

Abstract
A familial behavioral variant frontotemporal dementia associated with astrocyte-predominant tauopathy is described in 2 sisters born from consanguineous parents. The neuropathological examination revealed massive accumulation of abnormally hyper-phosphorylated, conformational, truncated at aspartic acid 421, ubiquitinated, and nitrated tau at Tyr29 in cortical astrocyte (including their perivascular foot processes) and Bergmann glia. Smaller amounts of abnormal tau were observed in neurons and rarely in oligodendrocytes. There was decreased expression of glial glutamate transporter in the majority of tau-positive astrocytes. Gel electrophoresis of sarkosyl-insoluble fractions showed 2 bands of 64 and 60 kDa and a doublet of 67–70 kDa (which are different from those seen in Alzheimer disease and in typical 4R and 3R tauopathies) together with several bands of lower molecular weight indicative of truncated tau. Analysis of expression of MAPT isoforms further revealed altered splicing and representation of tau isoforms involving exons 2, 3 and 10. Genetic testing revealed no known mutations in PSEN1, PSEN2, APP, MAPT, GRN, FUS, and TARDBP, and no pathological expansion in C9ORF72. However, a novel rare heterozygous sequence variant (p.Q140H) of uncertain significance was identified in FUS in both siblings.
Keywords: Astrocyte, Astrocyte-predominant tauopathy, Familial tauopathy, Frontotemporal lobar dementia, FUS, MAPT, Splicing, Tau
INTRODUCTION
Tauopathies are neurodegenerative diseases characterized by the accumulation of abnormally hyper-phosphorylated tau in neurons and glial cells. Pick disease, progressive supranuclear palsy, corticobasal degeneration and argyrophilic grain disease are the most common sporadic tauopathies (1–6). Glial globular tauopathies and tauopathy in the elderly have recently been included in the list (7, 8). Familial tauopathies are principally linked to mutations in MAPT, which encodes microtubule-associated tau protein (9–12). These are usually manifested as frontotemporal dementia (FTD) with or without parkinsonism. Rare cases of familial tauopathy with the presence of Pick bodies are linked to mutations in PS-1, which encodes presenilin 1 and which is usually causative of early onset familial Alzheimer disease (AD) (13–15).
Hyper-phosphorylated tau accumulates in astrocytes in the vast majority of tauopathies (16), but massive tau accumulation in subcortical astrocytes can occur in particular cases in combination or not with corticobasal degeneration (17). Widespread accumulation of tau-positive protoplasmic astroglia in the cerebral cortex, tau-positive coiled bodies and threads in the subcortical white matter, as well as tau-positive pre-tangle neurons in the subcortical white matter and brainstem nuclei, are characteristic of astrocyte-predominant familial tauopathy linked to N296H mutation in the exon 10 of MAPT (18).
We previously reported the clinical findings of a 46-year-old woman who presented with a 2-year history of aprosodic speech together with apathy and disinhibition, followed by inability to make decisions, poor personal hygiene, lack of empathy and progressive mental deterioration; she died accidentally 6 years after the appearance of the first neurological symptoms. Neuropathological examination revealed massive tau deposition in the brain, mainly involving astrocytes. Her parents were first-degree cousins. No mutations in the MAPT gene were detected at that time (19).
This woman’s sister died 3 years later. The first clinical manifestations were psychiatric symptoms followed by cognitive impairment and dementia. Neuropathological examination revealed similar alterations to those briefly described for her sister.
The present study is a detailed description of the neuropathological and molecular characteristics of tau deposition in this familial tauopathy, which is consistent clinically with behavioral variant frontotemporal dementia and neuropathologically with generalized astrocyte-predominant tauopathy. Genetic studies revealed no known mutations in genes associated with FTLD and other tauopathies but a variant of uncertain significance within the FUS gene.
MATERIALS AND METHODS
Clinical Summary
The parents were first cousins and died at the age 90 years without clinical history of neurological disease. They had 5 daughters. The first 2 women did not suffer from neurological disease. The patients were the third daughter (case 2) and 1 of 2 fraternal twins (case 1).
The first patient (Case 1) was a 46-year-old woman who had suffered from depression the last 2 years of life along with aprosodic speech, loss of phonetic and semantic fluency, and apathy. Magnetic resonance imaging (MRI) showed symmetrical subcortical hyper-intensities in the insular regions. The patient worsened during the following 4 years showing childish behavior and inability to make decisions. She was careless about her personal hygiene. Brain single-photon emission computed tomography showed bilateral frontotemporal hypo-perfusion. Analytical data in blood and serum, cerebrospinal fluid and urine were normal. She was institutionalized and died at the age of 62 years. Medical care during the whole years of admission in the asylum was restricted to measures geared to preserve wellbeing and to minimize general clinical complications. Detailed neurological examination was lacking.
The second patient (Case 2) was visited for the first time at the age of 58 years due to a psychiatric disorder, first labeled as paranoid schizophrenia, and that was characterized by depression, progressive deterioration of social behavior with little self-care, shabbiness, disorientation in time but not in space, lack of self-awareness (delusions of grandeur), confabulations (runner of marathons, possible arsonist), moderate loss of recent memory, difficulties in vocabulary, difficulties in self-organization and lack of control of emotions with a tendency to dramatize. The patient was admitted to an asylum due to the constant need for care caused by cognitive impairment and delirium. The MRI disclosed bilateral involvement of the frontal and temporal lobes with altered intensity of the subcortical white matter mainly involving the frontal and temporal lobes and the external capsules. Neurological examination during the last 2 years in the asylum was very limited and no record of ataxia and cerebellar signs was found in the files. The patient died at the age of 63 years.
