

Abstract
We present the synthesis of a series of six new glycoluril derived molecular clips and acyclic CB[n]-type molecular containers (1 – 3) that all feature SO3− solubilizing groups but differ in the number of glycoluril rings between the two terminal dialkoxyaromatic sidewalls. We report the x-ray crystal structure of 3b which shows that its dialkoxynaphthalene sidewalls actively define a hydrophobic cavity with high potential to engage in π–π interactions with insoluble aromatic guests. Compounds 1 – 3 possess very good solubility characteristics (≥ 38 mM) and undergo only very weak self-association (Ks < 92 M−1) in water. The weak self-association is attributed to unfavorable SO3−•••SO3− electrostatic interactions in the putative dimers 12 – 42. Accordingly, we created phase solubility diagrams to study their ability to act as solubilizing agents for four water insoluble drugs (PBS-1086, camptothecin, β-estradiol, and ziprasidone). We find that the containers 3a and 3b which feature three glycoluril rings between the terminal dialkoxy-o-xylylene and dialkoxynaphthalene sidewalls are less efficient solubilizing agents than 4a and 4b because of their smaller hydrophobic cavities. Containers 1 and 2 behave as molecular clip type receptors and therefore possess the ability to bind to and thereby solubilize aromatic drugs like camptothecin and ziprasidone, and PBS-1086.
Introduction
A ubiquitous problem facing the pharmaceutical industry is that an estimated 40–70% of new drug candidates are so poorly soluble that they cannot be formulated on their own.1 Accordingly, there is a real need for the development of new tools that improve the solubility of poorly soluble drugs that enable their formulation. To date, a wide variety of methods that enhance the rate and extent of dissolution have been developed including solid dispersions and nanocrystalline solid forms of the drug.2 Other methods that have been demonstrated to improve solubility include the preparation of salts, higher solubility prodrugs, dendrimer-drug systems, and designed co-crystalline forms of the drug.3 However, the most attractive approach to improving solubility of insoluble drugs from the point of view of supramolecular chemists relies on the use of cyclodextrin molecular containers (e.g. HP-β-CD, SBE-β-CD, Figure 1) as solubilizing excipients.4 Cyclodextrin derivatives are currently used to formulate a number of drugs that are administered to humans.
Figure 1.

Structures of solubilizing molecular containers.
In recent years, we and others, have been actively investigating the synthesis, molecular recognition properties, and applications of an alternative class of molecular container compounds known as cucurbit[n]urils (CB[n], n = 5, 6, 7, 8, 10, 14; Figure 1).5 The great interest in CB[n] compounds arises from the availability of a homologous series of hosts that display both high affinity (Ka up to 1017 M−1) and high selectivity toward their guests in water.6 In addition, the inherent stimuli responsiveness (pH, chemical, electrochemical, photochemical) of CB[n]•guest complexes have made CB[n] popular components to create functional systems including molecular machines, chemical sensors, affinity capture phases, and gas purification materials.7 Accordingly, workers in the CB[n] field envisioned that CB[n] compounds – particularly CB[7] with its good water solubility – might be good substitutes for cyclodextrins in pharmaceutical applications. In recent years, the macrocyclic CB[n] compounds were demonstrated to have low in vitro and in vivo toxicity8 and have been used to solubilize, protect, activate, and deliver pharmaceutical agents.9 CB[8] has been used as a glue to create polymer hydrogels and nanocapsules based on ternary complex formation.10 Derivatives of macrocyclic CB[n] have even been used for targeted drug delivery applications.11
Over the years, both the Isaacs and Sindelar groups have worked toward an improved understanding of the mechanism of CB[n] formation – especially with regard to the intermediate methylene bridged glycoluril dimers and higher oligomers12 – and the preparation of new members of the cucurbit[n]uril family. These studies have resulted in the preparation of glycoluril dimer based molecular clips,13 CB[n] analogues,14 inverted CB[n],15 nor-seco-CB[n],16 bambus[n]urils,17 CB[n] derivatives,11a,12e,18 CB[n] dimers,19 and acyclic CB[n]-type receptors.12d,12g,12j,20 Recently, the Isaacs group used acyclic CB[n] containers as solubilizing excipients21 for insoluble drugs and reported the influence of the nature of the solubilizing groups22 and aromatic sidewalls23 on their function. In this paper we explore the influence of the length of the central glycoluril oligomer on their ability of acyclic CB[n]-type receptors 1 – 4 to solubilize four insoluble drugs (Figure 2).
Figure 2.
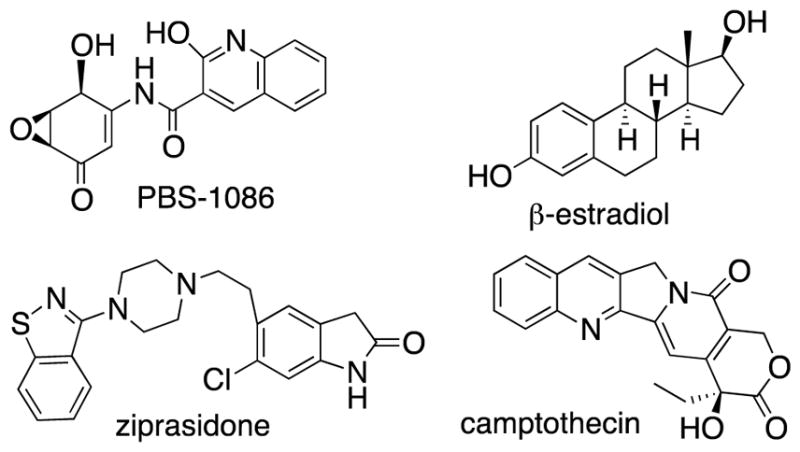
Structures of insoluble drugs used in this study.
Results and discussion
This results and discussion section is organized as follows. First, we present the synthesis of six new containers (1 – 3), studies of their inherent solubility, and studies of their tendency toward self-association. Next, we present the x-ray crystal structure of 3b and compare it to the x-ray crystal structures of previously reported glycoluril monomer, dimer, and tetramer derived containers. Next, we report the ability of 1 – 3 to act as solubilizing agents for four insoluble drugs. Finally, we compare the trends in the solubilization data for 1 – 3 with that of previously reported 4a and 4b.
Synthesis of Containers 1 – 3
Scheme 1 shows the synthesis of new compounds 1 – 3. For the preparation of molecular clips 1a and 1b we followed the path blazed by Nolte and coworkers and allowed glycoluril bis(cyclic ether) 1CE to react with the corresponding dialkoxyaromatic walls (5 and 6) at 70 °C in TFA/Ac2O to deliver molecular clips 1a and 1b in high yield.21,24 To prepare molecular clips 2a and 2b we first transformed the known glycoluril dimer 2NH25 into bis(cyclic ether) 2CE by treatment with paraformaldehyde in TFA at reflux (40%). Subsequently, we reacted 2CE with 5 or 6 in TFA/Ac2O at 70 °C to yield molecular clips 2a and 2b in good yield. For the preparation of acyclic CB[n] compounds 3a and 3b we first needed to prepare the glycoluril trimer 3CE. The preparation of 3CE was accomplished by reaction of glycoluril (7) with 1CE in methanesulfonic acid with careful temperature control (10 °C for 2h and then 23 °C for 2h). Subsequently, we allowed 3CE to react with 5 or 6 in TFA/Ac2O at 70 °C to give 3a(48%) and 3b (59%) in good yields.
Scheme 1.

Synthesis of new containers 1 – 3. Conditions: a) TFA/Ac2O (1:1), 70 °C, 5; b) TFA/Ac2O (1:1), 70 °C, 6; c) TFA, paraformaldehyde, reflux; d) 1CE, MeSO3H, 10°C to 23 °C.
We were fortunate to obtain the x-ray crystal structure of acyclic CB[n]-type receptor 3b (Figure 3). Figure 3 also shows the x-ray crystal structure of previously prepared molecular clips 1ester and 2ester (Figure 4) and acyclic CB[n]-type receptor 4b to illustrate the geometrical change that occurs across the homologous series of receptors 1 – 4 upon elongation of the glycoluril oligomer backbone. As can be readily seen, as one increases the number of glycoluril rings the receptors change from molecular clips with divergent to nearly parallel aromatic sidewalls (1 and 2) to containers with a well defined hydrophobic cavity despite their acyclic nature (3 and 4). Overall, as a result of the glycoluril trimer backbone, 3b is C-shaped and the two naphthalene rings help to define a cavity that is occupied by a molecule of acetone in the crystal. The mean planes of the aromatic rings of 3b are oriented at an average angle of 65.5° (range over 4 independent molecules in the crystal: 110.6° to 116.6°) and do not undergo either π–π stacking or edge-to-face CH-π interactions with each other. For example, molecular clips based on glycolurils (e.g. relatives of 1) feature outwardly oriented aromatic rings (35.9° interplanar angle) whereas those based on glycoluril dimers (e.g. relative of 2) feature aromatic rings that are close to parallel to one another (18.3° interplanar angle). In contrast, the x-ray crystal structure of 4b reported previously shows an 113.3° angle between the naphthalene rings and direct CH-π interactions which helps to define a hydrophobic box.
