base count in a gene covered by uniquely mapped reads to the total base count for that gene.
To judge the significance of differences in expressed genes, FDR ≤ 0.001 and the absolute value of log2 Ratio ≥ 1 were used as the threshold. A total of 1,252 significantly differentially expressed genes were identified between the two libraries, with 859 genes upregulated and 393 genes downregulated at Hetian sheep compared with Qira black sheep (S2 Table and Fig. 3). Among these genes, 9 genes 13 genes were found only expressed in the Qira black sheep library and Hetian sheep library respectively. The breakdown of fold-change among the DEGs was showed in Table 3. Interestingly, the number of genes having greater than four-fold difference and two-fold difference accounted for 7.03% and 29.79% of the total DEGs respectively.
Fig 3. Comparison of gene expression levels between the two libraries.

The expression levels are estimated by RPKM value.
Table 3. Fold change differences in expression of genes differentially expressed between the two sheep at FDR < 0.001.
| Fold change | ||||
|---|---|---|---|---|
| > 6 | 4–6 | 2–4 | < 2 | |
| Genes over-expressed in Hetian sheep | 23 | 42 | 217 | 577 |
| Genes over-expressed in Qira black sheep | 11 | 12 | 68 | 302 |
GO and pathway enrichment analysis
The enrichment of DEGs in GO terms was tested to gain insights into the biological implications. And several GO terms significantly enriched for DEGs were found and shown in Fig. 4. The GO annotation indicated that the DEGs were involved in many biological processes, such as reproduction, cellular process, metabolic process, biological regulation, developmental process, signaling, growth, and immune function. The main functional groups of DEGs in cellular component are cell, cell part, membrane and organelle, and in molecular function are catalytic activity and binding.
Fig 4. GO analysis of differentially expressed genes between the two breeds.

The differentially expressed genes are classified into three categories: cellular component, molecular function, and biological process. The left axis shows the percentage of genes in a category, and the right axis the number of genes.
Based on the KEGG pathway database, the pathway analysis was performed to predict the significantly enriched metabolic pathways and signal transduction pathways in DEGs. After pathway enrichment analysis, the significantly enriched pathways were listed in S3 Table. In total, 623 DEGs had KEGG pathway annotations. The results showed that the significant signaling pathways included 218 pathways, for example metabolic pathways, cell adhesion molecules, steroid (hormone) biosynthesis, TGFβ signaling pathway, oxidative phosphorylation, PPAR signaling pathway, and Wnt signaling pathway.
QRT-PCR confirmation
In the same RNA samples, the expression fold changes (2−ΔΔCt) of eight genes were tested by using qRT-PCR to validate the RNA-seq results. And the expression patterns of eight genes were general consistent with the RNA-seq results (Table 4), suggesting that the results of the RNA-seq experiments was efficient and accurate.
Table 4. Validation of selected RNA-seq based gene expression by real-time RT-PCR analysis.
| Gene | Description | Hetian sheep/Qira black sheep | Primer sequence | |
|---|---|---|---|---|
| RNA-seq | QRT-PCR | |||
| CYP19A1 | gi|172088165|ref|NP_001116472.1|/0/cytochrome P450 19A1 | 3.38 | 7.89 | F: TCGTCCTGGTCACCCTTCTG |
| R: CGGTCTCTGGTCTCGTCTGG | ||||
| ESR2 | gi|57619192|ref|NP_001009737.1|/0/estrogen receptor beta | −1.58 | −0.96 | F: TGATGTCCTTGACCAAGCTG |
| R: CACTTGGTCGTACAGGCTGA | ||||
| FSTL3 | gi|115496578|ref|NP_001069178.1|/6.39084e-111/follistatin-related protein 3 precursor | 1.86 | 1.96 | F: GGCTGAATGCTGTGCCTCTG |
| R: CCCTCGCACGAATCTTTG | ||||