Neuropathology
Formaldehyde-fixed, paraffin-embedded 4-μm-thick sections were stained with hematoxylin and eosin and Klüver-Barrera, and for immunohistochemistry using antibodies against β-amyloid, phosphorylated tau (clone AT8), 3Rtau, 4Rtau, α-synuclein, TDP-43 (clone 2E2-D3), phosphorylated neurofilaments (clone RT97), α-internexin, αB-crystallin (clone G2JF), glial fibrillary acidic protein (GFAP) and clone CD68 for microglia, as reported elsewhere (20).
Anti-RNA-binding protein Fused in Sarcoma (FUS) immunohistochemistry was performed using 2 different antibodies: anti-FUS (Sigma Aldrich HPA008784, St. Louis, MO; pc, dilution 1:1000, incubated overnight at 4°C after heat-induced epitope retrieval in EDTA at pH 9), and anti-FUS (Lifespan Biosciences, Seattle, WA; pc, dilution 1:500, incubated overnight at 4°C after heat-induced epitope retrieval in citrate buffer at pH 6). The reported antigenic peptide of the Sigma antibody maps to a region between residues 90 and 220 whereas the reported antigenic peptide of the Lifespan antibody maps to a region between residues 200 and 250 of human fusion (involved in t (12;16) in malignant liposarcoma) using the numbering given in SwissProt entry P35637 (GeneID 2521) (20).
Tau immunohistochemistry, double-labeling immunofluorescence and confocal microscopy
De-waxed sections, 4 μm thick, of the frontal cortex, temporal cortex and cerebellum were processed for specific immunohistochemistry against several anti-tau antibodies and anti-ubiquitin. The sections were boiled in citrate buffer (20 minutes) to retrieve tau antigenicity. Endogenous peroxidases were blocked by incubation in 10% methanol-1% H2O2 solution (15 minutes) followed by 3% normal horse serum solution. Then the sections were incubated at 4°C overnight with the primary monoclonal antibodies against 4Rtau, 3Rtau, goat polyclonal anti-tau N-terminal, rabbit polyclonal specific phospho-tau Thr181, Ser199, Thr231, Ser262 and Ser422, double-phosphorylation sites Ser202-Thr205 (clone AT8) and Ser396-404 (PHF1), conformational tau modifications at amino acids 5-15 (Alz50) and amino acids 312-322 (MC-1), mouse monoclonal against tau truncated at aspartic acid 421 (tau-C3), rabbit polyclonal against ubiquitin, and nitrated tau at Tyr29 used as detailed elsewhere (16). Following incubation with the primary antibody, the sections were incubated with EnVision + system peroxidase (Dako, Glostrup, Denmark) for 30 minutes at room temperature. The peroxidase reaction was visualized with diaminobenzidine H2O2 or with diaminobenzidine, NH4NiSO4, and H2O2. The immunoreaction results in a brown and blue-grey precipitate, respectively. Control of the immunostaining included omission of the primary antibody; no signal was obtained following incubation with only the secondary antibody.
Double-labeling immunofluorescence and confocal microscopy were carried out as detailed (16). Tissue sections were incubated with combinations of primary antibodies against phospho-tau Thr181 and tau-C3. Other sections were incubated with monoclonal antibody AT8 and rabbit polyclonal anti-GFAP (Dako, diluted 1:250) or with AT8 and guinea pig anti-glial glutamate transporter (GLT-1) (Millipore, Billerica, MA, diluted 1:50). Finally, other sections were incubated with anti-phosphorylated tau antibodies and anti-TDP-43. After washing, the sections were incubated with Alexa488 or Alexa546 fluorescence secondary antibodies against the corresponding host species. Nuclei were stained with DRAQ5™. After washing, the sections were mounted in Immuno-Fluore mounting medium, sealed, and dried overnight. Sections were examined with a Leica TCS-SL confocal microscope (16).
Tau analysis in sarkosyl-insoluble fractions
Fresh samples from the frontal cortex of AD (used as a control), cerebral cortex and white matter of the temporal lobe, and hippocampus (~1 g) were homogenized in a glass tissue grinder in 10 vol (w/v) of cold suspension buffer consisting of 10 mM Tris–HCl (pH =7.4), 0.8 M NaCl, 1 mM EGTA, 10% sucrose, 0.1 mM phenylmethylsulfonyl fluoride, 2 Ag/ml aprotinin, 10 Ag/ml leupeptin, and 5 Ag/ml pepstatin. The homogenates were first centrifuged at 20,000 × g, and the supernatant (S1) was retained. The pellet (P1) was re-homogenized in 5 vol of homogenization buffer and re-centrifuged. The 2 supernatants (S1 + S2) were then mixed and incubated with N-lauroylsarcosinate 10% for 1 hour at room temperature while shaking. Samples were then centrifuged for 1 hour at 10,000g in a Ti 70 Beckman rotor. Sarkosyl-insoluble pellets (P3) were re-suspended (0.2 ml/g of starting material) in 50 mM Tris–HCl (pH = 7.4). Protein concentrations were determined with the BCA method and 10% sodium dodecyl sulphatepoliacrylamide gel electrophoresis (SDS-PAGE) was run. Fifty μg of protein was loaded in each lane with loading buffer containing 0.125 M Tris (pH = 6.8), 20% glycerol, 10% mercaptoethanol, 4% SDS, and 0.002% bromophenol blue. Samples were heated at 95°C for 5 minutes prior to gel loading. The proteins were then transferred to nitrocellulose membranes (Amersham, GE Healthcare, Buckinghamshire, UK) using an electrophoretic chamber system (Trans-Blot Electrophoretic Transfer Cell, Bio-Rad, Hercules, CA). Non-specific binding sites were blocked with Tris-buffered saline solution pH = 7.4 with 0.1% Tween-20 (TBST) containing 5% skimmed milk for 30 minutes, and incubated with anti-phospho-tau Ser422 and clone AT8. After washing, the membranes were incubated with the secondary antibody labeled with horseradish peroxidase (Dako) diluted 1:1,000 for 1 hour at room temperature, washed again, and developed with the chemiluminescence ECL Western Blotting system (Amersham). Membranes were then exposed to autoradiographic film (Hyperfilm ECL, Amersham).
