

Abstract
Mice with epidermal deletion of JunB transcription factor displayed a psoriasis-like inflammation. The relevance of these findings to humans and the mechanisms mediating JunB function are not fully understood. Here, we demonstrate that impaired JunB function via gene silencing or overexpression of a dominant negative mutant increased human keratinocyte cell proliferation but decreased cell barrier function. RNA-seq revealed over 500 genes affected by JunB loss-of-function which included an upregulation of an array of proinflammatory molecules relevant to psoriasis. Among these were TNFα, CCL2, CXCL10, IL6R and SQSTM1, an adaptor protein involved in NF-κB activation. ChIP-Seq and gene reporter analyses showed that JunB directly suppressed SQSTM1 through binding to a consensus AP-1 cis-element located around 2 Kb upstream of SQSTM1-trasncription start site. Similar to JunB loss-of-function, SQSTM1-overexpression induced TNFα, CCL2 and CXCL10. Conversely, NF-κB-inhibition genetically with a mutant IκBα or pharmacologically with PDTC prevented cytokine, but not IL6R, induction by JunB-deficiency. Taken together, our findings indicate that JunB controls epidermal growth, barrier formation and proinflammatory responses through direct and indirect mechanisms, pinpointing SQSTM1 as a key mediator of JunB-suppression of NF-κB-dependent inflammation.
Keywords: JunB, SQSTM1/p62, NF-κB, keratinocyte, proinflammatory cytokine
Introduction
Psoriasis and psoriatic arthritis affect nearly 3% of the US population(Helmick et al., 2014; Rachakonda et al., 2014). While the etiology of the process is known to be multifold involving multiple cell types and various genetic and environmental factors, it is clear that a disarrayed T-cell driven immune response is critical for the pathogenesis of psoriasis (Clark, 2010). Current therapies are mostly designed to target immune cells, and treatment results are often mixed and unpredictable (Belge et al., 2014). Recently, it has become apparent that defects in epidermal cells, the predominant barrier cells of the skin, can initiate a wide spectrum of immune responses. This was best demonstrated using conditional genetic animal models. Of particular interest, mice with epidermal deletion of JunB, an AP-1 family transcription factor, alone or together with c-Jun, developed joint and skin lesions that bared a remarkable resemblance to human arthritis and psoriasis, respectively (Zenz et al., 2005). Additionally, targeted JunB deletion in epidermal cells led to a multi-organ inflammation involving blood, kidney and lymph nodes (Meixner et al., 2008). These studies establish that JunB plays an important role in epidermal cell-mediated innate and adaptive immune responses in mice.
In humans, JunB is located in the previously identified psoriasis susceptibility locus PSOR6 (19p13) (Huffmeier et al., 2009), though the significance of which has not been validated by later GWAS studies. Interestingly, JunB mRNA levels were found increased in psoriatic lesions as compared to nonlesional skin (Haider et al., 2006; Johansen et al., 2004; Kulski et al., 2005), whereas JunB protein levels were reduced in the basal and spinous layers of the lesional epidermis (Park et al., 2009; Zenz et al., 2005). The discrepancy between the mRNA and protein levels may be due to the presence of lymphocytes in the lesional skin or a posttranslational effect. Regardless, the reduced protein expression suggests that JunB-hypofunction in epidermal cells may have a causal effect on epidermal cell proliferation and inflammation. In agreement with this possibility, our recent studies have shown that JunB inhibits, whereas its dominant negative mutant promotes, epidermal growth(Jin et al., 2011).
In this study, we demonstrated that gene silencing of JunB in normal epidermal cells resulted in increased cell proliferation and compromised epidermal barrier function. By using global RNA-seq and ChIP-seq, we found that JunB regulated expression of a wide-range of molecules involved in cell growth, interaction, differentiation and inflammation through both direct and indirect mechanisms. In particular, JunB directly interacted with the consensus AP-1 response element to suppress gene transcription of SQSTM1/p62, an adaptor protein linked to Paget’s disease of bone and neurodegenerative disorders, and also upregulated in psoriasis (Rea et al., 2014; Sundaram et al., 2011). SQSTM1/p62 has been characterized as a key regulator involved in NF-κB activation and autophagy (Wooten et al., 2005; Zotti et al., 2014). We found that NF-κB, but not autophagy, is essential for the expression of various psoriasis-relevant chemokines induced by decreased JunB-function. These findings demonstrate that JunB-hypofunction impairs epidermal barrier function, and triggers a proinflammatory response through direct and indirect target gene regulation, and identified SQSTM1 and NF-κB signaling axis as a potential therapeutic target for skin diseases such as psoriasis.