| LTBP4 | gi|426243822|ref|XP_004015745.1|/4.81709e-99/PREDICTED: LOW QUALITY PROTEIN: latent-transforming growth factor beta-binding protein 4 | −1.59 | −0.84 | F: GTCGGTGTTGGTACAGATG |
| R: GCAGACGTGAACGAGTG | ||||
| PRLR | gi|21263860|sp|O46561.1|PRLR_SHEEP/0/RecName: Full = Prolactin receptor; Short = PRL-R | 5.40 | 2.94 | F: CTTACCACAACATTGCTGACG |
| R: GTTTAGCAGAGAACAAGGGG | ||||
| SCARB1 | gi|426247178|ref|XP_004017363.1|/5.24272e-135/PREDICTED: scavenger receptor class B member 1 | 2.09 | 1.01 | F: TGGCCTCCATCCTAACG |
| R: TCGAACCACATTAGCGG | ||||
| STAR | gi|57163979|ref|NP_001009243.1|/6.49941e-160/steroidogenic acute regulatory protein, mitochondrial precursor | 6.47 | 2.61 | F: TCCATGTGCGTGCTGGCTGG |
| R: GGAGCACCATGCAGGTGGGG | ||||
| TGFβ1I1 | gi|466003579|ref|XP_004268779.1|/1.70899e-154/PREDICTED: transforming growth factor beta-1-induced transcript 1 protein | −1.42 | −0.74 | F: CAGGTCTTTCGAGGTGG |
| R: CTTCACCTGCACCTTCTG | ||||
| β-actin | F: TCCGCAAAGACCTCTACG | |||
| R: CGGAGTACTTGCGCTCA | ||||
Discussion
The RNA-seq technology is a powerful approach for identification the expression levels of thousands of genes simultaneously in various tissues including ovary [20]. In this study, differences in gene expression between Qira black sheep and Hetian sheep ovaries were analyzed using RNA-Seq technology. The Qira black sheep and Hetian sheep were the similar age, body size and came from the same region, but their fecundity was very different (the average lambing rate of Qira black sheep was 215.5% and Hetian sheep was 102.5%). The different fecundity may be associated with the different regulation of gene expression, which is involved in the ovulation or follicular development [23]. The results of differential gene expression are more likely to reflect the differences between Qira black sheep and Hetian sheep, and will be valuable for future studies on the identification of major genes or novel genes that affect sheep fecundity.
In this study, A total of 1,252 genes were significantly differentially expressed, some of the DGEs corresponding to genes previously associated with prolificacy processes, such as prolactin receptor (PRLR), progesterone receptor (PGR), estrogen receptor beta (ESR2), cytochrome P450 19A1 (CYP19A1), cytochrome P450 11A1 (CYP11A1), estradiol 17-beta-dehydrogenase 12 (HSD17B12), inhibin beta A (INHBA), while other genes may be involved in the follicular development or control of ovulation. For example, we identified members of vascular endothelial growth factor (VEGF) pathway including VEGF receptor 2 (VEGFR2) upregulated in Hetian sheep, and VEGF B isoforms (VEGFB) upregulated in Qira black sheep. Angiogenesis is critical for the female ovulatory cycle including follicular development, ovulation, and corpora lutea formation [33]. It has been demonstrated that VEGF system constitute the most important signaling pathway in angiogenesis [34], and play an important role in the process of follicular development through the regulation of thecal angiogenesis [34–35]. In particular, several studies reported that VEGF can increase rat follicular cell proliferation and inhibit the apoptosis [34], stimulate ovarian follicular development [35–36], and control follicle progression and luteogenesis [37]. Another example is STAR, which is involved in the regulation of steroid hormone biosynthesis and follicular development in the mammalian ovary [38–39]. In this study, the expression differences between the Qira black sheep and Hetian sheep ranged from 13.96- to 1.0-fold. The expression differences of 879 DEGs were relatively small (less than 2-fold), but some of them may have large effects on prolificacy processes involved in follicular development, ovulation, litter size, hormone biosynthesis, or others [40].