MAPT gene sequencing
DNA was extracted from blood cells in case 1 and 2 followed by gene amplification of exons 2-6 and 8-12 of MAPT according to the reference sequence NM_005910.3 of the National Centre of Biotechnology Information using double-sense automatic sequencing with an ABI3130 (Applied Biosystems, Foster City, CA) and analysis of the sequence with the software SeqScape v2.5.
Exome sequencing
Exome sequencing was performed in both the proband and her sister. Genomic DNA was extracted from peripheral blood and fragmented by sonication using a Covaris instrument (E210, Covaris Inc., Woburn, MA) to achieve an average fragment size of 200 base pairs. One μg of DNA from each individual was used for the construction of a paired-end library and exome capture was performed using the TruSeq DNA Sample Prep Kit (Illumina, San Diego, CA). Sequencing was performed on an Illumina Genome Analyzer HiSeq2500 with100-bp-long, paired-end reads, following the manufacturer’s instructions. Raw image files were processed with the Illumina Pipeline software (CASAVA 1.8) (Illumina, San Diego, CA). The sequencing reads were aligned to the National Center for Biotechnology Information human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner (BWA). Variant calling was performed using Genome Analysis Toolkit (GATK) and the Ingenuity Variant Analysis software (Qiagen, Redwood City CA) was used for variant annotation and filtering. Filtering was carried out applying a series of steps: low-quality variants were filtered out using the Illumina Qscore threshold of 20; in addition, variants present in dbSNP138 and with a minimum allele frequency higher than 1% in 1000 Genomes Project (http://www.1000genomes.org/) and Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/) databases were filtered out; finally, we focused on predicted missense, frame-shift, stop gain/loss and splice site variants in brain-expressed genes that were shared between the two siblings. We also checked variant frequency in the recently developed ExAC database, a collection of sequenced exomes from population and disease-specific studies (http://exac.broadinstitute.org). Variants of interest were confirmed using standard Sanger sequencing.
C9ORF72 expansion detection
The presence of expanded GGGGCC hexanucleotide repeats within C9ORF72 was detected using the repeat-primed PCR reaction as described (21). PCR products were run on an ABI3730 DNA Analyzer and analyzed using the Peak Scanner Software.
Semiquantitative expression analysis of MAPT isoforms
Total RNA was extracted starting from 30 mg of postmortem temporal lobe specimen of the proband using the RNeasy Mini kit (Qiagen). Human Brain, Temporal Lobe Total RNA (Clontech, cat.636564) was used as control RNA. cDNA was obtained using the SuperScript II Reverse Transcriptase (Life Technologies, Carlsbad, CA) starting from 1 μg of total RNA. Specific primers were designed (MAPT_E1-85-For TACACCATGCACCAAGACCA and MAPT_E4-330-Rev GTCTCCAATGCCTGCTTCTT; MAPT_E8-1384-F GCTGATGGTAAAACGAAGATCG and MAPT_E11-1915-R ACTTGGAGGTCACCTTGCTC) and PCR reaction was performed on cDNA in order to amplify the regions containing the coding exon2-3 and exon10 of MAPT transcript (NM_016835). PCR products were run in a 2% agarose gel and the intensities of the bands were measured using GelQuant software (http://biochemlabsolutions.com/GelQuantNET.html).
RESULTS
General neuropathological findings
The 2 cases had similar neuropathological alterations and are described together. Marked neuron loss was found in the frontal and temporal cortices, and moderate loss of neurons in the insula, amygdala, caudate, putamen and thalamus. Spongiosis resulting from neuron loss was encountered in the upper layers of the frontal cortex, and to a lesser extent in the upper layers of the temporal cortex; certain areas of the frontal cortex also showed spongiosis in the inner cortical layers. The entorhinal cortex, subiculum and hippocampus showed marked neuron loss consistent with mesial gliosis in the second case, but neuron loss in these regions was moderate in the first case. Discrete losses of Purkinje cells, neurons of the dentate nucleus and inferior olive were noted in both cases. These changes were accompanied by astrocytic gliosis, more marked in parallel with neuron loss, and activated microglia. Moderate myelin pallor was found in the white matter of the centrum semiovale. Small blood vessel disease occurred in the older sister but was absent in the younger.
The most dramatic change was revealed with anti-phosphorylated tau antibody clone AT8. Hyper-phosphorylated tau deposits were massive in astrocytes of the cerebral cortex, hippocampus, subiculum, entorhinal cortex, caudate, putamen, globus pallidus, thalamus, sub-thalamus, and nuclei of the midbrain, pons and medulla oblongata. The morphology of astrocytes was reminiscent of reactive astrocytes but in addition to the cytoplasm, perivascular foot processes around the majority of cortical blood vessels were decorated with heavy deposits of hyper-phosphorylated tau. Cerebellar Bergmann glia were also heavily stained with the AT8 antibody.
Tufted and tufted-like astrocytes, astrocytic plaques, thorn astrocytes and astrocytes with fine and varicose processes, and globular glial inclusions (as described in other tauopathies) were absent. Astrocytes were also stained with anti-4Rtau and anti-3Rtau antibodies (Figs. 1, 2).
Figure 1.

Astrocyte-predominant familial tauopathy (case 1). (A) cerebral cortex; (B) striatum; (C) amygdala; (D) cerebellum; (E) neurons in the upper cerebral cortex; (F) astrocytes in the inner cerebral cortex; (G) cerebellum. (A–G) AT8; (H) 4Rtau; (I) 3Rtau. Note the morphology of reactive astrocytes and the presence of tau immunoreactivity in perivascular astrocyte foot processes surrounding blood vessels. Paraffin sections, with DAB chromogen (brown deposits) and lightly counterstained with hematoxylin. A–D, I, bar in I = 50 μm; E–H, bar in H = 25 μm.