Figure 3.

Cross eyed stereoviews of the x-ray crystal structures of: a) 1ester,26 b) 2ester,27 c) 3b, d) 4b.21 Color code: C, gray; H, white; N, blue; O, red; S, yellow.
Figure 4.
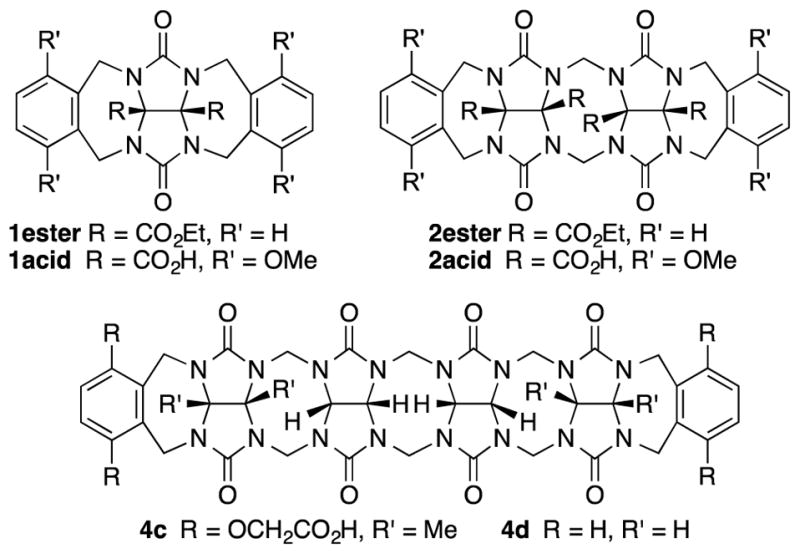
Structures of comparison compounds.
The packing of 3b in the crystal is intriguing (Figure 5). Analogous to what is observed for molecular clips based on glycoluril or glycoluril dimers,28 the individual molecules of 3b interact with one another via CH-π and π–π interactions between their naphthalene sidewalls in a head-to-tail fashion to yield linear tape like assemblies. The distance between the mean planes of the naphthalene rings is averages 3.66 Å (range 3.60 to 3.78 Å). These tapes extend along both the b-axis and the c-axis; they are alternately arranged in a criss-cross fashion reminiscent of building method of a log cabin home as one extends along the a-axis. The channels defined by the packing of the linear assemblies are filled with the solubilizing arms and solvating water molecules.
Figure 5.
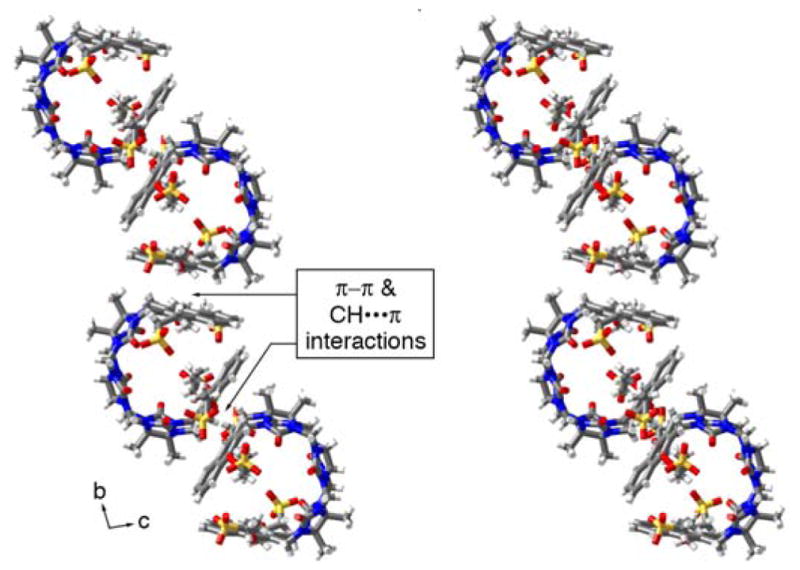
Illustration of the packing of molecules of 3b into linear assemblies along the b-axis. Color code: C, gray; H, white; N, blue; O, red; S, yellow.
Inherent Solubility of 1 – 4
Given our interest in assessing the performance of 1 – 3 as solubilizing excipients for insoluble drugs we first sought to measure their inherent solubility. For this purpose, we stir an excess of container with 20 mM phosphate buffered D2O and readjust the pD to 7.4 until equilibrium is reached and then remove excess insoluble container by centrifugation and filtration. An aliquot of the filtrate is diluted by a known factor and combined with a solution of benzene-1,3,5-tricarboxylic acid as internal standard (1.00 mM); the relative integrals of diagnostic resonances in the 1H NMR spectrum for the container (1a: 6.87 ppm, 1b: 7.56 ppm, 2a: 6.99 ppm, 2b: 7.84 ppm, 3a: 7.02 ppm, 3b: 7.95 ppm) versus those of the internal standard (8.38 ppm) were used to determine the inherent solubility of each container. The results are summarized in Table 1. As can be readily seen, the solubility of all the containers bearing dialkoxy-o-xylylene sidewalls is higher than 100 mM which is advantageous for their use as solubilizing excipients. In contrast, the solubility of containers with naphthalene sidewalls are variable. Containers 2b and 3b display high solubility (> 300 mM) whereas 1b and 4b are modest (38 and 14 mM, respectively).
Table 1.
Inherent solubility values of 1 – 4 and their self-association constant Ks determined in 20 mM sodium phosphate buffer, pD 7.4.
| 1a | 1b | 2a | 2b | 3a | 3b | 4aa) | 4ba) | |
|---|---|---|---|---|---|---|---|---|
| Solub. (mM) | 466 | 38 | 396 | 343 | 102 | 336 | 105 | 14 |
| Ks (M−1) | 30 | 92 | 12 | 6 | 3 | 49 | 47 | 624 |
Values from the literature.21
Containers 1 – 3 Do Not Self-associate Strongly
In order for molecular containers to have high potential for use as solubilizing excipients they must not be strongly self-associated which would reduce their ability to form container•drug complexes. Accordingly, we studied the self-association of 1 – 3 by preparing solutions of different concentrations of 1 – 3 and monitoring the changes in the 1H NMR chemical shifts. The chemical shifts of the protons on the aromatic sidewall were particularly sensitive to self-association. Figure 6 shows the changes in chemical shift of Ha, Hb, and Hc as a function of [2a]. The changes in chemical shift were fitted to the standard two-fold self-association model21,29 implemented within Scientist 3.0™ (Supporting Information) which yielded a self-association constant (Ks) of 12 M−1. Table 1 gives the self-association constants measured for 1 – 3 (Supporting Information) and also presents the known values for 4 from the literature21 for comparison. As expected, the Ks values measured for acyclic CB[n]-type receptors 3a and 3b are quite low (< 50 M−1) which is advantageous for their use as solubilizing agents. Given the well-known propensity for glycoluril and glycoluril dimer derived molecular clips to undergo self-association,13a,26,28a,28d,30 we were surprised that the Ks values for 1 and 2 were low (< 100 M−1). For purposes of comparison, the structures and self-association constants measured previously for compounds 1acid (Ks = 1840 M−1)26 and 2acid (Ks = 41700 M−1)13a which differ from 1a and 2a in the spatial orientation of their solubilizing groups are shown in Figure 4. Because the CO2− solubilizing groups of 1acid and 2acid are on their convex face, unfavorable carboxylate-carboxylate electrostatic interactions are avoided upon formation of dimers 1acid2 and 2acid2. In contrast, the SO3− groups of 1 – 4 are directed toward each other within the putative dimeric species 12 – 42 which results in unfavorable electrostatic sulfonate-sulfonate interactions which decreases the propensity of 1 – 4 to dimerize. The self-association constant of 1a (2a) is 61-fold (3500-fold) lower than that measured for 1acid (2acid) which amounts to a destabilization of the dimer by 2.4 (4.8) kcal mol−1 which we attribute to electrostatics. The recognition of the importance of electrostatic interactions that discourage self-association of 1 – 4 provides a rationale for the superior binding constants observed previously for 4a and 4c relative to 4d12g,12j which
Figure 6.