Results
JunB loss-of-function leads to an increased keratinocyte cell proliferation but reduced barrier function
To determine the role of JunB in human skin, we first examined JunB loss-of-function effects on epidermal growth and barrier function via siRNA-mediated gene silencing in primary human keratinocytes. The efficiency of gene silencing was confirmed by RT-PCR and Western blotting (Fig. 1a). Cell growth analyses were performed in the absence or presence of TNFα which has been shown to inhibit normal keratinocyte growth in culture (Detmar and Orfanos, 1990). JunB gene silencing induced a significantly higher rate of cell proliferation, and diminished the growth inhibitory effects of TNFα (Fig. 1b). To assess effects on barrier function, we plated cells on permeable membrane inserts for induction of differentiation with 1.2 mM Ca++, and then measured the transepithelial electrical resistance (TER) as described (Sumigray et al., 2012). A higher TER value corresponds to a healthier barrier function. Gene silencing of JunB significantly reduced the TER at both 24h and 48h time-points (Fig. 1c), indicating that JunB is important for the epidermal cell barrier formation.
Figure 1. JunB loss-of-function leads to increased epidermal cell proliferation and reduced barrier function.

(a) Confirmation of gene silencing via real-time RT-PCR and Western blotting. Relative expression levels of JunB were shown below each band via densitometry. (b) Cell growth of human keratinocytes treated with or without 50ng/ml TNFα for 72h. (c) Barrier functions as measured by the Transepithelial Electrical Resistance (TER). Keratinocytes were induced to differentiate with 1.2 mM Ca++on permeable membranes for 24h and 48h prior to TER measurement. (d) Confirmation of gene expression via immunoblotting. (e) Cell growth analysis. (f) Immunostaining of 6-week old skin grafts generated on SCID mice with human keratinocytes transduced to express genes as indicated. Ki-67 [orange] and nuclei [blue]. Graphs represent the average values of triplicate samples + SD. P-values were obtained via two-tiered student T-test. Scale bar=100μm.
To confirm the growth effects observed with gene silencing, we transduced keratinocytes with retrovirus for stable overexpression of JunB or the dominant negative deletion mutant of JunB (DNJunB) (Fig.1d). Monolayer cell growth analysis revealed that overexpression of JunB significantly reduced cell proliferation, while DNJunB induced a modest increase of cell growth (Fig. 1e). Consistently, skin grafts regenerated on immunodeficient SCID mice with the transduced keratinocytes showed that JunB reduced epidermal proliferation, whereas DNJunB induced hyperproliferation as indicated by the over 2-fold changes of the number of Ki-67-positive cells (Fig. 1f). These findings indicate that JunB is involved in controlling epidermal cell proliferation and barrier function.
Global RNA-seq analysis reveals an important role for JunB in regulation of epidermal inflammation and cell adhesion
To examine how JunB regulates epidermal homeostasis, we performed RNA-seq with mRNA isolated from primary human keratinocytes that had been transfected with siRNA oligonucleotides for JunB gene silencing. Around 8-20 million sequence reads passed quality control for each sample, accounting for over 15,000 genes that were expressed at a detectable level in normal human keratinocytes. In response to JunB gene silencing, about 500 genes displayed significant changes with 138 decreased and 84 increased by over 2-fold (Table S1-S2). The JunB responsive genes include many of those involved in inflammation and cell adhesion as shown by the heatmap analysis (Fig. 2a-b). Of the 29 inflammation related genes, over 70% of them were found increased in patient psoriatic lesions in a recent large scale RNA-seq data set (GSE54456) (Li et al., 2014). Additionally, a majority of these molecules have been previously linked to psoriasis and psoriatic arthritis. These include CCL2, CXCL10, CXCL11 and TNFα, as well as receptors such as IL6R and the leukocyte interaction molecule ICAM1(Antonelli et al., 2009; Danik et al., 2009; Fraticelli et al., 2001; Paukkonen et al., 1995; Plant et al., 2006). Similarly, 70% of the 16 downregulated cell-adhesion molecules were found decreased in patient psoriatic lesions (GSE54456).
Figure 2. RNA-seq reveals JunB-targets involved in inflammation and cell adhesion.

(a-b) Heat map analysis of gene expression of 3 independent sets of human keratinocytes underwent gene-transfection with siRNA oligonucelotides targeting JunB (siJunB) or nonsilecning control (siCon). (c) Real-time RT-PCR for JunB, TNFα, IL6R, CCL2, CXCL10 and SQSTM-1. Graph represents average fold changes of triplicate samples + SD. (d) Cytokine analysis of cell culture conditioned media. Graph represents average of tetrad samples + SD.