Figure 2.

Astrocyte-predominant familial tauopathy (case 2). (A) Upper layers cerebral cortex; (B) inner layers cerebral cortex; (C) CA1; (D) thalamus; (E) cerebellum; (F) pons; (G) inferior olive; (H) cerebral cortex; (I) dentate gyrus; (J–L) cerebellum. (A–J) AT8; (K) 4Rtau; (L) 3Rtau. Similar features as seen in case 1 showing the morphology tau-immunoreactive reactive astrocytes and perivascular immunoreactivity due to phosphor-tau deposition in astrocyte foot processes. Bergmann glia are heavily immunostained with anti-phospho-tau antibodies. Note the relatively small number of tau-containing neurons vs. the large number of tau-containing astrocytes. Paraffin sections, with DAB chromogen (brown deposits), lightly counterstained with hematoxylin; A–G, bar in G = 50 μm; H–L, bar in L = 25 μm.
Some astrocytes were stained with anti-αB-crystallin antibodies. Hyper-phosphorylated tau deposition was also present, although in less numbers, in neurons forming tangles and very rarely pre-tangles in the same regions including the cerebral cortex, diencephalic nuclei, pontine nuclei and medial reticular formation. No tau deposits were found in neurons of the cerebellum including Purkinje cells. Pick bodies were absent. No β-amyloid deposits were seen in any region. α-synuclein deposits (either in the form of Lewy bodies, Lewy neurites or glial inclusions) were absent. Phosphorylated-neurofilament- and α-internexin-immunoreactive neuronal inclusions were not present. TDP-43-immunoreactive filaments and granular intracytoplasmic neuronal inclusions were scattered in the frontal cortex, insula, hippocampus, amygdala and entorhinal cortex. TDP-43 pathology was not related to hyper-phosphorylated tau-immunoreactive intracytoplasmic inclusions, as revealed by double-labeling immunofluorescence and confocal microscopy. FUS immunoreactivity, as revealed with 2 distinct antibodies, was restricted to the nuclei; extra-nuclear inclusions were not seen; no evidence of FUS pathology was observed on neuropathological examination. With respect to oligodendrocytes, a few coiled bodies were scattered in the cerebral white matter of the frontal and temporal lobes.
Specific characteristics of tau pathology and effects on glial glutamate transporter expression
In addition to the data reported for AT8 and 4Rtau and 3Rtau, tau inclusions in neurons and astrocytes in the brain, brainstem and cerebellum in both cases were immunoreactive with the following antibodies: tau N-terminal, phospho-tau Thr181, phospho-tau Ser 199/202, phospho-tau Thr 231, phospho-tau Ser 262, phospho-tau Ser 422, clone PHF1 (phosphorylation sites Ser396-404), tau 5, tau conformational antibodies Alz50 (amino acids 5-15) and MC1 (amino acids 312-322). Most astrocytes in the cerebral cortex and all Bergmann glia were positive with the antibody tau-C3 (which recognizes truncated tau at aspartic acid 421) and anti-ubiquitin; the inclusions were positive with an antibody anti-nitrated tau protein at Tyr29 (Supplementary Figs. 1, 2).
Double-labeling immunofluorescence and confocal microscopy disclosed variable distortion of the GFAP cytoskeleton in cortical astrocytes and Bergmann glia in astrocyte-predominant familial tauopathy (Supplementary Fig. 3). Phospho-tau Thr181colocalized with tau-C3 in the vast majority of, if not all, Bergmann glia in both cases (Fig. 3).
Figure 3.

(A–C) Double-labeling immunofluorescence and confocal microscopy to phospho-tau Thr181 (green) and antibody tau-C3, which recognizes truncated tau at aspartic acid 421. Note colocalization of phospho-tau and truncated tau. (D–H) Double-labeling immunofluorescence and confocal microscopy to phospho-tau clone AT8 (green) and glial glutamate transporter (red) in cerebral cortex (D–F) and cerebellum (G–I). Loss of glutamate transporter immunoreactivity is found in some (arrows) but not all phospho-tau-containing astrocytes (D–F) and Bergmann glia (G–I). Nuclei are stained with DRAQ5™ (blue). A–C, bar = 16 μm; D–F, bar = 40 μm; G–I, 16 μm.
Finally, to learn whether abnormal tau deposition might affect glutamate transport, double-labeling immunofluorescence and confocal microscopy of phospho-tau and glial glutamate transporter was used in both cases. Glial glutamate transporter immunoreactivity was altered in cortical astrocytes and Bergmann glia where glial glutamate transporter immunoreactivity was preserved in some but was markedly decreased in other neighboring astrocytes showing equal expression of phospho-tau protein (Fig. 3).
Molecular profile of tau in sarkosyl-insoluble fractions
Sarkosyl-insoluble fractions of the temporal cortex, temporal white matter and hippocampus in astrocyte-predominant familial tauopathy blotted with antibodies anti-phosphorylated tau Ser 422 and clone AT8 revealed 2 main bands of 64 and 60 kDa and a doublet of ~67–70 kDa. This pattern differed from that seen in the temporal cortex of cases with AD stage V of Braak and Braak processed in parallel in which the characteristic hyper-phosphorylated tau band pattern of 68, 64 and 60 kDa, in addition to a higher weak band of 73 kDa, was clearly obtained. Several bands of lower molecular weight were found in AD and astrocyte-predominant familial tauopathy (Fig. 4).
Figure 4.