To investigate the biological functions of the DEGs, we performed the GO annotation and KEGG pathway analysis. This study clearly revealed that some hormone related genes involved in steroid biosynthesis and steroid hormone biosynthesis pathways toward upregulated in Hetian sheep. For steroid biosynthesis pathway, we found seven genes assigned to almost all steps from lanosterol 14-alpha demethylase-like to lathosterol oxidase. Eight of nine genes (CYP11A1, CYP19A1, HSD17B12, hydroxyl-delta-5-steroid dehydrogenase, and others) involved in the steroid hormone biosynthesis pathway were upregulated in Hetian sheep, and one gene (estradiol 17-beta-dehydrogenase 1) was upregulated in Qira black sheep. These genes might be an indirect or direct effect on steroid hormone biosynthesis, and their expression profiles in this study were consistent with the expression profiles of SCARB1, low-density lipoprotein receptor (LDLR), very low-density lipoprotein receptor isoform 2 (VLDLR) and STAR. SCARB1, LDLR, and VLDLR have been shown to be involved in biosynthesis of steroid hormones through effects on the absorption of cholesterol substrates from circulating lipoproteins [41–43]. And STAR has been shown to be involved in the transportation of intracellular cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane, thereby regulating the biosynthesis of steroid hormones [42–43]. Other genes related to steroid hormones and upregulated in Qira black sheep are ESR2 and PGR, which are considered important ovarian factors in regulation of female reproduction [3].
As the important intraovarian growth factors, some members of the transforming growth factor-beta (TGFβ) superfamily have emerged as essential regulators of sheep ovarian follicles growth, differentiation and ovulation [44]. Follistatin-related protein 3 is a circulating glycoprotein, and acts as an endogenous inhibitor of TGFβ ligands such as bone morphogenetic proteins and activin [45–46]. Numerous studies have shown the roles of activins in many reproductive processes, such as activins has been shown to promote ovine preantral follicle and oocyte growth in vitro [47], stimulate the rodent ovarian follicular development, and inhibit oocyte apoptosis [48]. One study has shown the crucial roles of follistatin-related protein 3 in limiting testis organ size and promoting age-related testicular regression [49]. We identified FSTL3 upregulated in Hetian sheep, which implied that FSTL3 may play a negative effect on fertility in Hetian sheep by regulating activin and members of the BMP subfamily [50].
The RNA-seq and qRT-PCR results showed that several members of TGFβ associated genes were upregulated in Qira black sheep, implied that TGFβ played important roles in high prolificacy of Qira black sheep. Previous work had suggested that latent TGFβ binding proteins (LTBPs) might play an important role in TGFβ activation [51]. Miao et al. (2013) have proved that LTBP4 gene and TGFβ1I1 gene were highly upregulated in Small-tail Han sheep (high-fecundity; with FecB BB or ++ genotype) compared with Surabaya fur sheep (low-fecundity) [20]. In our study, LTBP4 gene and TGFβ1I1 gene were highly upregulated in Qira black sheep, implying that they might play critical roles for high prolificacy. And the results suggested that other members of TGFβ superfamily were also involved in the female fertility, such as thrombospondin-2 (THBS2) and proteolipid protein 2 (PLP2). THBS2 is highly expressed in bovine ovarian small follicles than either medium or large follicles [52] as well as in small atretic follicles than healthy follicles [53].
Furthermore, the mitogen-activated protein kinases (MAPK), insulin, Wnt, gonadotropin releasing hormone (GnRH), and Notch pathways have also been identified, suggesting that some molecules of these pathways might also involved in sheep fertility or maintaining physiological activity of the ovary [54–56]. It has been suggested that oocyte quality, embryo development, and male fertility are affected by mitochondrial haplotypes [57–58]. Our data indicated that plenty of DEGs were linked to mitochondrial, such as glutaredoxin-2 mitochondrial glutamate carrier 1, and mitochondrial import inner membrane translocase subunit TIM44. In addition, proteins required for transport are also differentially expressed, such as zinc transporter 3, zinc transporter ZIP8 isoform 1, intraflagellar transport protein 57 homolog, and receptor-transporting protein 4. In summary, all of these results suggest that plenty of the DEGs are potentially critical for regulating sheep fecundity.