To validate the RNA-seq data, we performed RT-PCR for a number of proinflammatory molecules, including TNFα, CXCL10, IL6R and SQSTM1, which were increased in response to reduced JunB expression (Fig. 2c), To further examine whether the altered mRNA levels were reflected at protein levels, we collected conditioned keratinocyte culture media for cytokine analysis. We found that CCL2 and CXCL10 were significantly upregulated in cells with reduced JunB expression (Fig. 2d). In agreement with results observed with gene silencing, overexpression of the dominant negative JunB (DNJunB) also increased the expression levels of multiple cytokines, such as TNFα, CXCL10 and CCL2, as demonstrated by RT-PCR or cytokine analysis (Fig. S1a-b). Additionally, we confirmed by RT-PCR that the cell adhesion molecule CNTN1 was reduced, whereas MMP7, MMP9 and ICAM1 was upregulated, whereas the terminal differentiation marker filaggrin was not changed in response to reduced JunB expression (Fig. S2a). Western blotting further validated the increased expression of ICAM1 in response to JunB gene silencing (Fig. S2b). These results indicate that JunB loss-of-function induces cell adhesion defects and proinflammatory responses reminiscent of psoriasis.
ChIP-Seq analysis identifies SQSTM1 as a direct JunB target
Next, we performed ChIP-seq analysis to identify JunB direct target genes. To do this, genomic DNA was isolated from primary human keratinocytes for chromosomal immunoprecipitation (ChIP) with an antibody against JunB and then subjected to ChIP-seq analyses. Peak calling using Chipseeqer identified a total of 2377 sequences. Alignment with the RefSeq database revealed that 43% of these sequences were located in gene promoter regions with 17% in the proximal region (2kb+/−) and 26% in the distal region (2-50kb) (Fig. 3a). The rest were located in the introns (32.7%), intergenic regions (20.4%) and exons (0.2%). Motif analysis with ChipSeeqerFIRE demonstrated that AP-1 consensus elements were present with a high confidence score of 10 out of 10 (Giannopoulou and Elemento, 2011) (Fig. 3b), confirming that JunB ChIP achieved a satisfactory enrichment of the putative AP-1 target genes.
Figure 3. ChIP-seq identifies p62/SQSTM1 as a JunB-target gene.

(a) Percent of ChIP-seq distribution. (b) Motif analysis with ChIPseeqerFire. (c) A representative ChIP-seq peak located on SQSTM1. (d) DNA sequence of the SQSTM1-promoter (Promoter ID: 33839; http://rulai.cshl.edu/cgi-bin/TRED). The consensus AP-1 element -5′TGACTCA3′- was underlined in red. (e) ChIP-PCR with primers marked in (d). Graph represents the average fold enrichment of triplicate samples by JunB-ChIP over the input DNA + SD. (f) Dual luciferase gene reporter analysis of 293T cells transfected with the firefly luciferase reporter constructs containing the WT SQSTM1-promoter or the mutant with a defective AP-1 element along with the CMV-renilla luciferase and JunB expression constructs. Graph represents the average of relative luciferase reading units + SD.
Focused peak analysis on UCSC genome browser (hg19) identified a peak on chromosome 5 at about 1.8 kb upstream of the transcription start site of SQSTM1 (Fig. 3c), which encodes an adaptor protein important for NF-κB activation and autophagy (Sanz et al., 2000), and was recently found upregulated in psoriatic lesions (Lee et al., 2011). In agreement with SQSTM1 being an actively transcribed gene, its promoter region was enriched with H3K27Ac modification, a marker for active enhancers, and DNase-sensitive, an indicator for open chromatin state. In addition, within the peak harbored a putative AP-1-response element (TGACTCA) (Fig. 3e). To validate the ChIP-seq result, we performed quantitative ChIP-PCR with primers designed to cover the 5′ and 3′ends of the AP-1 cis-element. As expected, JunB ChIP achieved an over 10-fold enrichment of SQSTM1-sequence (Fig. 3e). To further examine the importance of the AP-1 cis-element, we performed luciferase gene reporter analysis, and found that JunB inhibited gene expression driven by the WT SQSTM1-promoter but not the mutant containing a defective AP-1 response-element (Fig.3f). Thus, JunB suppresses SQSTM1 expression via the AP-1 cis-element.
SQSTM1 mediates chemokine expression
Interestingly, among the JunB-responsive genes, only 27 of them were enriched by JunB ChIP-seq (Table S3), which suggests that some of those responsive genes were caused by secondary effects. Specifically, we predicted that SQSTM1 is involved in mediating the upregulation of the inflammatory cytokines. To test this idea, we first confirmed that SQSTM1 was increased at the protein level by JunB gene silencing (Fig. 4a). We then performed gene silencing or retrovirally mediated overexpression of exogenous SQSTM1 (Fig. 4b-c). Quantitative RT-PCR revealed that exogenous expression of SQSTM1 increased CXCL10 and CCL2 mRNA levels as compared to LacZ control (Fig. 4d). Conversely, gene silencing of SQSTM1 prevented the induction of both chemokines by reduced JunB expression (Fig. 4e). These results indicate that SQSTM1 positively regulates chemokine expression.