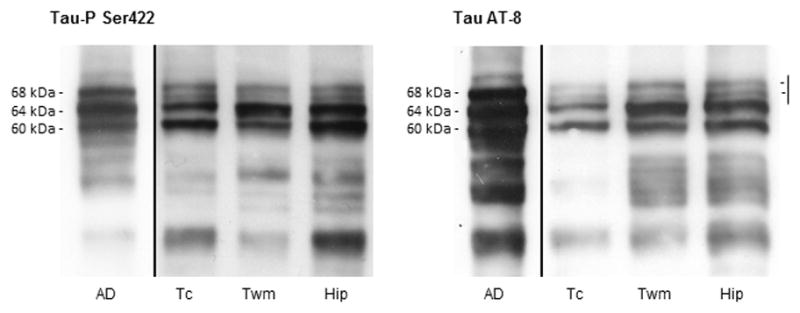
Tau band pattern in sarkosyl-insoluble fractions of the temporal cortex (Tc), temporal white matter (Twm), and hippocampus (Hip) in astrocyte-predominant familial tauopathy processed in parallel with a case of Alzheimer disease (AD) stage V of Braak and Braak for comparison. Two main bands of 64 and 60 kDa, as revealed with antibodies Tau-P Ser422 and AT8, are observed in temporal cortex and white matter, and hippocampus in astrocyte-predominant familial tauopathy in contrast with the three bands of 68, 64 and 60 kDa characteristically found in AD. In addition, a doublet between 67 and 70 kDa, instead of the band of 68 kDa of AD, is found in astrocyte-predominant familial tauopathy. This doublet differs from the additional thin weak band of 73 kDa characteristically occurring in AD. Finally, several bands of lower molecular weight are indicative of several truncated tau species.
MAPT sequencing
No known mutations were observed in exons 2-6 and 8-12 of MAPT. Previously known common polymorphisms were identified, but none was shared between the two subjects.
Exome sequencing
No pathological expansion was detected in C9ORF72 and whole-exome sequencing was performed in the proband (case 1) and her sister (case 2). 116.9 × 106 100 base pair, paired end reads were obtained in case 1, and 107.1 × 106 in case 2, with a mean depth of coverage in the targeted exome of 62x and 60x per base, respectively.
A total of 39,707 variants were identified in case 1 and 39,269 variants for case 2, and 26,833 were shared between the 2 subjects. A series of filtering steps was applied to reduce the number of candidate variants. First, we filtered out variants present in dbSNP138 and with a frequency lower than 1% in 1000 Genomes Project and ESP databases. Second, we focused on missense, splice site, stop gain/loss and frame-shift variants shared by the 2 siblings in genes associated with dementia or expressed in brain. No homozygous, known pathogenic, or rare variants were identified in genes associated with dementia (including PSEN1, PSEN2, APP, MAPT, GRN, FUS, and TARDBP) or expressed in brain applying a recessive model. One hundred thirty-eight shared variants were novel (not present in dbSNP138 database), coding, and non-synonymous, and 113 shared heterozygous variants in brain-expressed genes were filtered applying a dominant model, and only 1 was within the coding sequence of a gene previously associated with dementia, the previously unreported variant within the FUS gene c.420G>C, predicted to result in a p.Q140H substitution. This substitution was not reported in online repositories of human variation, including the ExAC database, collecting ~60,000 sequenced exomes. The variant falls within the SYGQ-rich domain and it is predicted to be benign by SIFT and Polyphen2. Sanger sequencing confirmed the heterozygous variant in both samples.
MAPT isoform expression analysis
FUS is known to alter MAPT isoform levels (22, 23). In order to test a potential effect of the FUS p.Q140H variant on its function, we performed a semiquantitative analysis on MAPT isoform expression. RNA was extracted from temporal lobe pathological samples from case 1 and expression levels of MAPT isoforms containing exons 2, 3 and 10 were compared to RNA from a normal control using semiquantitative PCR (Fig. 5).
Figure 5.
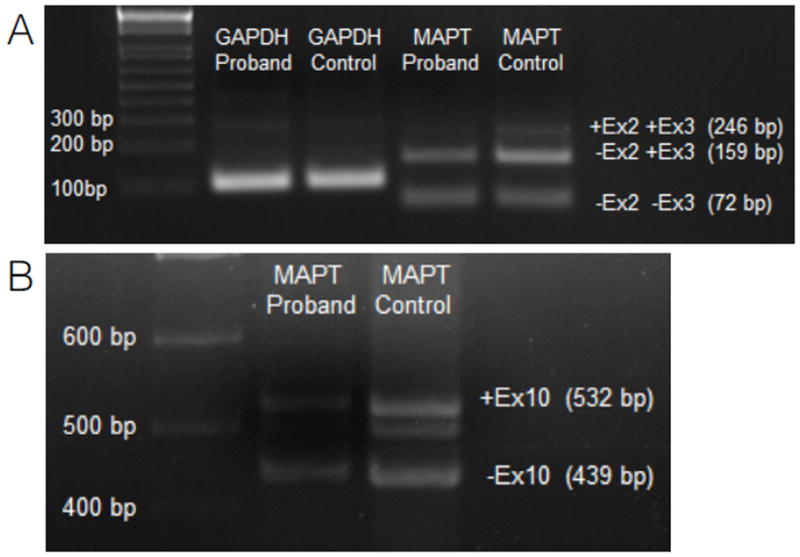
MAPT splicing analysis by semiquantitative PCR. (A) Isoforms including exon 2 and 3. +Ex2 +Ex3, isoforms containing both exon 2 and exon 3; −Ex2 +Ex3, isoforms missing exon 2; −Ex2 −Ex3, isoform missing both exon 2 and exon 3. (B) Isoforms including exon 10. +Ex10, isoforms containing exon 10; −Ex10, isoforms missing exon 10. GAPDH, glyceraldehyde 3-phosphate dehydrogenase (housekeeping gene).