Previous studies demonstrated that growth differentiation factor 9 gene (GDF9), bone morphogenetic protein 15 gene (BMP15), and bone morphogenetic protein 1B receptor gene (BMPR1B) are the major genes affecting the hyper-prolificacy in sheep [59–61]. Similarly, a previous study showed that BMPR-1B gene and GDF9 gene might be two candidate genes of fecundity in Qira black sheep [21]. However, the BMP15, BMPR1B, GDF9 genes were not differentially expressed between Qira black sheep and Hetian sheep in this study. One potential reason for this finding is that there might be different genes present that change the gene expression patterns of BMPR1B, BMP15, GDF9 and related genes or pathways affecting sheep fecundity. Another possible explanation is that there may be a result of using the whole ovary as material to determine differentially expressed genes. And these results are consistent with previous report on Small-tail Han sheep and Surabaya fur sheep ovaries using RNA-seq technology [20]. Anyway, our work will provide a valuable foundation for further molecular research of the sheep reproduction. Nonetheless, further studies are required to investigate the functional characterization of some selected DEGs, such as the expression profiling in ovarian follicles (or specific cell lines) at different cyclic stages in each breed, the functions in sheep follicular development, ovulation, hormone secretion and the regulatory networks.
Conclusion
In this study, RNA-Seq technology was employed to analyze the DEGs in Qira black sheep and Hetian sheep in a genome-wide level. We found that 1,252 genes were significantly differentially expressed between the two libraries. KEGG pathway analysis indicated that some of these genes are involved in steroid biosynthesis, steroid hormone biosynthesis, TGFβ, Insulin, Wnt, Notch, and other signaling pathways. Our data indicated that there were many differences of ovary gene expression patterns between Qira black sheep and Hetian sheep. And the present study provides a very useful genetic resource that may contribute to a better understanding of the mechanisms involved in sheep litter size variations.
Supporting Information
(XLS)
(XLS)
(XLS)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Natural Science Foundation of China (31260536) and Special Launching Funds for High-Level Talents of Shihezi University (RCZX201202). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Javanmard A, Azadzadeh N, Esmailizadeh AK. Mutations in bone morphogenetic protein 15 and growth differentiation factor 9 genes are associated with increased litter size in fat-tailed sheep breeds. Vet Res Commun. 2011; 35: 157–167. 10.1007/s11259-011-9467-9 [DOI] [PubMed] [Google Scholar]
- 2. Ibeagha-Awemu EM, Kgwatalala P, Zhao X. A critical analysis of production-associated DNA polymorphisms in the genes of cattle, goat, sheep, and pig. Mamm Genome 2008; 19: 591–617. 10.1007/s00335-008-9141-x [DOI] [PubMed] [Google Scholar]
- 3. Fernandez-Rodriguez A, Munoz M, Fernandez A, Pena RN, Tomas A, Noguera JL, et al. Differential Gene Expression in Ovaries of Pregnant Pigs with High and Low Prolificacy Levels and Identification of Candidate Genes for Litter Size. Biology of Reproduction 2011; 84: 299–307. 10.1095/biolreprod.110.085589 [DOI] [PubMed] [Google Scholar]
- 4. Mardis ER. The impact of next-generation sequencing technology on genetics. Trends in Genetics 2008; 24: 133–141. 10.1016/j.tig.2007.12.007 [DOI] [PubMed] [Google Scholar]
- 5. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 2008; 5: 621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 6. Geng R, Yuan C, Chen Y. Exploring Differentially Expressed Genes by RNA-Seq in Cashmere goat (Capra hircus) Skin during Hair Follicle Development and Cycling. PLoS ONE 2013; 8(4): e62704 10.1371/journal.pone.0062704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu T, Guo X, Wang H, Hao F, Du X, Gao X, et al. Differential gene expression analysis between anagen and telogen of Capra hircus skin based on the de novo assembled transcriptome sequence. Gene 2013; 520(1): 30–38. 10.1016/j.gene.2013.01.068 [DOI] [PubMed] [Google Scholar]