Figure 4. SQSTM1 positively regulates chemokine expression.

(a-c) Immunoblotting of SQSTM1, JunB, Actin or GAPDH with protein lysates collected from human keratinocytes at 48h post gene transduction. Relative expression levels of SQSTM1 and JunB were shown below each band. Note: Results obtained with two different siRNA oligonucleotides (Invitrogen S7776 and S7662) targeting JunB were comparable. Most studies were performed with the second oligonucleotide. (d) Relative mRNA levels of CXCL10 by real-time RT-PCR. Graph represents relative mRNA levels of triplicates + SD with GAPDH used as the internal control. (e) Cytokine analysis of cell culture conditioned media. Graph represents average of tetrad samples + SD.
NF-κB is required for the induction of CCL2 and CXCL10
Since SQSTM1 is a key mediator of NF-κB activation by TRAF6/IKKγ signaling axis (Wooten et al., 2005; Zotti et al., 2014), we predicted that NF-κB is responsible for the increased cytokine expression. In agreement with this possibility, NF-κB activation was induced by gene silencing of JunB as indicated by the increased levels of pIKKα/β and pIκBα and NF-κB-driven luciferase expression (Fig. 5a-b). Inhibition of NF-κB via expression of the IκBα mutant (IκBαM), a well-known super-repressor of NF-κB abolished the induction of CXCL10 by SQSTM1 (Fig. 5c). To further test whether NF-κB is required for the cytokine expression, we treated cells with the pharmacological NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) or autophagy inhibitor chloroquine (CQ) at 24h post-gene silencing. The conditioned culture media were collected for cytokine analyses at the 72h time-point. We found that PDTC but not CQ markedly reduced the protein levels of CCL2 and CXCL10 (Fig. 5d). In line with the results obtained with the pharmacological agents, expression of IκBαM significantly reduced the mRNA levels of TNFα, CCL2 and CXCL10, as shown by quantitative RT-PCR (Fig. 5e). In contrast, IL6R, a direct target of JunB which was previously validated in mouse keratinocytes (Zenz et al., 2005), was not affected by IκBαM. In agreement with the responsiveness to NF-κB, TNFα, CCL2 and CXCL10, but not IL6R, all contained one or more putative NF-κB cis-elements on the promoter region (Fig. S2). These findings indicate that NF-κB is required for the induction of the proinflammatory cytokines.
Figure 5. NF-κB is required for cytokine induction.

(a) Immunoblotting for pIKKα, pIκBα, p62 and Actin of protein lysates isolated from keratinocytes at 48h post-transfection. (b) Dual luciferase NF-κB-gene reporter assay. (c) Relative mRNA levels of SQSTM1 and CXCL10 in human keratinocytes at 48h post-gene transduction as determined by real-time RT-PCR with 18S RNA used as the internal control. (d) Cytokine analysis of keratinocyte conditioned culture media collected at 48h after treatment with 20uM PDTC or 10uM chloroquine (CQ). (e) Relative mRNA levels of TNFα, CXCL10, CCL2 and IL6R analyzed by RT-PCR as in described above. Graphs represent the average values of triplicate or tetrad samples + SD. P-values were obtained via two-tiered student T-test.
Taken together, our findings support a working model in which JunB-hypofunction leads to increased expression of SQSTM1 and consequent NF-κB-dependent induction of proinflammatory cytokines (Fig. 6). Additionally, JunB loss-of-function increases cell proliferation, and compromises barrier functions through direct and indirect target gene regulations. Future studies are needed to determine whether the proinflammatory cytokines indirectly affect cell growth and barrier function.
Figure 6. Working model.
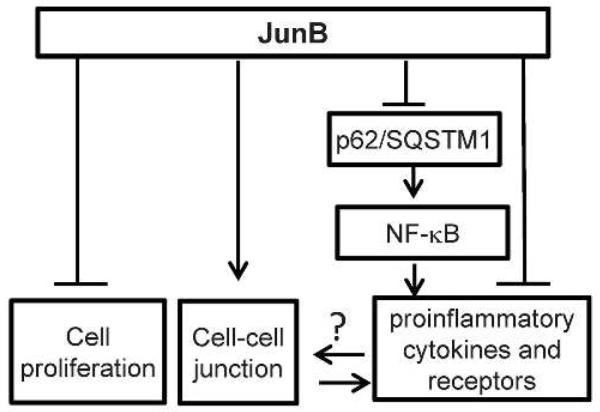
JunB-suppression of inflammation. JunB regulates epidermal cell proliferation, cell adhesion and inflammation through direct and indirect target genes. In particular, JunB-hypofunction leads to increased expression of SQSTM1 and consequently NF-κB-dependent expression of multiple inflammatory cytokines.