The expression of the isoforms containing both exon2 and exon3 was decreased 10-fold compared to control, isoforms missing exon2 were 50% less expressed and isoforms missing both exon2 and exon3 showed no changes in expression levels when compared to control.
In addition, the relative balance among isoforms showed alterations in the affected sample compared to the control. In the sample from the case 1, isoforms containing exon 2 and 3 represented 1.3% of the total MAPT, isoforms missing exon 2 were 51.6% and the isoforms missing both exon 2 and 3 were 47.1%, respectively. In the control sample MAPT isoforms containing exon2 and 3 are the 8.6%, isoforms missing exon2 were the 60.9% and MAPT isoforms missing both exon2 and 3 are the 30.4% of the total transcribed.
We detected a 5-fold reduction in expression levels of isoforms containing exon10, while isoforms missing exon10 are reduced 50% compared to the control. Isoforms containing and missing exon10 in controls were 53.7% and 46.3% of the total, respectively, in controls and, 35.6% and 64.4% in the patient. Taken together, these results support an alteration of the relative amounts of tau isoforms in case 1 compared with a control RNA sample.
DISCUSSION
The present familial cases are clinically manifested by frontotemporal dementia with atypical onset, the first one starting with aprosodic speech and insular hyper-intensities, and the second one, an older sister, with paranoid schizophrenia. The twin sister of the younger patient was unaffected. The parents were cousins. The postmortem neuropathological examination results were similar in the 2 affected cases and the main neuropathological findings are consistent with a generalized, severe frontotemporal lobar degeneration with massive abnormal tau deposition in astrocytes in the cerebrum, cerebellum and Bergmann glia, and to a lesser degree in selected neurons and a few oligodendrocytes. The morphology of astrocytic inclusions differs from that seen in common tauopathies including tufted and tufted-like astrocytes, astrocytic plaques, thorn-like astrocytes, astrocytes with fine and varicose cellular processes, and globular glial inclusions. Astrocytes bearing abnormal tau are similar to non-specific reactive astrocytes showing partial disruption of the astroglial cytoskeleton and unique accumulation of abnormal tau in astrocyte foot processes, thus resulting in marked perivascular tau deposition. Variegated astroglial, and neuronal and oligodendroglial inclusions, are found in frontotemporal lobar degeneration linked to mutations in MAPT (11), but in no case does tau deposition in astrocytes have a profile like that seen in the present astrocyte-predominant familial tauopathy.
Moreover, the complexity of abnormal tau deposition in astrocytes (including their perivascular foot processes) was revealed with antibodies anti-tau N-terminal and tau-5; with phospho-specific anti-phosphorylated tau antibodies against different sites including Thr 181, tau Ser 199/202, Thr 231, Ser 262, Ser 422, phosphorylation sites Ser 202-Thr 205 (as revealed by clone AT8), Ser 396-404); with anti-tau conformational antibodies Alz50 (amino acids 5-15) and MC1 (amino acids 312-322); with the antibody tau-C3 (which recognizes truncated tau at aspartic acid 421); and with an anti-ubiquitin antibody. All Bergmann glia and a~90% of cortical astrocytes containing abnormal tau deposits are positive with all these antibodies. Immunostaining with all these antibodies indicates a complex end-stage alteration of tau deposition in astrocytes in the present astrocyte-predominant familial tauopathy (16). Abnormal tau deposition also contains nitrated tau at Tyr29, which has been associated with neurodegeneration (24).
Immunohistochemistry also showed 4Rtau and 3Rtau in astroglial, neuronal and oligodendroglial deposits. Therefore, the present astrocyte-predominant familial tauopathy can be categorized as a 4Rtau-3Rtau tauopathy. The effects of abnormal tau deposition in astrocytes, in addition to the distortion of the cytoskeleton, are barely known in most tauopathies. In a transgenic mouse model of astrocytic tau pathology generated by expressing the human tau protein driven by the glial fibrillary acidic protein promoter (25). These mice accumulated hyper-phosphorylated tau in astroglial cytoplasm, which appeared as ubiquitin-immunoreactive inclusions that redistribute within the astroglial cytoskeleton network (25). These characteristics are similar to those seen in cortical astrocytes and Bergmann glia in the present astrocyte-predominant familial tauopathy. Interestingly, additional studies demonstrated impaired glial glutamate-aspartate transporter in the same transgenic mouse model (26). Based on these observations, we examined the expression of the glial glutamate transporter with double-labeling immunofluorescence and confocal microscopy and identified reduced glial glutamate transporter immunoreactivity in some, but not all, tau-bearing astrocytes in astrocyte-predominant familial tauopathy, suggesting that tau pathology in astrocytes may jeopardize glutamate transport in this particular human tauopathy.
Examination of sarkosyl-insoluble fractions identifies particular hyper-phosphorylated tau patterns in AD and tauopathies. Three bands of 68, 64 and 60 kDa together with a weak upper band of ~73 kDa are characteristically found in AD. Two bands of 68 and 64 kDa characterize most 4Rtau tauopathies, including progressive supranuclear palsy, corticobasal degeneration, argyrophilic grain disease, GGT and argyrophilic grain disease; and 2 bands of 64 and 60 kDa distinguish 3Rtau tauopathies represented by Pick disease (27–30). Regarding familial tauopathies, different patterns of tau hyper-phosphorylation are found depending on the MAPT mutation: a band pattern similar to that seen in AD (3 bands of 68, 64 and 60 kDa together with a weak band of 73 kDa); a band pattern similar to that seen in Pick disease (bands of 64 and 60 kDa) but commonly associated with weak upper bands of 68 and 73 kDa; and a band pattern reminiscent of that seen in 4Rtau tauopathies (68 and 64 kDa), often with a weak band of 73 kDa (11).