- 8. Esteve-Codina A, Kofler R, Palmieri N, Bussotti G, Notredame C, Pérez-Enciso M. Exploring the gonad transcriptome of two extreme male pigs with RNA-seq. BMC Genomics 2011; 12: 552 10.1186/1471-2164-12-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McCabe M, Waters S, Morris D, Kenny D, Lynn D, Creevey C. RNA-seq analysis of differential gene expression in liver from lactating dairy cows divergent in negative energy balance. BMC Genomics 2012; 13: 193 10.1186/1471-2164-13-193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang C, Wang G, Wang J, Ji Z, Liu Z, Pi X, et al. Characterization and Comparative Analyses of Muscle Transcriptomes in Dorper and Small-Tailed Han Sheep Using RNA-Seq Technique. PLOS ONE 2013; 8 (8): e72686 10.1371/journal.pone.0072686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jäger M, Ott CE, Grunhagen J, Hecht J, Schell H, Mundlos S, et al. Composite transcriptome assembly of RNA-seq data in a sheep model for delayed bone healing. BMC Genomics 2011; 12: 158 10.1186/1471-2164-12-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ott CE, Jäger M, Grünhagen J, Hecht J, Schell H, Mundlos S, et al. Extension of the sheep sequence space using composite transcriptome assembly of RNA-Seq data in a sheep model for delayed bone healing. Bone 2011; 48: S162–S162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du ZQ, Eisley CJ, Onteru SK, Madsen O, Groenen MAM, Ross JW, et al. Identification of species-specific novel transcripts in pig reproductive tissues using RNA-seq. Animal Genetics 2014; 45(2): 198–204. 10.1111/age.12124 [DOI] [PubMed] [Google Scholar]
- 14. Samborski A, Graf A, Krebs S, Kessler B, Bauersachs S. Deep Sequencing of the Porcine Endometrial Transcriptome on Day 14 of Pregnancy. Biology of Reproduction 2013; 84: 1–13. [DOI] [PubMed] [Google Scholar]
- 15. Driver AM, Peñagaricano F, Huang W, Ahmad KR, Hackbart KS, Wiltbank MC, et al. RNA-Seq analysis uncovers transcriptomic variations between morphologically similar in vivo- and in vitro-derived bovine blastocysts. BMC Genomics 2012; 13: 118 10.1186/1471-2164-13-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chitwood JL, Rincon G, Kaiser GG, Medrano JF, Ross PJ. RNA-seq analysis of single bovine blastocysts. BMC Genomics 2013; 14: 350 10.1186/1471-2164-14-350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatzirodos N, Irving-Rodgers HF, Hummitzsch K, Harland ML, Morris SE, Rodgers RJ. Transcriptome profiling of granulosa cells of bovine ovarian follicles during growth from small to large antral sizes. BMC Genomics 2014; 15: 24 10.1186/1471-2164-15-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu XP. Genome-wide transcriptome analysis of ovaries between Jining grey goat and Laiwu black goat using RNA-Seq. M.Sc. Thesis, Chinese Academy of Agricultural Sciences Dissertation. 2013.
- 19. Bonnet A, Cabau C, Bouchez O, Sarry J, Marsaud N, Foissac S, et al. An overview of gene expression dynamics during early ovarian folliculogenesis: specificity of follicular compartments and bi-directional dialog. BMC Genomics 2013; 14: 904 10.1186/1471-2164-14-904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miao X, Luo Q. Genome-wide transcriptome analysis between small-tail Han sheep and the Surabaya fur sheep using high-throughput RNA sequencing. Reproduction 2013; 6: 587–96. [DOI] [PubMed] [Google Scholar]
- 21. Shi HC, Bai J, Niu ZG, Muniresha, Fen LJ, Jia B. Study on Candidate Gene for Fecundity Traits in Xingjiang Cele Black Sheep. African Journal of Biotechnology 2010; 9(49): 8498–8505. [Google Scholar]
- 22. Yu JG, Dong H, Ji YG, Kong YL, Guligena. Conservate measures and current situation of hetian sheep. Grass-Feeding Livestock 2011; 2: 7–9. (in Chinese with English abstract) [Google Scholar]
- 23.Bai J. Study On DNA Molecular Markers On Fecundity Traits In Cele Black sheep And Duolang sheep. M.Sc. Thesis, The University of Shihezi. 2007.