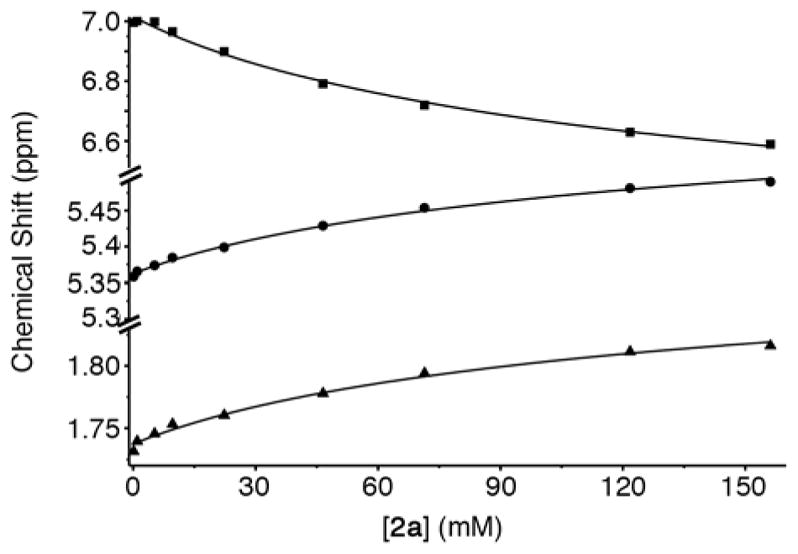
Plot of chemical shift versus [2a]. The solid line represents the best global fit of the data to a two-fold self-association model with Ks = 12 M−1. Conditions: 20 mM sodium phosphate buffered D2O, pD 7.4, room temperature. Key: Ha, ■; Hb, ●; Hc, ▲.
| (1) |
lacks the anionic solubilizing groups which translates into the superior solubilizing abilities of 4a and 4b.23
Construction of Phase Solubility Diagrams for 1 – 3 with Insoluble Drugs
In order to assess the ability of 1 – 3 as solubilizing agents for insoluble drugs we created phase solubility diagrams (PSD)29,31 for the six new containers 1 – 3. Phase solubility diagrams are plots of concentration of container on the x-axis versus concentration of solubilized poorly soluble drug on the y-axis. Several types of PSDs are possible (AL, AP, AN; Figure 7) although linear (AL-type) PSDs are most common for cyclodextrin molecular containers. Containers that display AL-type PSDs behave according to equation 1 where S0 is the inherent solubility of the drug, Ka is the binding constant for the container•drug complex, and slope is the slope of the PSD.29 In this paper, we generally observed AL-type PSDs except for 2a and 2b with ziprasidone and 2a with PBS-1086 which displayed AP-type behaviour. We consider PSDs with slopes ≥0.5 (e.g. a 50 mM solution of container solubilizes 25 mM drug) as indication that a given container is a very good solubilizing agent for a given drug. If we substitute slope = 0.5 into equation 1 then it is easy to show that Ka × S0 = 1.23 Alternatively, if we want to solubilize a drug with inherent solubility of 1 × 10−5 M (1 × 10−6 M) then the container must display a binding constant of 1 × 105 M−1 (1 × 106 M−1) to achieve a slope of 0.5.
Figure 7.
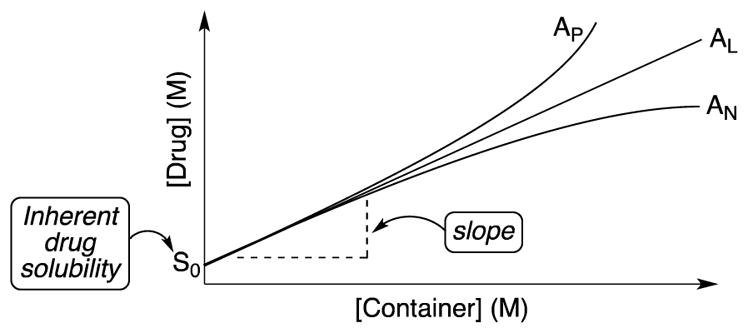
Idealized phase solubility diagrams.
In this section we report the phase solubility behavior of 1 – 3 with four insoluble drugs (camptothecin, PBS-1086, β-estradiol, and ziprasidone) which we have previously studied with 4a and 4b.21,23 To create the PSDs we prepare a solution of container of known concentration and stir it with an excess of solid insoluble drug. After equilibrium is reached, the excess of insoluble drug is removed by centrifugation and filtration. The concentration of drug in the filtrate is determined by 1H NMR spectroscopy after addition of benzene-1,3,5- tricarboxylic acid (1.00 mM) as a non-binding internal standard using the relative integrals of diagnostic resonances of the drug versus those of the internal standard. Figure 8a shows the PSDs for solutions of containers 1 – 4 with the insoluble drug β-estradiol. As can be readily seen most of the PSDs are of the AL-type and it is therefore appropriate to perform a linear fitting of the data to determine the value of the slope. The slope values range from 0 to 0.92 for 4b which indicates that the affinity of the various containers toward β-estradiol differ significantly. Next, we substituted the measured slope values and the known inherent solubilities (S0)23 into equation 1 to determine the affinity constant (Ka, M−1) for the interaction of each container•β-estradiol complex (Table 2). In Table 2, the values of slope and Ka and their uncertainty are given to two significant figures which reflects the limitations of phase solubility measurements. Similar experiments were performed for camptothecin with containers 1 – 3 and compared with the data previously reported for 4 (Figure 8b, c and Table 2).21,23 Figure 8c shows that the PSD for 4a and camptothecin displays a plateau region above [4a] = 10 mM which reflects the limited solubility of the 4a•camptothecin complex. The data points shown in red (Figure 8c) are excluded from the linear fitting used to calculate slope and Ka. Using the same procedures, we generated PSDs for systems comprising containers 1 – 3 with ziprasidone and the developmental anti-cancer agent PBS-108632 and calculated Ka values (Table 1 and Supporting Information) for the container•drug complexes.
Figure 8.

Phase solubility diagrams created for: a) 1 – 4 with β-estradiol, b) 1b and 4b with camptothecin, and c) 1a, 2, 3, and 4a with camptothecin. Conditions: 20 mM sodium phosphate buffer, room temperature, pD 7.4. Symbols: 1a, ◆; 1b, ◇; 2a, ■; 2b, □; 3a, ▲; 3b, △; 4a, ●; 4b, ○. Data points colored red were not used for linear fitting.
Table 2.
Inherent solubility (S0, μM) and slope values calculated from the linear region of the PSDs for containers 1 – 4 with the four poorly soluble drugs. The corresponding Ka (M−1) values were calculated using equation 1.
| Camptothecin | Ziprasidone | PBS-1086 | β-estradiol | ||
|---|---|---|---|---|---|
| S0 | 54±3.9 | 63±3.5 | 4.5±0.90 | 8.8±0.42 | |
|
| |||||
| 1a | slope | - | - | - | - |
| Ka | - | - | - | - | |
|
| |||||
| 1b | slope | 0.54±0.032 | - | - | 0.022±0.0026 |
| Ka | 2.2(±0.16)×104 | - | - | 2.6(±0.32)×103 | |
|
| |||||
| 2a | slope | 0.16±0.0043 | NL | NL | - |
| Ka | 3.5(±0.27)×103 | - | - | - | |
|
| |||||
| 2b | slope | 0.087±0.0012 | NL | 0.046±0.0040 | - |
| Ka | 1.8(±0.13)×103 | - | 1.1(±0.24)×104 | - | |
|
| |||||
| 3a | slope | 0.045±0.0051 | 0.070±0.012 | 0.032±0.0018 | 0.013±0.0011 |
| Ka | 8.7(±1.2)×102 | 1.2(±0.22)×103 | 7.4(±1.5)×103 | 1.5(±0.14)×103 | |
|
| |||||
| 3b | slope | 0.10±0.0051 | 0.38±0.026 | 0.14±0.0056 | 0.11±0.0058 |
| Ka | 2.0(±0.18)×103 | 9.8(±0.95)×103 | 3.6(±0.74)×104 | 1.4(±0.10)×104 | |
|
| |||||
| 4a a) | slope | 0.14±0.0070 | 1.1±0.19 | 0.71±0.027 | 0.35±0.019 |
| Ka | 2.9(±0.26)×103 | TL | 5.5(±1.2)×105 | 6.2(±0.48)×104 | |
|
| |||||
| 4ba) | slope | 1.1 ± 0.059 | 0.43±0.052 | 0.89 ± 0.0043 | 0.92±0.053 |
| Ka | TL | 1.2(±0.20)×104 | 1.9(±0.38)×106 | 1.3(±0.86)×106 | |
Interpretation of the Trends in the Ka values and Solubilization Efficiency
This section discusses the trends in the Ka values as a function of the structural variables of the containers and the guests.