Interestingly, the pattern of hyper-phosphorylated tau in the present astrocyte-predominant familial tauopathy is distinguished by 2 bands of 64 and 60 kDa and a weak doublet of 67-70 kDa in the temporal cortex and temporal white matter, and in the hippocampus, the only regions analyzed. The Western blot pattern in this patient has similarities to those seen in 3Rtau tauopathies with predominant bands of 64 and 60 kDa but with a component, consistent of a doublet of 67-70 kDa, that substitutes the band of 68 kDa associated to 4Rtau tauopathies (10, 11).
Genetic studies showed no known mutations in PSEN1, PSEN2, APP, MAPT, GRN, FUS, and TARDBP and no pathological expansion has been detected in C9ORF72. However, a previously unreported variant was identified in FUS, which encodes the nuclear protein FUS. The very rare variant c.420G>C, present in heterozygosity for both samples, is predicted to result in a p.Q140H substitution, which falls within the SYGQ-rich domain and it is predicted to be benign by both SIFT and Polyphen2. Mutations in FUS are linked to familial and sporadic cases of amyotrophic lateral sclerosis, and, rarely, frontotemporal lobar degeneration with FUS inclusions (31–37). Tau-negative, TDP-43-negative and FUS-immunoreactive cytoplasmic inclusions are characteristically reported in all of these diseases. In addition, FUS-immunoreactive intraneuronal cytoplasmic inclusions are observed in rare sporadic frontotemporal dementias such as atypical FTLD-U, neuronal intermediate filament inclusion disease, and basophilic inclusion body disease (20, 38–43). FUS-immunoreactive inclusions were absent in the present cases.
The link between abnormal FUS and altered tau splicing, and abnormally hyper-phosphorylated, conformational and truncated tau deposition in the present astrocyte-predominant familial tauopathy has no precedent. However, recent studies have shown that FUS directly binds to tau pre-mRNA, and knockdown of FUS in hippocampal neurons leads to preferential inclusion of Tau exons 3 and 10 (44, 45).
The analysis of expression of MAPT isoforms further reveals altered splicing and representation of tau isoforms involving exons 2, 3 and 10. The expression of the isoforms containing both exon2 and exon3 results to be decreased 10-fold compared to control; isoforms missing exon2 are 50% less expressed; isoforms containing exon10 reduced 5-fold; and isoforms missing exon10 expressed 50% less compared to the controls. In addition, the percentage of isoforms containing or lacking exons 2, 3 and 10 are largely altered when compared to controls.
Whether the FUS variant observed in the present cases is the cause of altered MAPT splicing and the origin of this new tauopathy is unclear. However, the present data suggest that the FUS variant carried by both cases is linked to altered tau mRNA splicing and production of abnormal tau, which is prone to hyper-phosphorylation, altered conformation, truncation and ubiquitination, and accumulates mainly in astrocytes and astrocyte processes, and to a lesser extent in neurons and oligodendroglia causing a new astrocyte-predominant familial tauopathy. Whether this FUS variant confers a gain of function that promotes 3R MAPT splicing is not known. Further studies will be needed to conclusively link this variant to the observed changes in splicing in these subjects.
Supplementary Material
Acknowledgments
We thank Dr. Peter Davies, Bronx, NY, for providing antibodies PHF1, Alz50 and tau MC-1, and T. Yohannan for editorial help. We thank Dr. Mochtar Pribadi (Coppola lab) for testing the presence of a pathological C9ORF72 expansion
This study was funded by the Seventh Framework Programme of the European Commission, grant agreement 278486: DEVELAGE and by the Instituto de Salud Carlos III FIS PI14/00757.We acknowledge the support of the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691).
Footnotes
Financial disclosure and conflict of interests: No relevant data.
References
- 1.Lowe J, Mirra S, Hyman BT, et al. Ageing and dementias. In: Love S, Louis DN, Ellison DW, editors. Greenfield’s Neuropathology. 8. Vol. 1. London: Hodder Arnold; 2008. pp. 1031–1152. [Google Scholar]
- 2.Ferrer I, Santpere G, van Leeuwen FW. Argyrophilic grain disease. Brain. 2008;131:1416–32. doi: 10.1093/brain/awm305. [DOI] [PubMed] [Google Scholar]
- 3.Goedert M, Spillantini MG. Pathogenesis of the tauopathies. J Mol Neurosci. 2011;45:425–31. doi: 10.1007/s12031-011-9593-4. [DOI] [PubMed] [Google Scholar]
- 4.Dickson DW, Haw JJ, Agid Y, et al. Progressive supranuclear palsy and corticobasal degeneration. In: Dickson DW, Weller RO, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. Oxford: Wiley-Blackwell; 2011. pp. 135–55. [Google Scholar]
- 5.Muñoz DG, Morris HR, Rossor M. Pick’s disease. In: Dickson DW, Weller RO, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. Oxford: Wiley-Blackwell; 2011. pp. 156–64. [Google Scholar]
- 6.Tolnay M, Braak H. Argyrophilic grain disease. In: Dickson DW, Weller RO, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. Oxford: Wiley-Blackwell; 2011. pp. 165–70. [Google Scholar]
- 7.Kovacs GG, Molnár K, Lászlo L, et al. A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol. 2011;122:205–22. doi: 10.1007/s00401-011-0819-x. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed Z, Bigio EH, Budka H, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropath. 2013;126:537–44. doi: 10.1007/s00401-013-1171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spillantini MG, Goedert M, Crowther RA, et al. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A. 1997;94:4113–8. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muñoz DG, Ferrer I. Neuropathology of hereditary forms of frontotemporal dementia and parkinsonism. In: Duyckaerts C, Litvan I, editors. Handbook of Clinical Neurology: Dementias. London, New York, Oxford, Philadelphia, St. Louis, Sydney, Toronto, Edinburgh: Elsevier; 2008. pp. 393–414. [DOI] [PubMed] [Google Scholar]
- 11.Ghetti B, Wszolek Z, Boeve BF, et al. Frontotemporal dementia and parkinsonism linked to chromosome 17. In: Dickson DW, Weller RO, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. Oxford: Wiley-Blackwell; 2011. pp. 110–34. [Google Scholar]