Discussion
Our studies demonstrate that reduced JunB expression affects multiple cellular processes through direct and indirect regulation of an array of target genes. The most pronounced consequences of JunB reduction is the increased expression of various proinflammatory chemokines and reduced expression of cell adhesion molecules. SQSTM1, as a direct downstream target of JunB, serves as a critical mediator to the uncontrolled NF-κB activation and subsequent proinflammatory responses.
Our findings suggest that the reduced JunB protein expression in psoriatic lesions, as demonstrated in earlier studies (Park et al., 2009; Zenz et al., 2005), may contribute to the development of the lesions due to the uncontrolled expression of the inflammatory cytokines. In this respect, JunB may be important for the inhibition of the disease onset or progression. Given that JunB mRNA level is increased in psoriatic lesions (Haider et al., 2006; Johansen et al., 2004; Kulski et al., 2005), it is conceivable that the reduced JunB protein level in the lesional skin is due to a posttranscriptional deregulation. Future studies will be necessary to determine whether and how JunB protein translation or stability is altered in psoriatic lesions.
Current therapies for psoriasis are mainly focused on targeting T-cell mediated disease processes (Belge et al., 2014). A chosen treatment option may produce good outcomes for some patients but be less efficient for others, which is likely due to the complex nature of the etiology involving genetic and environmental heterogeneities. Our findings suggest that it might be possible to block the disease at an early stage by interfering keratinocyte-mediated proinflammatory responses. Strategies that modulate the SQSTM1/NF-κB signaling axis in keratinocytes might be applied for disease prevention.
SQSTM1 is implicated in cancers of various organs, including kidney(Li et al., 2013), tuberous sclerosis complex (Parkhitko et al., 2011), pancreatic (Ling et al., 2012), prostate (Chang et al., 2014), lung(Inoue et al., 2012), and oral (Inui et al., 2013; Kuo et al., 2014). However, in cutaneous squamous cell carcinoma, SQSTM1 displays an inverse correlation with malignant progression (Yoshihara et al., 2014), suggesting that SQSTM1 may have epidermis-specific functions. Similar to SQSTM1, NF-κB is known to stimulate inflammation and tumorigenesis in a wide range of tissues. On the other hand, persistent NF-κB activation promotes epidermal aging and inhibits cutaneous neoplasia (Adler et al., 2007; Dajee et al., 2003; Zhang et al., 2004; Zhang et al., 2005b). Thus, NF-κB-inhibition predicts both beneficial and harmful results (Wullaert et al., 2011). It will be important to determine whether SQSTM1 is involved in the regulation of the functional dichotomy of NF-κB, and may be exploited to maximize the beneficial effects of NF-κB-targeted therapy.
Materials and Methods
Cell culture, gene transfection and barrier function analysis
Primary human keratinocytes were isolated from surgically discarded foreskin samples obtained from Duke Children’s Hospital in accordance to in institutionally approved IRB protocol. Patient consent for experiments was not required because French laws consider human tissue left over from surgery as discarded material. Cells were grown and passaged in keratinocyte serum free media (Invitrogen, Grand Island, NY) at 37 °C with 5% CO2, and used at passage 2-3. 293T cells were obtained from ATCC and cultured in 10%FBS/DMEM. Gene transfection was performed with Genjet transfection reagent (SignaGen Laboratories, Rockville, MD). siRNA oligonucleotides targeting JunB (S7776 and S7662), SQSTM1 (HSS113116) and non-silencing control were purchased from Invitrogen and used at 100nM concentrations. Unless noted, siJunB were performed with S7662. The SQSTM1 cDNA expression construct was obtained from Addgene (Fan et al., 2010), and subcloned into the LZRS retroviral construct. For barrier function analysis, cells were seeded onto to Costar Transwell Permeable membrane inserts at 24h post-transfection, and induced with 1.2 mM calcium for 24h and 48h. TER was measured by using a Millipore Millicell ERS-2 ohm meter. Readings were normalized against wells with no cells (set as zero resistance) and to insert area. A higher resistance indicates better barrier function. The retroviral LZRS expression constructs for JunB and DNJunB were described previously (Jin et al., 2011).