Influence of the Number of Glycoluril Rings
As shown in Figure 3, the crystal structures of 1 – 4 demonstrate that compounds 1 and 2 are molecular clips with divergent and slightly convergent aromatic sidewalls, respectively. In contrast, containers 3 and 4 are more properly defined as acyclic CB[n]-type receptors because they feature more fully formed ureidyl C=O portals and closed cavities that are fully defined by the glycoluril rings and the aromatic sidewalls. Accordingly, it is appropriate to compare the solubilization properties of 1 and 2 and separately, 3 and 4. We find that container 1a is uniformly unsuccessful as a solubilizing agent, whereas container 2a is able to solubilize camptothecin, ziprasidone, and PBS-1086. We attribute the better solubilizing ability of 2a to the more nearly parallel alignment of its aromatic walls (centroid to centroid distance = 7.40 Å) which preorganizes it to engage in π–πinteractions with drugs that contain aromatic rings. In contrast, the behavior of containers 1b and 2b toward the four drugs is drug dependent. For example, container 1b performs better for camptothecin and β-estradiol but 2b performs better for ziprasidone and PBS-1086. In this regard it is noteworthy that the PSDs for 2a and 2b with ziprasidone are non-linear which is indicative of higher order containern•drug complexes. The behavior of acyclic CB[n]-type containers 3 and 4 toward the four drugs exhibit clear trends. For example, container 3a binds less strongly (Ka ≈ 103 – 104 M−1) to all four drugs than 4a does (2.9 × 103 < Ka < 5.5 × 105 M−1). We attribute this result to the larger hydrophobic cavity of 4a relative to 3a and the more fully formed ureidyl C=O portals of 4a which results in stronger ion-dipole interactions with cationic drugs (e.g. ziprasidone). Similarly, container 3b (2.0 × 103 < Ka < 3.6 × 104 M−1 does not perform as well as 4b (1.2 × 104 < Ka < 1.9 × 106 M−1) as a solubilizing agent for these four drugs. Accordingly, we conclude that container 4 is a more efficient solubilizing agent that container 3 although the relative in vivo efficacy of drugs formulated by the different containers will depend on the ability of the drugs to be released by dilution and competition from endogenous cationic small molecules (e.g. spermine).33
Influence of Aromatic Sidewall
The influence of the nature of the aromatic sidewall (e.g. benzene versus naphthalene) is clear cut for containers 1, 3, 4. In most cases the containers with the longer aromatic sidewall (e.g. naphthalene) display higher Ka values toward the drugs. For example, container 1b binds nicely (Ka = 2.2 × 104 and 2.6 × 103 M−1) toward camptothecin and β-estradiol whereas the binding of 1a to these drugs could not be detected. Similarly, the ratio of the Ka values of 3b versus 3a toward camptothecin (2.3), ziprasidone (8.2), PBS-1086 (4.9), and β-estradiol (9.3) indicate that 3b is a significantly better host than 3a. Containers 4b and 4a display a similar trend with the ratio of Ka values for 4b versus 4a toward camptothecin (>6.4), PBS-1086 (3.5), and β-estradiol (21). Interestingly, ziprasidone which is both narrow and cationic at pD 7.4 prefers to bind to the smaller cavity of container 4a relative to 4b. We conclude that the containers with substituted naphthalene sidewalls generally outperform those with o-xylylene sidewalls because they possess larger hydrophobic cavities which should increase the number of solvating H2O molecules expelled upon binding6d,34 and result in the formation of π–π interactions between larger π-surfaces particularly for insoluble drugs that contain aromatic rings.
Conclusions
In summary, we have reported the synthesis of a series of molecular container compounds (1 – 3) that differ in the number of glycoluril rings that connect the two terminal substituted o-xylylene or naphthalene sidewalls. Compounds 1 – 3 display very good aqueous solubility (> 38 mM) and somewhat surprisingly do not undergo significant self-association in aqueous solution (Ks ≤ 92 M−1). We trace the low values of Ks observed for 1 and 2 to unfavorable sulfonate-sulfonate electrostatic interactions (2.4 to 4.8 kcal mol−1) that would occur in the putative dimeric complexes 12 and 22. The x-ray crystal structure of acyclic CB[n] type receptor 3b shows a cavity shaped by its two naphthalene walls that do not engage in π–π or CH•••π interactions. PSDs were created for 1 – 3 with four insoluble drugs (camptothecin, ziprasidone, PBS-1086, and β-estradiol) and compared with the PSDs measured previously for 4a and 4b. We find that the acyclic CB[n]-type containers containing larger numbers of glycoluril rings (e.g. 4) and the larger naphthalene sidewalls (e.g. 4a versus 4b) are generally superior solubilizing agents for insoluble drugs. For the molecular clip receptors 1 and 2 the results are less clear cut although the naphthalene walled compounds often display higher Ka values than the o-xylylene walled analogues and more often exhibit the desirable AL-type PSDs. When combined with our previous studies on the influence of the nature of the solubilizing group (e.g. anionic versus neutral versus cationic) and the aromatic sidewalls (benzene, two isomeric naphthalenes, alkylated variants), this study allows us to conclude that compounds 4a and 4b are most efficient as solubilizing agents for insoluble drugs and are best positioned for further development as solubilizing excipients for real world pharmaceutical applications. In this regard the higher values of Ka exhibited by acyclic CB[n]-type receptors compared to HP-β-cyclodextrin promises to broaden the range of drugs that can be efficiently solubilized using molecular container technology and to do so at lower concentrations of container.
Experimental
General Experimental
Starting materials were purchased from commercial suppliers and were used without further purification or were prepared by literature procedures. Compounds 1CE and 2NH were prepared according to literature procedures.21,25 Melting points were measured on a Meltemp apparatus in open capillary tubes and are uncorrected. IR spectra were measured on a JASCO FT/IR 4100 spectrometer and are reported in cm−1. NMR spectra were measured on commercial spectrometers operating at 600 MHz and 400 MHz for 1H and 125 MHz for 13C. Mass spectrometry was performed using a JEOL AccuTOF electrospray instrument (ESI) or on a Bruker 12T Apex IV FT-ICR mass spectrometer.