- 12.Dickson DW, Kouri N, Murray ME, et al. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-Tau) J Mol Neurosci. 2011;45:384–9. doi: 10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dermaut B, Kumar-Singh S, Engelborghs S, et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol. 2004;55:617–26. doi: 10.1002/ana.20083. [DOI] [PubMed] [Google Scholar]
- 14.Halliday GM, Song YJ, Lepar G, et al. Pick bodies in a family with presenilin-1 Alzheimer’s disease. Ann Neurol. 2005;57:139–43. doi: 10.1002/ana.20366. [DOI] [PubMed] [Google Scholar]
- 15.Riudavets MA, Bartoloni L, Troncoso JC, et al. Familial dementia with frontotemporal features associated with M146V presenilin-1 mutation. Brain Pathol. 2013;23:595–600. doi: 10.1111/bpa.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrer I, López-González I, Carmona M, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol. 2014;73:81–97. doi: 10.1097/NEN.0000000000000030. [DOI] [PubMed] [Google Scholar]
- 17.Sakai K, Piao YS, Kikugawa K, et al. Corticobasal degeneration with focal, massive tau accumulation in the subcortical white matter astrocytes. Acta Neuropathol. 2006;12:341–48. doi: 10.1007/s00401-006-0093-5. [DOI] [PubMed] [Google Scholar]
- 18.Iseki E, Matsumura T, Marui W, et al. Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol. 2001;102:285–92. doi: 10.1007/s004010000333. [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Beldarrain M, Ruiz Ojeda J, Ferrer I, et al. Aprosodic speech with insular hyperintensities and 4R tau pathology on autopsy. Neurocase. 2013;19:583–6. doi: 10.1080/13554794.2012.713489. [DOI] [PubMed] [Google Scholar]
- 20.Gelpi E, Lladó A, Clarimón J, et al. Phenotypic variability within the inclusion body spectrum of basophilic inclusion body disease and neuronal intermediate filament inclusion disease in frontotemporal lobar degenerations with FUS-positive inclusions. J Neuropathol Exp Neurol. 2012;71:795–805. doi: 10.1097/NEN.0b013e318266efb1. [DOI] [PubMed] [Google Scholar]
- 21.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishigaki S, Masuda A, Fujioka Y, et al. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep. 2012;2:529. doi: 10.1038/srep00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujioka Y, Ishigaki S, Masuda A, et al. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci Rep. 2013;3:2388. doi: 10.1038/srep02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reyes JF, Geula C, Vana L, et al. Selective tau tyrosine nitration in non-AD tauopathies. Acta Neuropathol. 2012;123:119–32. doi: 10.1007/s00401-011-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forman MS, Lal D, Zhang B, et al. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci. 2005;25:3539–50. doi: 10.1523/JNEUROSCI.0081-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dabir DV, Robinson MB, Swanson E, et al. Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J Neurosci. 2006;26:644–54. doi: 10.1523/JNEUROSCI.3861-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goedert M, Spillantini MG, Cairns NJ, et al. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–68. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 28.Delacourte A, Robitaille Y, Sergeant N, et al. Specific pathological Tau protein variants characterize Pick’s disease. J Neuropathol Exp Neurol. 1996;55:159–68. doi: 10.1097/00005072-199602000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Buée Scherrer V, Hof PR, Buée L, et al. Hyperphosphorylated tau proteins differentiate corticobasal degeneration and Pick’s disease. Acta Neuropathol. 1996;91:351–9. doi: 10.1007/s004010050436. [DOI] [PubMed] [Google Scholar]
- 30.Buée L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999;9:681–93. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 32.Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann M, Rademakers R, Roeber S, et al. Anew subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–31. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai SL, Abramzon Y, Schymick JC, et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2011;32:550.e1–4. doi: 10.1016/j.neurobiolaging.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belzil VV, St-Onge J, Daoud H, et al. Identification of a FUS splicing mutation in a large family with amyotrophic lateral sclerosis. J Hum Genet. 2011;56:247–9. doi: 10.1038/jhg.2010.162. [DOI] [PubMed] [Google Scholar]
- 36.Sproviero W, La Bella V, Mazzei R, et al. FUS mutations in sporadic amyotrophic lateral sclerosis: clinical and genetic analysis. Neurobiol Aging. 2012;33:837.e1–5. doi: 10.1016/j.neurobiolaging.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–26. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- 38.Neumann M, Roeber S, Kretzschmar HA, et al. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009b;118:605–16. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muñoz DG, Neumann M, Kusaka H, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118:617–27. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- 40.Urwin H, Josephs KA, Rohrer JD, et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120:33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armstrong RA, Gearing M, Bigio EH, et al. The spectrum and severity of FUS-immunoreactive inclusions in the frontal and temporal lobes of ten cases of neuronal intermediate filament inclusion disease. Acta Neuropathol. 2001;121:219–28. doi: 10.1007/s00401-010-0753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snowden JS, Hu Q, Rollinson S, et al. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol. 2011;122:99–110. doi: 10.1007/s00401-011-0816-0. [DOI] [PubMed] [Google Scholar]
- 43.Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative disease. Nature Rev Neurology. 2014;10:337–48. doi: 10.1038/nrneurol.2014.78. [DOI] [PubMed] [Google Scholar]
- 44.Orozco D, Tahirovic S, Rentzsch K, et al. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 2012;13:759–64. doi: 10.1038/embor.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orozco D, Edbauer D. FUS-mediated alternative splicing in the nervous system: consequences for ALS and FTLD. J Mol Med. 2013;91:1343–54. doi: 10.1007/s00109-013-1077-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.