RNA-Seq and RT-PCR
For RNA-seq, total RNA was isolated from triplicate sets of human keratinocytes at 48h after transfection using RNAeasy column purification (Qiagen, Germantown, MD). cDNA library was prepared via oligo-dT-directed reverse transcription (Ambion, Grand Island, NY), and subject to deep sequencing on Illumina HiSeq2000 (50bp single-read sequencing) and analyzed at Duke Genome Sequencing & Analysis Resource. The analysis pipeline includes the initial QC to remove sequencing adaptors and low quality bases to facilitate mapping using fastx toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). High quality reads were mapped to the human reference genome (hg19) using tophat (Trapnell et al., 2009). The gene differential expression analysis was performed between control and treatment using cuffdiff with upper quartile normalization, multi-hit correction and 0.5% FDR(Trapnell et al., 2013). Three biological replicates were used for statistical analysis of differential expression for each comparison. Pathway analyses were performed using WEB-based GEne SeT AnaLysis Toolkit (Wang et al., 2013; Zhang et al., 2005a). SYBR green-based real-time RT-PCR was performed in Bio-Rad iCycler with primers listed in (Table S4). Ribosomal 18S RNA or GAPDH was used as an internal control.
ChIP-Seq and ChIP-PCR
ChIP was performed as described (Guo et al., 2012). Briefly, primary keratinocytes cultured in eight 30-cm plates were scraped off the dish with PBS at about 60-80% confluence, and cross-linked with 1% formaldehyde for 8 minutes, and then treated with 1.25 M glycine for 5 minutes to stop crosslink. Cells were pelleted by centrifuging 5 minutes at 1500 rpm, and lysed with ChIP lysis buffer (Santa Cruz Biotechnology, Santa CruZ, CA) for cell nuclei isolation. DNA fragmentation was performed by sonication with Branson Sonifier. The fragmented DNA underwent IP with the antibody against JunB (Cell Signaling Technology, Beverly, MA) followed by purification with MinElute DNA kit (Qiagen) and library construction with the NEBNext® DNA library preparation kit (New England BioLabs). The library was sequenced using Illumina Hiseq 2000 platform and analyzed at Duke Genome Sequencing & Analysis Resource. The analysis pipeline includes the initial QC to remove sequencing adaptors and low quality bases to facilitate mapping using fastx toolkit (Martin, 2011). High quality reads were mapped to the human reference genome (hg19) using bwa (Li and Durbin, 2009), and genes lists were from the Ensemble Gene database (http://www.ensemble.org) and the RefSeq (www.ncbi.nlm.nih.gov/RefSeq). Distal promoter and terminator regions of the genes were defined as those sequences that mapped +/−2 kb upstream or +/− 50 kb from transcription “start” and “end” sites, respectively. Peak detection, annotation, de novo motif discovery and pathway analysis were performed using ChIPseeqer tool (Giannopoulou and Elemento, 2011), and visualized by UCSC genome browser (Kent et al., 2002). Motif analysis was performed with FIRESeq as described(Mathelier and Wasserman, 2013). ChIP-PCR was performed with primers listed in (Table S4). The raw data and additional information of ChIP-seq and RNA-seq are available from NCBI Gene Expression Omnibus (Accession number GSE63081).
Multiplex cytokine analysis
For cytokine analysis, human keratinocytes cultured in 12-well plates were transfected in duplicates with 100nM siRNA oligonucleotides targeting non-silencing control and JunB (Invitrogen). At 24h post-transfection, cells were treated with DMSO control, Pyrrolidine Dithiocarbamate (PDTC) or Chloroquine (Sigma, St Louis, MO) for 48h. Cell culture conditioned media were analyzed in tetrads with custom ordered Luminex Screening Assay System (R&D Systems, Pittsburgh, PA). The readings were obtained on a Bioplex reader-200 (Bio-Rad, Hercules, CA) at a flow rate of 60ul/min and 50ul/sample.
Skin regeneration
Human skin regeneration was performed as described in our previous studies in accordance with protocols approved by the Duke Animal Care and Use Committee (Jin et al., 2011). Briefly, primary human keratinocytes were transduced with retrovirus encoding genes of interest, and seeded onto devitalized human cadaver dermis (National Disease Research Institute) for grafting onto the back of CB17.SCID mice (n=3-5 per group, purchased from National Cancer Institute). Animals were euthanized at 6 weeks post-grafting for tissue collection.
Western blotting and immunostaining
For western blotting, 20ug protein lysates of primary human keratinocytes collected at 48-72h post transfection were separated on SDS-PAGE, and immunobloted with antibodies against JunB, SQSTM1, IκBα and actin, followed by IR-dye-conjugated secondary antibodies (Invitrogen). The blots were scanned on the Odyssey imaging system (Li-COR, Lincoln, NE). For immunostaining, 5um frozen tissue sections were fixed with cold methanol, incubated with primary antibodies against Ki-67, and detected with a secondary antibody conjugated to Alexa 5 (Invitrogen) and counterstaining with Hoechst 33342. The images were obtained with the Olympus imaging system.
Gene reporter analysis
NF-κB-luciferase (Promega) and SQSTM1-luciferase reporter constructs were transfected together with the CMV-renilla luciferase and genes of interest into 293T cells using Genjet transfection reagent. Dual luciferase assay was performed at 24h post-transfection. The relative luciferase reading unit (RLU) was obtained by normalizing the readings of firefly luciferase to that of renilla luciferase, and then to that of the control cells. The p-values were obtained in Excel with two-tiered student T-test.