Compound 1a
A solution of dimethylglycoluril bis(cyclic ether) 1CE (0.650 g, 2.56 mmol) in TFA (7 mL) was mixed with compound 5 (3.96 g, 10.2 mmol). The mixture was stirred and heated at 70 °C for 3 h and then was poured into MeOH (70 mL). The solid was collected by filtration and was dried under high vacuum. The crude solid was recrystallized two times from a mixture of water and acetone (1:2, v/v, 20 mL). The solid was dissolved in water (10 mL) and adjusted to pH = 7 by adding 1 M aqueous NaOH. The solvent was removed by rotary evaporation and then the solid was further dried under high vacuum to yield compound 1a (2.16 g, 83%) as a white solid. M.p. > 280 °C. IR (ATR, cm−1): 2925w, 1684s, 1477s, 1437m, 1355w, 1311m, 1261s, 1195s, 1095m, 1051s, 800m, 768m, 616m, 532m. 1H NMR (600 MHz, D2O): δ 6.85 (s, 4H), 5.18 (d, J = 16.2, 4H), 4.14 (d, J = 16.2, 4H), 4.05 – 3.90 (m, 8H), 3.15 – 2.95 (m, 8H), 2.20 – 2.05 (m, 8H), 1.77 (s, 6H). 13C NMR (125 MHz, D2O, 1, 4-dioxane as internal reference): δ 157.2, 149.4, 127.6, 114.4, 78.2, 68.2, 47.4, 34.4, 23.2, 15.0 (10 out of the 12 expected resonances are observed). High-Res MS (ESI): m/z 925.1607 ([M – 4Na + 3H]−, C34H45N4O18S4, calculated for 925.1612)
Compound 1b
A solution of dimethylglycouril bis(cyclic ether) 1CE (0.650 g, 2.56 mmol) in TFA (7 mL) was mixed with compound 6 (4.57 g, 10.2 mmol). The mixture was stirred and heated at 70 °C for 3 h and then was poured into MeOH (70 mL). The solid was collected by filtration and was dried under high vacuum. The crude solid was recrystallized twice from a mixture of water and acetone (1:2, v/v, 20 mL). The solid was dissolved in water (10 mL) and adjusted to pH = 7 by adding 1 M aqueous NaOH. The solvent was removed by rotary evaporation and then the solid was further dried under high vacuum to yield compound 1b (2.60 g, 91%) as a white solid. M.p. > 280 °C. IR (ATR, cm−1): 2939w, 2858w, 1690m, 1468s, 1426w, 1344m, 1307w, 1181s, 1047s, 1029s, 952w, 766m, 738m. 1H NMR (600 MHz, D2O): δ 8.00 – 7.95 (m, 4H), 7.60 – 7.55 (m, 4H), 5.20 (d, J = 16.2, 4H), 4.39 (d, J = 16.2, 4H), 3.65 – 3.55 (m, 8H), 2.95 – 2.75 (m, 8H), 2.15 – 2.05 (m, 8H), 1.81 (s, 6H). 13C NMR (125 MHz, D2O, 1, 4-dioxane as internal reference): δ 157.1, 147.8, 126.8, 125.9, 121.3, 78.2, 73.7, 47.6, 36.0, 24.6, 15.5 (11 out of the 14 expected resonances were observed). High-Res MS (ESI): m/z 1091.1392 ([M – Na]−, C42H46N4Na3O18S4, calculated for 1091.1377).
Compound 2CE
Compound 2NH (2.12 g, 5.81mmol) was dissolved in TFA (58 mL). Paraformaldehyde (872 mg, 29.04 mmol) was added and the reaction mixture stirred and heated at 75–80 °C for 20 h in a pre-heated oil bath. The reaction mixture was poured into MeOH (400 mL) and the resulting precipitate was filtered and washed with MeOH. Compound 2CE was obtained as white solid (1.04 g, 2.33 mmol, 40%). M.p. > 300°C. IR (ATR, cm−1): 2999w, 2960w, 2875w, 1722s, 1468m, 1440s, 1376m, 1302s, 1232m, 1093m, 1067m, 1001m, 949m, 917m, 881m, 768m, 737m, 668w. 1H NMR (400 MHz, DMSO-d6): 5.47 (d, J = 15.8, 2H), 5.15 (d, J = 11.0, 4H), 4.87 (d, J = 11.0, 4H), 4.33 (d, J = 15.8, 2H), 1.83 (s, 6H), 1.65 (s, 6H). 13C NMR (125 MHz, DMSO-d6): 154.5, 76.4, 72.2, 70.6, 44.3, 17.9, 16.7. HR-MS (ESI): m/z 449.1891 ([M + H]+), C18H24N8O6, calcd. 449.1897.
Compound 3CE
Compound 7 (1.196 g, 8.4 mmol) was dissolved in methane sulfonic acid (40 mL) under N2 atmosphere. The solution was cooled to 8–12 °C in an ice bath and then 1CE (3.996 g, 15.7 mmol) was added at once. The reaction mixture was stirred at 8–12 °C for 2 h and then the ice bath was removed at the reaction was stirred at room temperature for another 2 h. The reaction was poured into precooled (5 °C) acetone (700 mL) and the resulting precipitate was obtained by filtration. The solid was washed with about 100 mL acetone and then transferred while still wet into a mixture of acetonitrile and water (1:1 (v:v), 100 mL) and sonicated. The resulting precipitate was filtered and suspended in formic acid (2 mL). Finally the solid was thoroughly dried on the frit, washed with water and methanol and dried under vacuum to give 3CE as a white solid (204 mg, 0.332 mmol, 4%). M.p. > 300 °C. IR (ATR, cm−1): 3551w, 3455w, 3384w, 2990w, 2949w, 1718s, 1702s, 1656w, 1465m, 1425m, 1315s, 1271m, 1239s, 1181m, 1074m, 1013s, 918m, 851s, 788m, 735m, 663w. 1H NMR (400 MHz, DMSO-d6, 30 °C): 5.58 – 5.52 (m, 6H), 5.16 (d, J = 10.9 Hz, 2H), 4.85 (d, J = 11.0 Hz, 4H), 4.24 (d, J = 15.1 Hz, 4H), 1.81 (s, 6H), 1.64 (s, 6H). 13C NMR (125 MHz, DMSO-d6): δ 155.2, 154.8, 154.7, 77.1, 72.4, 70.5, 69.9, 48.5, 17.7, 15.7. HR-MS (ESI): m/z 625.2363 ([M + H]+), C24H30N12O8, calcd. 615.2388.
Compound 2a
Compound 2CE (448 mg, 1.0 mmol) was dissolved in TFA/Ac2O (1:1 (v:v), 8.0 mL). Compound 5 (916 mg, 2.30 mmol) was added. The reaction was stirred and heated in a preheated oil bath for 3h at 70 °C. Then the mixture was poured into acetone (75 mL). The precipitate was collected by centrifugation. The solid was dissolved in water (10 mL) and precipitated by the addition of EtOH (75 mL). The precipitate was collected by centrifugation, then redissolved in water (40 mL) and reprecipitated by the addition of acetone (120 mL). The precipitate was collected by filtration, dissolved in water and the pH of the solution adjusted to 7 by addition of 1 M aqueous NaOH. The solvent was removed under reduced pressure and the residue dried under vacuum. Compound 2a was obtained as brownish solid (533 mg, 0.475 mmol, 48%). M.p. > 300 °C. IR (ATR, cm−1): 3450w, 2946w, 2876w, 1706s, 1464s, 1391w, 1298m, 1179s, 1039s, 883w, 800m, 754m, 657w, 594m. 1H NMR (400 MHz, D2O): 6.98 (s, 4H), 5.35 (d, J = 16.2, 2H), 5.22 (d, J = 16.2, 4H), 4.32 (d, J = 16.2, 2H), 4.11 (d, J = 16.2, 4H), 4.10 – 4.00 (m, 4H), 3.90 – 3.80 (m, 4H), 3.20 – 3.00 (m, 8H), 2.25 – 2.10 (m, 8H), 1.74 (s, 6H), 1.67 (s, 6H). 13C NMR (125 MHz, D2O, 1,4-dioxane as internal reference): δ 156.5, 150.9, 128.9, 116.7, 79.4, 77.5, 70.3, 48.8, 44.3, 35.5, 25.3, 16.2, 16.1. HR-MS (ESI): m/z ([M - Na]−), 1185.1880, C42H52N8O20S4Na3, calcd. 1185.1868.
Compound 2b
Compound 2CE (448.4 mg, 1.00 mmol) was dissolved in TFA/Ac2O (1:1 (v:v), 6.5 mL). Compound 6 (1.79 g, 4.00 mmol) was added and the reaction mixture stirred and heated for 3 h at 75–80 °C in a pre-heated oil bath. The solution was poured into MeOH (75 mL) and filtered. The residue was dissolved in water (10 mL) and then acetone (50 mL) was added. The precipitate was obtained by filtration and then dissolved in water. The pH of the solution was adjusted to 7 by the addition of 1 M aqueous NaOH. The solvent was removed under reduced pressure and the residue was dried in high vacuum yielding compound 2b as an off-white solid (752 mg, 0.57 mmol, 57%). M.p. > 300 °C. IR (ATR, cm−1): 3446m, 2945w, 2884w, 1715s, 1463s, 1344m, 1308m, 1267w, 1176s, 1098m, 1033s, 901w, 820w, 756m, 595m. 1H NMR (400 MHz, D2O): 7.90 – 7.75 (m, 8H), 5.35 (d, J = 16.3, 2H), 5.18 (d, J = 16.2, 4H), 4.34 (d, J = 16.3, 2H), 4.23 (d, J = 16.2, 4H), 3.75 – 3.65 (m, 4H), 3.30 – 3.20 (m, 4H), 3.15 – 3.05 (m, 4H), 3.00 – 2.85 (m, 4H), 2.10 – 1.95 (m, 4H), 1.90 – 1.75 (m, 4H), 1.75 (s, 6H), 1.70 (s, 6H). 13C NMR (150 MHz, D2O, 1,4-dioxane as internal reference): δ 156.2, 148.2, 128.2, 128.1, 127.4, 123.4, 78.8, 77.4, 74.0, 48.7, 44.0, 36.5, 25.9, 16.5, 16.4. HR-MS (ESI): m/z 677.0957 ([M + Na]2+), C50H56N8O20S4Na5, calcd. 677.0940.