- 24. Li RQ, Yu C, Li YR, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009; 25(15): 1966–1967. 10.1093/bioinformatics/btp336 [DOI] [PubMed] [Google Scholar]
- 25. Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 2008; 5(7): 621–8. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 26. Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997; 7(10): 986–95. [DOI] [PubMed] [Google Scholar]
- 27. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. The Annals of Statistics 2001; 29: 1165–1188. [Google Scholar]
- 28. Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-Seq data. Bioinformatics 2010; 26: 136–138. 10.1093/bioinformatics/btp612 [DOI] [PubMed] [Google Scholar]
- 29. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000; 25(1): 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zheng Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006; 34(Web Server issue): W293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008; 36 (Database issue): D480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C (T) method. Nat Protoc. 2008; 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 33. Fraser HM. Regulation of the ovarian follicular vasculature. Reproductive Biology and Endocrinology 2006; 4: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Irusta G, Abramovich D, Parborell F, Tesone M. Direct survival role of vascular endothelial growth factor (VEGF) on rat ovarian follicular cells. Molecular and Cellular Endocrinology 2010; 325: 93–100. 10.1016/j.mce.2010.04.018 [DOI] [PubMed] [Google Scholar]
- 35. Shimizu T. Promotion of ovarian follicular development by injecting vascular endothelial growth factor (VEGF) and growth differentiation factor 9 (GDF-9) genes. Journal of Reproduction and Development 2006; 52: 23–32. [DOI] [PubMed] [Google Scholar]
- 36. Yang MY, Fortune JE. Vascular endothelial growth factor stimulates the primary to secondary follicle transition in bovine follicles in vitro. Mol Reprod Dev. 2007; 74(9): 1095–1104. [DOI] [PubMed] [Google Scholar]
- 37. Qiu Y, Seager M, Osman A, Castle-Miller J, Bevan H, Tortonese DJ, et al. Ovarian VEGF165b expression regulates follicular development, corpus luteum function and fertility. Reproduction 2012; 143: 501–511. 10.1530/REP-11-0091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manna PR, Huhtaniemi LT, Stocco DM. Detection of hCG Responsive Expression of the Steroidogenic Acute Regulatory Protein in Mouse Leydig Cells. Biol Proced. 2004; 6(1): 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamashita H, Murayama C, Takasugi R, Miyamoto A, Shimizu T. BMP-4 suppresses progesterone production by inhibiting histone H3 acetylation of StAR in bovine granulosa cells in vitro. Molecular and Cellular Biochemistry 2011; 348(1–2): 183–190. 10.1007/s11010-010-0654-8 [DOI] [PubMed] [Google Scholar]
- 40. Garosi P, De Filippo C, van Erk M, Rocca-Serra P, Sansone SA, Elliott R. Defining best practice for microarray analyses in nutrigenomic studies. Br J Nutr. 2005; 93: 425–432. [DOI] [PubMed] [Google Scholar]
- 41. Argov N, Sklan D. Expression of mRNA of lipoprotein receptor related protein 8, low density lipoprotein receptor, and very low density lipoprotein receptor in bovine ovarian cells during follicular development and corpus luteum formation and regression. Mol Reprod Dev. 2004; 68: 169–175. [DOI] [PubMed] [Google Scholar]
- 42. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011; 32: 81–151. 10.1210/er.2010-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ying SJ, Xiao SH, Wang CL, Zhong BS, Zhang GM, Wang ZY, et al. Effect of nutrition on plasma lipid profile and mRNA levels of ovarian genes involved in steroid hormone synthesis in Hu sheep during luteal phase. J Anim Sci. 2013; 91: 5229–5239. 10.2527/jas.2013-6450 [DOI] [PubMed] [Google Scholar]