Supplementary Material
Acknowledgement
This work was supported by NIH/NIAMS R01 to Jennifer Zhang. We thank Olivier Fedrigo, Yiping He and Changcun Guo of Duke University, Howard Change and Paul Khavari of Stanford University for their advice on ChIP-seq experiments, and Terry Lechler of Duke University for assistance in barrier function analysis. We also thank Mitsuyasu Kato of University of Tsukuba for the cDNA expression constructs of JunB and DNJunB, and Paul Chiao of Texas University for the SQSTM1-promoter driven luciferase expression construct.
Footnotes
Conflict of Interest
Authors declare no conflict of interest
References
- Adler AS, Sinha S, Kawahara TL, et al. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21:3244–57. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli A, Fallahi P, Delle Sedie A, et al. High values of Th1 (CXCL10) and Th2 (CCL2) chemokines in patients with psoriatic arthtritis. Clin Exp Rheumatol. 2009;27:22–7. [PubMed] [Google Scholar]
- Belge K, Bruck J, Ghoreschi K. Advances in treating psoriasis. F1000prime reports. 2014;6:4. doi: 10.12703/P6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MA, Morgado M, Warren CR, et al. p62/SQSTM1 Is Required for Cell Survival of Apoptosis-Resistant Bone Metastatic Prostate Cancer Cell Lines. Prostate. 2014;74:149–63. doi: 10.1002/pros.22737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA. Skin-resident T cells: the ups and downs of on site immunity. J Invest Dermatol. 2010;130:362–70. doi: 10.1038/jid.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–43. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Danik JS, Pare G, Chasman DI, et al. Novel Loci, Including Those Related to Crohn Disease, Psoriasis, and Inflammation, Identified in a Genome-Wide Association Study of Fibrinogen in 17 686 Women The Women’s Genome Health Study. Circ-Cardiovasc Gene. 2009;2:134–41. doi: 10.1161/CIRCGENETICS.108.825273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmar M, Orfanos CE. Tumor necrosis factor-alpha inhibits cell proliferation and induces class II antigens and cell adhesion molecules in cultured normal human keratinocytes in vitro. Arch Dermatol Res. 1990;282:238–45. doi: 10.1007/BF00371643. [DOI] [PubMed] [Google Scholar]
- Fan W, Tang Z, Chen D, et al. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy. 2010;6:614–21. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraticelli P, Sironi M, Bianchi G, et al. Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. Journal of Clinical Investigation. 2001;107:1173–81. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannopoulou EG, Elemento O. An integrated ChIP-seq analysis platform with customizable workflows. BMC bioinformatics. 2011;12:277. doi: 10.1186/1471-2105-12-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Chang CC, Wortham M, et al. Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc Natl Acad Sci U S A. 2012;109:17603–8. doi: 10.1073/pnas.1208807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider AS, Duculan J, Whynot JA, et al. Increased JunB mRNA and protein expression in psoriasis vulgaris lesions. Journal of Investigative Dermatology. 2006;126:912–4. doi: 10.1038/sj.jid.5700183. [DOI] [PubMed] [Google Scholar]
- Helmick CG, Lee-Han H, Hirsch SC, et al. Prevalence of psoriasis among adults in the U.S.: 2003-2006 and 2009-2010 National Health and Nutrition Examination Surveys. American journal of preventive medicine. 2014;47:37–45. doi: 10.1016/j.amepre.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffmeier U, Lascorz J, Becker T, et al. Characterization of Psoriasis Susceptibility Locus 6 (PSORS6) in Patients with Early Onset Psoriasis and Evidence for Interaction with PSORS1. Exp Dermatol. 2009;18:285. doi: 10.1136/jmg.2008.065029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Suzuki T, Mitsuishi Y, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–6. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui T, Chano T, Takikita-Suzuki M, et al. Association of p62/SQSTM1 Excess and Oral Carcinogenesis. Plos One. 2013;8 doi: 10.1371/journal.pone.0074398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JY, Ke H, Hall RP, et al. c-Jun promotes whereas JunB inhibits epidermal neoplasia. J Invest Dermatol. 2011;131:1149–58. doi: 10.1038/jid.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen C, Kragballe K, Rasmussen M, et al. Activator protein 1 DNA binding activity is decreased in lesional psoriatic skin compared with nonlesional psoriatic skin. Brit J Dermatol. 2004;151:600–7. doi: 10.1111/j.1365-2133.2004.06088.x. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulski JK, Kenworthy W, Bellgard M, et al. Gene expression profiling of Japanese psoriatic skin reveals an increased activity in molecular stress and immune response signals. J Mol Med-Jmm. 2005;83:964–75. doi: 10.1007/s00109-005-0721-x. [DOI] [PubMed] [Google Scholar]