Compound 3a
To a solution of compound 3CE (0.91 g, 1.48 mmol) in TFA/Ac2O (1:1 (v:v), 20.8 mL) compound 5 (1.37 g, 3.44 mmol) was added. The mixture was stirred and heated at 75–80 °C for 3 h and then poured into MeOH (75 mL). The solid was collected by centrifugation. The residue was washed twice with MeOH (45 mL) and dried under vacuum. The off-white solid was dissolved in water and the pH was adjusted to 7 by addition of 1 M aqueous NaOH. The solvent was removed under reduced pressure. The residue was mixed with acetone (100 mL) and the solid collected by centrifugation. The solid was washed once with acetone (45 mL) and MeOH (45 mL) and then dissolved in water. The pH was checked to be at 7. The solvent was removed under reduced pressure and the residue dried under vacuum to yield compound 3a as a yellow solid (983 mg, 0.715 mmol, 48%). M.p. > 300 °C. IR (ATR, cm−1): 3444w, 2941w, 1713s, 1466s, 1380w, 1316m, 1237m, 1183s, 1091m, 1037s, 976w, 922w, 841m, 789m, 729w, 593m. 1H NMR (D2O, 400 MHz): 7.02 (s, 4H), 5.51 (d, J = 15.8, 4H), 5.40 (s, 2H), 5.30 (d, J = 16.3, 4H), 4.25 (d, J = 15.8, 4H), 4.19 (d, J = 16.3, 4H), 4.15 – 4.00 (m, 8H), 3.20 – 3.05 (m, 8H), 2.25 – 2.15 (m, 8H), 1.77 (s, 6H), 1.74 (s, 6H). 13C NMR (125 MHz, D2O dioxane as internal reference): δ 157.6, 157.00, 150.9, 128.8, 116.0, 79.6, 78.3, 71.8, 69.7, 49.0, 48.9, 35.6, 25.3, 16.6, 15.8. HR-MS (ESI): m/z 1353.2513 ([M – Na + 2H]+), C48H60N12O22S4Na3, calcd. 1353.2521.
Compound 3b
Compound 3CE (614.6 mg, 1.00 mmol) was dissolved in TFA/Ac2O (1:1 (v:v), 6.5 mL). Compound 6 (1.79 g, 4.00 mmol) was added and the reaction mixture stirred and heated for 3 h at 72–80 °C in a pre-heated oil bath. The suspension was filtered using a glass frit and the residue washed with MeOH. The solid was dissolved in water (20 mL) and precipitated by the addition of acetone (80 mL). The precipitate was obtained by filtration, and the solid was washed with acetone and then dissolved in water. The pH of the solution was adjusted to 7 by addition of 1 M aqueous NaOH. The solvent was removed under reduced pressure. Finally, the yellowish solid was dried under vacuum to yield compound 3b as a yellowish solid (867 mg, 58.8 mmol, 59%). M.p. > 300 °C, IR (ATR, cm−1): 3441w, 2943w, 2881w, 1716s, 1465s, 1382w, 1345m, 1313m, 1177s, 1079m, 1035s, 950m, 881w, 827m, 788m, 724m, 668m. 1H NMR (400 MHz, D2O): 8.00 – 7.90 (m, 8H), 5.50 (d, J = 15.9, 4H), 5.41 (s, 2H), 5.24 (d, J = 16.4, 4H), 4.30 – 4.20 (m, 8H), 4.00–3.90 (br m, 4H), 3/55–3/45 (br m, 4H), 3.08 (t, J = 8.0, 8H), 2.10–1.95 (br m, 8H), 1.78 (s, 6H), 1.76 (s, 6H). 13C NMR (150 MHz, D2O, dioxane as internal reference) δ 157.5, 156.9, 148.8, 128.6, 128.5, 127.6, 123.8, 79.4, 78.1, 74.7, 71.6, 48.8, 36.9, 26.0, 16.5, 16.2 (16 of the 17 expected resonances were observed). HR-MS (ESI): m/z 1453.2811 ([M – Na + 2H]+), C56H64N12O22Na2, calcd. 1453.2834.
Supplementary Material
Acknowledgments
L.I. thanks the National Institutes of Health (CA168365 to L.I.) for financial support. V.S. thanks the Czech Science Foundation (13-15576S) and the Czech Ministry of Education (LM2011028 and LO1214) for financial support. B.Z. thanks the University of Maryland for a Wylie dissertation fellowship. L.G. thanks the program of Employment of Newly Graduated Doctors of Science for Scientific Excellence (Grant No. CZ.1.07/2.3.00/30.0009), co-financed by the European Social Fund and the Czech Republic’s state budget. We thank Timothy Fouts, Jeffrey Meshulam, and Kathryn Bobb (Rel-MD) for samples of PBS-1086.
Footnotes
Electronic Supplementary Information (ESI) available: Details of the synthesis and characterization of 1 – 3, solubility determinations, and phase solubility diagrams. Crystallographic information files (.cif) for 3b (CCDC1045907). See DOI: 10.1039/b000000x/
Notes and references
- 1.(a) Lipinski CA. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]; (b) Hauss DJ. Adv Drug Delivery Rev. 2007;59:667–676. doi: 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 2.(a) Leuner C, Dressman J. Eur J Pharmaceut Biopharmaceut. 2000;50:47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]; (b) Muller RH, Keck CM. J Biotechnol. 2004;113:151–170. doi: 10.1016/j.jbiotec.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 3.(a) Patri AK, Kukowska-Latallo JF, Baker JR. Adv Drug Delivery Rev. 2005;57:2203–2214. doi: 10.1016/j.addr.2005.09.014. [DOI] [PubMed] [Google Scholar]; (b) Blagden N, de Matas M, Gavan PT, York P. Adv Drug Delivery Rev. 2007;59:617–630. doi: 10.1016/j.addr.2007.05.011. [DOI] [PubMed] [Google Scholar]; (c) Serajuddin ATM. Adv Drug Delivery Rev. 2007;59:603–616. doi: 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]; (d) Stella VJ, Nti-Addae KW. Adv Drug Delivery Rev. 2007;59:677–694. doi: 10.1016/j.addr.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 4.(a) Okimoto K, Rajewski RA, Uekama K, Jona JA, Stella VJ. Pharm Res. 1996;13:256–264. doi: 10.1023/a:1016047215907. [DOI] [PubMed] [Google Scholar]; (b) Rajewski RA, Stella VJ. J Pharm Sci. 1996;85:1142–1169. doi: 10.1021/js960075u. [DOI] [PubMed] [Google Scholar]; (c) Stella VJ, Rajewski RA. Pharm Res. 1997;14:556–567. doi: 10.1023/a:1012136608249. [DOI] [PubMed] [Google Scholar]; (d) Rekharsky MV, Inoue Y. Chem Rev. 1998;98:1875–1917. doi: 10.1021/cr970015o. [DOI] [PubMed] [Google Scholar]
- 5.(a) Freeman WA, Mock WL, Shih NY. J Am Chem Soc. 1981;103:7367–7368. [Google Scholar]; (b) Kim J, Jung IS, Kim SY, Lee E, Kang JK, Sakamoto S, Yamaguchi K, Kim K. J Am Chem Soc. 2000;122:540–541. [Google Scholar]; (c) Day A, Arnold AP, Blanch RJ, Snushall B. J Org Chem. 2001;66:8094–8100. doi: 10.1021/jo015897c. [DOI] [PubMed] [Google Scholar]; (d) Liu S, Zavalij PY, Isaacs L. J Am Chem Soc. 2005;127:16798–16799. doi: 10.1021/ja056287n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Day AI, Blanch RJ, Arnold AP, Lorenzo S, Lewis GR, Dance I. Angew Chem, Int Ed. 2002;41:275–277. doi: 10.1002/1521-3773(20020118)41:2<275::aid-anie275>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]; (f) Cheng XJ, Liang LL, Chen K, Ji NN, Xiao X, Zhang JX, Zhang YQ, Xue SF, Zhu QJ, Ni XL, Tao Z. Angew Chem, Int Ed. 2013;52:7252–7255. doi: 10.1002/anie.201210267. [DOI] [PubMed] [Google Scholar]