- 44. Juengel JL, Bibby AH, Reader KL, Lun S, Quirke LD, Haydon LJ, et al. The role of transforming growth factor-beta (TGF-beta) during ovarian follicular development in sheep. Reproductive Biology and Endocrinology 2004; 2: 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robertson RD, Mukherjee A. Synexpression group analyses identify new functions of FSTL3, a TGFβ ligand inhibitor. Biochemical and Biophysical Research Communications 2012; 427(3): 568–573. 10.1016/j.bbrc.2012.09.098 [DOI] [PubMed] [Google Scholar]
- 46. Mcnatty KP, Galloway SM, Wilson T, Smith P, Hudson NL, O'Connell A, et al. Physiological effects of major genes affecting ovulation rate in sheep. Genet Sel Evol. 2005; 37 (Suppl. 1): S25–S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thomas FH, Armstrong DG, Telfer EE. Activin promotes oocyte development in ovine preantral follicles in vitro. Reproductive Biology and Endocrinology 2003; 1: 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cossigny DA, Findlay JK, Drummond AE. The effects of FSH and activin A on follicle development in vitro. Reproduction 2012; 143: 221–229. 10.1530/REP-11-0105 [DOI] [PubMed] [Google Scholar]
- 49. Oldknow KJ, Seebacher J, Goswami T, Villen J, Pitsillides AA, O'Shaughnessy PJ, et al. Follistatin-like 3 (FSTL3) mediated silencing of transforming growth factor β (TGFβ) signaling is essential for testicular aging and regulating testis size. Endocrinology 2013; 154(3): 1310–20. 10.1210/en.2012-1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Welt C, Sidis Y, Keutmann H, Schneyer A. Activins, inhibins, and follistatins: from endocrinology to signaling. A paradigm for the new millennium. Exp Biol Med. 2002; 227: 724–752. [DOI] [PubMed] [Google Scholar]
- 51. Sheppard D. Integrin-mediated activation of latent transforming growth factor β. Cancer and Metastasis Reviews 2005; 24: 395–402. [DOI] [PubMed] [Google Scholar]
- 52. Greenaway J, Gentry PA, Feige JJ, LaMarre J, Petrik JJ. Thrombospondin and vascular endothelial growth factor are cyclically expressed in an inverse pattern during bovine ovarian follicle development. Biol Reprod. 2005; 72: 1071–1078. [DOI] [PubMed] [Google Scholar]
- 53. Hatzirodos N, Hummitzsch K, Irving-Rodgers HF, Harland ML, Morris SE, Rodgers RJ. Transcriptome profiling of granulosa cells from bovine ovarian follicles during atresia. BMC Genomics 2014; 15:40 10.1186/1471-2164-15-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Harrouk W, Clarke HJ. Mitogen-activated protein (MAP) kinase during the acquisition of meiotic competence by growing oocytes of the mouse. Mol Reprod Dev. 1995; 41: 29–36. [DOI] [PubMed] [Google Scholar]
- 55. Harwood BN, Cross SK, Radford EE, Haac BE, De Vries WN. Members of the Wnt signaling pathways are widely expressed in mouse ovaries, oocytes, and cleavage stage embryos. Dev Dynam. 2008; 237: 1099–1111. 10.1002/dvdy.21491 [DOI] [PubMed] [Google Scholar]
- 56. Wang HX, Tekpetey FR, Kidder GM. Identification of Wnt/beta-catenin signaling pathway components in human cumulus cells. Mol Hum Reprod. 2009; 15: 11–17. 10.1093/molehr/gan070 [DOI] [PubMed] [Google Scholar]
- 57. Cannon MV, Takeda K, Pinkert CA. Mitochondrial biology in reproduction. Reprod Med Biol. 2011; 10: 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yee WK, Sutton KL, Dowling DK. In vivo male fertility is affected by naturally occurring mitochondrial haplotypes. Current Biology 2013; 3: R55–R56. 10.1530/EJE-12-0563 [DOI] [PubMed] [Google Scholar]
- 59. Shimasaki S, Zachow RJ, Li D, Kim H, Iemura S, Ueno N, et al. A functional bone morphogenetic protein system in the ovary. PNAS 1999; 96: 7282–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ibeagha-Awemu EM, Kgwatalala P, Zhao X. A critical analysis of production-associated DNA polymorphisms in the genes of cattle, goat, sheep, and pig. Mamm Genome 2008; 19: 591–617. 10.1007/s00335-008-9141-x [DOI] [PubMed] [Google Scholar]
- 61. Vinet A, Drouilhet L, Bodin L, Mulsant P, Fabre S, Phocas F. Genetic control of multiple births in low ovulating mammalian species. Mamm Genome 2012; 23: 727–740. 10.1007/s00335-012-9412-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS)
(XLS)
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.