- Kuo WL, Sharifi MN, Lingen MW, et al. p62/SQSTM1 accumulation in squamous cell carcinoma of head and neck predicts sensitivity to phosphatidylinositol 3-kinase pathway inhibitors. PLoS One. 2014;9:e90171. doi: 10.1371/journal.pone.0090171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HM, Shin DM, Yuk JM, et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol. 2011;186:1248–58. doi: 10.4049/jimmunol.1001954. [DOI] [PubMed] [Google Scholar]
- Li B, Tsoi LC, Swindell WR, et al. Transcriptome Analysis of Psoriasis in a Large Case-Control Sample: RNA-Seq Provides Insights into Disease Mechanisms. J Invest Dermatol. 2014 doi: 10.1038/jid.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LJ, Shen C, Nakamura E, et al. SQSTM1 Is a Pathogenic Target of 5q Copy Number Gains in Kidney Cancer. Cancer Cell. 2013;24:738–50. doi: 10.1016/j.ccr.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, Kang Y, Zhao R, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–20. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cut adapt removes adapter sequences from high-throughput sequencing reads, EMBnet. EMBnet. 2011;17:10–2. [Google Scholar]
- Mathelier A, Wasserman WW. The next generation of transcription factor binding site prediction. Plos Comput Biol. 2013;9:e1003214. doi: 10.1371/journal.pcbi.1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meixner A, Zenz R, Schonthaler HB, et al. Epidermal JunB represses G-CSF transcription and affects haematopoiesis and bone formation. Nat Cell Biol. 2008;10:1003–11. doi: 10.1038/ncb1761. [DOI] [PubMed] [Google Scholar]
- Park CC, Kim KJ, Woo SY, et al. Comparison of the Expression Profile of JunB, c-Jun, and S100A8 (Calgranulin A) in Psoriasis Vulgaris and Guttate Psoriasis. Ann Dermatol. 2009;21:35–8. doi: 10.5021/ad.2009.21.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhitko A, Myachina F, Morrison TA, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Pro Natl Acad Sci USA. 2011;108:12455–60. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paukkonen K, Naukkarinen A, Horsmanheimo M. The development of manifest psoriatic lesions is linked with the appearance of ICAM-1 positivity on keratinocytes. Arch Dermatol Res. 1995;287:165–70. doi: 10.1007/BF01262326. [DOI] [PubMed] [Google Scholar]
- Plant D, Young HS, Watson RE, et al. The CX3CL1-CX3CR1 system and psoriasis. Exp Dermatol. 2006;15:900–3. doi: 10.1111/j.1600-0625.2006.00486.x. [DOI] [PubMed] [Google Scholar]
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512–6. doi: 10.1016/j.jaad.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Rea SL, Majcher V, Searle MS, et al. SQSTM1 mutations--bridging Paget disease of bone and ALS/FTLD. Exp Cell Res. 2014;325:27–37. doi: 10.1016/j.yexcr.2014.01.020. [DOI] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, et al. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–86. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumigray KD, Foote HP, Lechler T. Noncentrosomal microtubules and type II myosins potentiate epidermal cell adhesion and barrier formation. J Cell Biol. 2012;199:513–25. doi: 10.1083/jcb.201206143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram K, Shanmugarajan S, Rao DS, et al. Mutant p62P392L stimulation of osteoclast differentiation in Paget’s disease of bone. Endocrinology. 2011;152:4180–9. doi: 10.1210/en.2011-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Duncan D, Shi Z, et al. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten MW, Geetha T, Seibenhener ML, et al. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625–9. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- Wullaert A, Bonnet MC, Pasparakis M. NF-kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011;21:146–58. doi: 10.1038/cr.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara N, Takagi A, Ueno T, et al. Inverse correlation between microtubule-associated protein 1A/1B-light chain 3 and p62/sequestosome-1 expression in the progression of cutaneous squamous cell carcinoma. J Dermatol. 2014;41:311–5. doi: 10.1111/1346-8138.12439. [DOI] [PubMed] [Google Scholar]
- Zenz R, Eferl R, Kenner L, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–75. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005a;33:W741–W8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Green CL, Tao S, et al. NF-kappaB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 2004;18:17–22. doi: 10.1101/gad.1160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Tao S, Kimmel R, et al. CDK4 regulation by TNFR1 and JNK is required for NF-kappaB-mediated epidermal growth control. J Cell Biol. 2005b;168:561–6. doi: 10.1083/jcb.200411060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotti T, Scudiero I, Settembre P, et al. TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Mol Immunol. 2014;58:27–31. doi: 10.1016/j.molimm.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.