- 6.(a) Mock WL, Shih NY. J Org Chem. 1986;51:4440–4446. [Google Scholar]; (b) Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L. J Am Chem Soc. 2005;127:15959–15967. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]; (c) Rekharsky MV, Mori T, Yang C, Ko YH, Selvapalam N, Kim H, Sobransingh D, Kaifer AE, Liu S, Isaacs L, Chen W, Moghaddam S, Gilson MK, Kim K, Inoue Y. Proc Natl Acad Sci U S A. 2007;104:20737–20742. doi: 10.1073/pnas.0706407105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Biedermann F, Uzunova VD, Scherman OA, Nau WM, De Simone A. J Am Chem Soc. 2012;134:15318–15323. doi: 10.1021/ja303309e. [DOI] [PubMed] [Google Scholar]; (e) Cao L, Sekutor M, Zavalij PY, Mlinaric-Majerski K, Glaser R, Isaacs L. Angew Chem, Int Ed. 2014;53:988–993. doi: 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]
- 7.(a) Isaacs L. Acc Chem Res. 2014;47:2052–2062. doi: 10.1021/ar500075g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ko YH, Kim E, Hwang I, Kim K. Chem Commun. 2007:1305–1315. doi: 10.1039/b615103e. [DOI] [PubMed] [Google Scholar]; (c) Hennig A, Bakirci H, Nau WM. Nature Methods. 2007;4:629–632. doi: 10.1038/nmeth1064. [DOI] [PubMed] [Google Scholar]; (d) Dsouza R, Hennig A, Nau W. Chem Eur J. 2012;18:3444–3459. doi: 10.1002/chem.201103364. [DOI] [PubMed] [Google Scholar]; (e) Lee DW, Park K, Banerjee M, Ha S, Lee T, Suh K, Paul S, Jung H, Kim J, Selvapalam N, Ryu S, Kim K. Nat Chem. 2011;3:154–159. doi: 10.1038/nchem.928. [DOI] [PubMed] [Google Scholar]; (f) Ahn Y, Jang Y, Selvapalam N, Yun G, Kim K. Angew Chem, Int Ed. 2013;52:3140–3144. doi: 10.1002/anie.201209382. [DOI] [PubMed] [Google Scholar]; (g) Miyahara Y, Abe K, Inazu T. Angew Chem, Int Ed. 2002;41:3020–3023. doi: 10.1002/1521-3773(20020816)41:16<3020::AID-ANIE3020>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (h) Del Barrio J, Horton P, Lairez D, Lloyd G, Toprakcioglu C, Scherman O. J Am Chem Soc. 2013;135:11760–11763. doi: 10.1021/ja406556h. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hettiarachchi G, Nguyen D, Wu J, Lucas D, Ma D, Isaacs L, Briken V. PLoS One. 2010;5:e10514. doi: 10.1371/journal.pone.0010514. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Uzunova VD, Cullinane C, Brix K, Nau WM, Day AI. Org Biomol Chem. 2010;8:2037–2042. doi: 10.1039/b925555a. [DOI] [PubMed] [Google Scholar]; (c) Oun R, Floriano RS, Isaacs L, Rowan EG, Wheate NJ. Toxicol Res. 2014;3:447–455. doi: 10.1039/C4TX00082J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Dong N, Xue SF, Zhu QJ, Tao Z, Zhao Y, Yang LX. Supramol Chem. 2008;20:659–665. [Google Scholar]; (b) Dong N, Wang X, Pan J, Tao Z. Acta Chim Sinica. 2011;69:1431–1437. [Google Scholar]; (c) Saleh N, Koner AL, Nau WM. Angew Chem Int Ed. 2008;47:5398–5401. doi: 10.1002/anie.200801054. [DOI] [PubMed] [Google Scholar]; (d) Walker S, Oun R, McInnes FJ, Wheate NJ. Isr J Chem. 2011;51:616–624. [Google Scholar]; (e) Jeon YJ, Kim SY, Ko YH, Sakamoto S, Yamaguchi K, Kim K. Org Biomol Chem. 2005;3:2122–2125. doi: 10.1039/b504487a. [DOI] [PubMed] [Google Scholar]
- 10.(a) Appel E, del Barrio J, Loh X, Scherman O. Chem Soc Rev. 2012;41:6195–6214. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]; (b) Zhang J, Coulston RJ, Jones ST, Geng J, Scherman OA, Abell C. Science. 2012;335:690–694. doi: 10.1126/science.1215416. [DOI] [PubMed] [Google Scholar]
- 11.(a) Cao L, Hettiarachchi G, Briken V, Isaacs L. Angew Chem, Int Ed. 2013;52:12033–12037. doi: 10.1002/anie.201305061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim E, Kim D, Jung H, Lee J, Paul S, Selvapalam N, Yang Y, Lim N, Park CG, Kim K. Angew Chem, Int Ed. 2010;49:4405–4408. doi: 10.1002/anie.201000818. [DOI] [PubMed] [Google Scholar]; (c) Jung H, Park KM, Yang JA, Oh EJ, Lee DW, Park K, Ryu SH, Hahn SK, Kim K. Biomaterials. 2011;32:7687–7694. doi: 10.1016/j.biomaterials.2011.06.060. [DOI] [PubMed] [Google Scholar]
- 12.(a) Stancl M, Svec J, Sindelar V. Isr J Chem. 2011;51:592–599. [Google Scholar]; (b) Isaacs L. Isr J Chem. 2011;51:578–591. [Google Scholar]; (c) Chakraborty A, Wu A, Witt D, Lagona J, Fettinger JC, Isaacs L. J Am Chem Soc. 2002;124:8297–8306. doi: 10.1021/ja025876f. [DOI] [PubMed] [Google Scholar]; (d) Huang WH, Zavalij PY, Isaacs L. J Am Chem Soc. 2008;130:8446–8454. doi: 10.1021/ja8013693. [DOI] [PubMed] [Google Scholar]; (e) Lucas D, Minami T, Iannuzzi G, Cao L, Wittenberg JB, Anzenbacher P, Isaacs L. J Am Chem Soc. 2011;133:17966–17976. doi: 10.1021/ja208229d. [DOI] [PubMed] [Google Scholar]; (f) Stancl M, Necas M, Taraba J, Sindelar V. J Org Chem. 2008;73:4671–4675. doi: 10.1021/jo800699s. [DOI] [PubMed] [Google Scholar]; (g) Stancl M, Hodan M, Sindelar V. Org Lett. 2009;11:4184–4187. doi: 10.1021/ol9017886. [DOI] [PubMed] [Google Scholar]; (h) Ma D, Gargulakova Z, Zavalij PY, Sindelar V, Isaacs L. J Org Chem. 2010;75:2934–2941. doi: 10.1021/jo100186q. [DOI] [PubMed] [Google Scholar]; (i) Stancl M, Gargulakova Z, Sindelar V. J Org Chem. 2012;77:10945–10948. doi: 10.1021/jo302063j. [DOI] [PubMed] [Google Scholar]; (j) Stancl M, Gilberg L, Ustrnul L, Necas M, Sindelar V. Supramol Chem. 2014;26:168–172. [Google Scholar]
- 13.(a) Isaacs L, Witt D, Lagona J. Org Lett. 2001;3:3221–3224. doi: 10.1021/ol016561s. [DOI] [PubMed] [Google Scholar]; (b) Isaacs L, Witt D. Angew Chem, Int Ed. 2002;41:1905–1907. [PubMed] [Google Scholar]; (c) Wu A, Chakraborty A, Fettinger JC, Flowers RA, II, Isaacs L. Angew Chem, Int Ed. 2002;41:4028–4031. doi: 10.1002/1521-3773(20021104)41:21<4028::AID-ANIE4028>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.(a) Lagona J, Fettinger JC, Isaacs L. Org Lett. 2003;5:3745–3747. doi: 10.1021/ol035468w. [DOI] [PubMed] [Google Scholar]; (b) Lagona J, Fettinger JC, Isaacs L. J Org Chem. 2005;70:10381–10392. doi: 10.1021/jo051655r. [DOI] [PubMed] [Google Scholar]
- 15.Isaacs L, Park SK, Liu S, Ko YH, Selvapalam N, Kim Y, Kim H, Zavalij PY, Kim GH, Lee HS, Kim K. J Am Chem Soc. 2005;127:18000–18001. doi: 10.1021/ja056988k. [DOI] [PubMed] [Google Scholar]
- 16.(a) Huang WH, Liu S, Zavalij PY, Isaacs L. J Am Chem Soc. 2006;128:14744–14745. doi: 10.1021/ja064776x. [DOI] [PubMed] [Google Scholar]; (b) Huang WH, Zavalij PY, Isaacs L. Angew Chem, Int Ed. 2007;46:7425–7427. doi: 10.1002/anie.200702189. [DOI] [PubMed] [Google Scholar]; (c) Huang WH, Zavalij PY, Isaacs L. Org Lett. 2008;10:2577–2580. doi: 10.1021/ol800893n. [DOI] [PubMed] [Google Scholar]
- 17.(a) Svec J, Necas M, Sindelar V. Angew Chem, Int Ed. 2010;49:2378–2381. doi: 10.1002/anie.201000420. [DOI] [PubMed] [Google Scholar]; (b) Havel V, Svec J, Wimmerova M, Dusek M, Pojarova M, Sindelar V. Org Lett. 2011;13:4000–4003. doi: 10.1021/ol201515c. [DOI] [PubMed] [Google Scholar]; (c) Svec J, Dusek M, Fejfarova K, Stacko P, Klan P, Kaifer AE, Li W, Hudeckova E, Sindelar V. Chem - Eur J. 2011;17:5605–5612. doi: 10.1002/chem.201003683. [DOI] [PubMed] [Google Scholar]; (d) Havel V, Sindelar V, Necas M, Kaifer AE. Chem Commun. 2014;50:1372–1374. doi: 10.1039/c3cc47828a. [DOI] [PubMed] [Google Scholar]; (e) Revesz A, Schroder D, Svec J, Wimmerova M, Sindelar V. J Phys Chem A. 2011;115:11378–11386. doi: 10.1021/jp205218k. [DOI] [PubMed] [Google Scholar]
- 18.(a) Gilberg L, Khan MSA, Enderesova M, Sindelar V. Org Lett. 2014;16:2446–2449. doi: 10.1021/ol500828k. [DOI] [PubMed] [Google Scholar]; (b) Cao L, Isaacs L. Org Lett. 2012;14:3072–3075. doi: 10.1021/ol3011425. [DOI] [PubMed] [Google Scholar]; (c) Vinciguerra B, Cao L, Cannon JR, Zavalij PY, Fenselau C, Isaacs L. J Am Chem Soc. 2012;134:13133–13140. doi: 10.1021/ja3058502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Wittenberg JB, Zavalij PY, Isaacs L. Angew Chem, Int Ed. 2013;52:3690–3694. doi: 10.1002/anie.201300404. [DOI] [PubMed] [Google Scholar]; (b) Zhang M, Cao L, Isaacs L. Chem Commun. 2014 doi: 10.1039/c4cc07268e. Ahead of Print. [DOI] [PubMed] [Google Scholar]
- 20.(a) Burnett CA, Witt D, Fettinger JC, Isaacs L. J Org Chem. 2003;68:6184–6191. doi: 10.1021/jo034399w. [DOI] [PubMed] [Google Scholar]; (b) Huang WH, Zavalij PY, Isaacs L. Org Lett. 2009;11:3918–3921. doi: 10.1021/ol901539q. [DOI] [PubMed] [Google Scholar]; (c) Ma D, Zavalij PY, Isaacs L. J Org Chem. 2010;75:4786–4795. doi: 10.1021/jo100760g. [DOI] [PubMed] [Google Scholar]; (d) Ma D, Zhang B, Hoffmann U, Sundrup MG, Eikermann M, Isaacs L. Angew Chem, Int Ed. 2012;51:11358–11362. doi: 10.1002/anie.201206031. [DOI] [PubMed] [Google Scholar]; (e) Minami T, Esipenko NA, Zhang B, Isaacs L, Nishiyabu R, Kubo Y, Anzenbacher P. J Am Chem Soc. 2012;134:20021–20024. doi: 10.1021/ja3102192. [DOI] [PubMed] [Google Scholar]; (f) Shen C, Ma D, Meany B, Isaacs L, Wang Y. J Am Chem Soc. 2012;134:7254–7257. doi: 10.1021/ja301462e. [DOI] [PubMed] [Google Scholar]; (g) Minami T, Esipenko N, Akdeniz A, Zhang B, Isaacs L, Anzenbacher P. J Am Chem Soc. 2013;135:15238–15243. doi: 10.1021/ja407722a. [DOI] [PubMed] [Google Scholar]
- 21.Ma D, Hettiarachchi G, Nguyen D, Zhang B, Wittenberg JB, Zavalij PY, Briken V, Isaacs L. Nat Chem. 2012;4:503–510. doi: 10.1038/nchem.1326. [DOI] [PubMed] [Google Scholar]
- 22.Zhang B, Zavalij PY, Isaacs L. Org Biomol Chem. 2014;12:2413–2422. doi: 10.1039/c3ob42603c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B, Isaacs L. J Med Chem. 2015;58 doi: 10.1021/jm501276u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sijbesma RP, Kentgens APM, Lutz ETG, van der Maas JH, Nolte RJM. J Am Chem Soc. 1993;115:8999–9005. [Google Scholar]
- 25.Zhou JJ, Yu X, Zhao YC, Xiao X, Zhang YQ, Zhu QJ, Xue SF, Zhang QJ, Liu JX, Tao Z. Tetrahedron. 2014;70:800–804. [Google Scholar]
- 26.Isaacs L, Witt D, Fettinger JC. Chem Commun. 1999:2549–2550. [Google Scholar]
- 27.Witt D, Lagona J, Damkaci F, Fettinger JC, Isaacs L. Org Lett. 2000;2:755–758. doi: 10.1021/ol991382k. [DOI] [PubMed] [Google Scholar]
- 28.(a) Wang ZG, Zhou BH, Chen YF, Yin GD, Li YT, Wu AX, Isaacs L. J Org Chem. 2006;71:4502–4508. doi: 10.1021/jo0603375. [DOI] [PubMed] [Google Scholar]; (b) She NF, Meng XG, Gao M, Wu AX, Isaacs L. Chem Commun. 2008:3133–3135. doi: 10.1039/b800785c. [DOI] [PubMed] [Google Scholar]; (c) She NF, Gao M, Meng XG, Yang GF, Elemans JAAW, Wu AX, Isaacs L. J Am Chem Soc. 2009;131:11695–11697. doi: 10.1021/ja904920r. [DOI] [PubMed] [Google Scholar]; (d) Wu A, Mukhopadhyay P, Chakraborty A, Fettinger JC, Isaacs L. J Am Chem Soc. 2004;126:10035–10043. doi: 10.1021/ja0486972. [DOI] [PubMed] [Google Scholar]; (e) Ghosh S, Wu A, Fettinger JC, Zavalij PY, Isaacs L. J Org Chem. 2008;73:5915–5925. doi: 10.1021/jo8009424. [DOI] [PubMed] [Google Scholar]
- 29.Connors KA. Binding constants. John Wiley & Sons; New York: 1987. [Google Scholar]
- 30.(a) Rowan AE, Elemans JAAW, Nolte RJM. Acc Chem Res. 1999;32:995–1006. [Google Scholar]; (b) Reek JNH, Elemans JAAW, de Gelder R, Beurskens PT, Rowan AE, Nolte RJM. Tetrahedron. 2002;59:175–185. [Google Scholar]
- 31.Higuchi T, Connors KA. Adv Anal Chem Inst. 1965;4:117–212. [Google Scholar]
- 32.(a) Fabre C, Mimura N, Bobb K, Kong SY, Gorgun G, Cirstea D, Hu J, Minami J, Ohguchi H, Zhang J, Meshulam J, Carrasco RD, Tai YT, Richardson PG, Hideshima T, Anderson KC. Clin Cancer Res. 2012;18:4669–4681. doi: 10.1158/1078-0432.CCR-12-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang J, Sliskovic DR, Ducker CE Profectus BioSciences, Inc., USA. WO 2010111460. 7-oxabicyclo[4.1.0]heptenyl heteroarylamide derivatives as inhibitors of nf-kb useful for treatment of cancer, inflammation, auto-immune diseases, diabetes, infections, cardiovascular diseases and ischemia-reperfusion injuries. 2010
- 33.Stella VJ, Rao VM, Zannou EA, Zia V. Adv Drug Delivery Rev. 1999;36:3–16. doi: 10.1016/s0169-409x(98)00052-0. [DOI] [PubMed] [Google Scholar]
- 34.Nau WM, Florea M, Assaf KI. Isr J Chem. 2011;51:559–577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.