

Abstract
Vitiligo is a common autoimmune disease of the skin that results in disfiguring white spots. There are no FDA-approved treatments, and current treatments are time-consuming, expensive, and have low efficacy. We sought to identify new treatments for vitiligo, and first considered repurposed medications because of the availability of safety data and expedited regulatory approval. We previously reported that the IFN-γ-induced chemokine CXCL10 is expressed in lesional skin from vitiligo patients, and that it is critical for the progression and maintenance of depigmentation in our mouse model of vitiligo. We hypothesized that targeting IFN-γ signaling might be an effective new treatment strategy. STAT1 activation is required for IFN-γ signaling and recent studies revealed that simvastatin, an FDA-approved cholesterol-lowering medication, inhibited STAT1 activation in vitro. Therefore, we hypothesized that simvastatin may be an effective treatment for vitiligo. We found that simvastatin both prevented and reversed depigmentation in our mouse model of vitiligo, and reduced the number of infiltrating autoreactive CD8+ T cells in the skin. Treatment of melanocyte-specific, CD8+ T cells in vitro decreased proliferation and IFN-γ production, suggesting additional effects of simvastatin directly on T cells. Based on these data, simvastatin may be a safe, targeted treatment option for patients with vitiligo.
Introduction
Vitiligo is a common, disfiguring autoimmune disease of the skin. Psychological consequences are severe, leading to depression, anxiety, sleep disturbances, sexual dysfunction, feelings of discrimination, and even suicidal thoughts/attempts. These emotional disturbances are comparable to those suffering from psoriasis and eczema (Linthorst Homan et al., 2009; Ongenae et al., 2006). The estimated prevalence of disease is 0.5–1% of the total population (Alikhan et al., 2011), or 1.5–3 million people in the United States (US) and 35–70 million in the world. The estimated direct health care cost burden of vitiligo in the US is $175 million each year (Bickers et al., 2006), a particularly high cost considering that there are few effective, and no systemic, treatments. The mechanisms of current treatments are untested and therefore unknown, although general, non-targeted immunosuppression is likely, and no treatments are effective for all patients. An orally available systemic treatment for vitiligo would be a helpful addition to currently limited treatment options.
Vitiligo pathogenesis incorporates both intrinsic defects within melanocytes that activate the cellular stress response, as well as autoimmune mechanisms that target these cells (Alikhan et al., 2011; Glassman, 2011; Laddha et al., 2013; Mosenson et al., 2012; Mosenson et al., 2013; Passeron and Ortonne, 2012; Schallreuter et al., 2008; Shah and Sinha, 2013; Spritz, 2012; Toosi et al., 2012; van den Boorn et al., 2011). Patients with vitiligo have increased numbers of autoreactive, melanocyte-specific CD8+ T cells in the skin and blood, and functional studies using human skin ex vivo support CD8+ T cells as critical for depigmentation (Ogg et al., 1998; van den Boorn et al., 2009). Recent advances in understanding the key cytokines that promote psoriasis and related autoimmune diseases have resulted in treatments with excellent efficacy and safety profiles, significantly improving patients’ quality of life (Baker et al., 2012). However, the cytokines that drive vitiligo pathogenesis are not shared with these diseases (Rashighi et al., 2014), and cytokine-targeted treatments for psoriasis have been ineffective for vitiligo (Alghamdi et al., 2011; Alghamdi et al., 2012; Dayel and Alghamdi, 2013).
We recently discovered that the IFN-γ-chemokine axis is active in human vitiligo, and functional studies in a vitiligo mouse model that we developed revealed that it is critical for both the progression and maintenance of vitiligo, implicating this pathway as a potential target for new treatments (Harris et al., 2012; Rashighi et al., 2014). IFN-γ signaling requires the activation of STAT1 to induce the transcription of downstream gene targets (Aaronson and Horvath, 2002), and humans deficient in STAT1 have severely impaired IFN-driven responses (Chapgier et al., 2006a; Chapgier et al., 2006b; Dupuis et al., 2001; Dupuis et al., 2003). Mice with selective STAT1-deficiency have impaired IFN-induced signaling and target gene expression (Meraz et al., 1996), although some IFN-induced genes may be STAT1-independent (Ramana et al., 2001). We reasoned that inhibitors of STAT1 activation might also interfere with IFN-γ signaling, and may prove to be effective treatments for vitiligo.
A recent study reported that simvastatin, an HMG-CoA reductase inhibitor approved by the FDA for the treatment of hypercholesterolemia, inhibited IFN-γ-induced STAT1 activation in vitro. This effect was specific for the HMG-CoA reductase pathway rather than an off-target effect, as it was rescued by the addition of mevalonate, a pathway intermediate downstream of HMG-CoA reductase but upstream of cholesterol (Zhao et al., 2011). Interestingly, high-dose simvastatin treatment of a patient with both vitiligo and hypercholesterolemia resulted in rapid repigmentation of his skin, supporting simvastatin treatment as a potential therapy for vitiligo (Noel et al., 2004). Therefore, we tested simvastatin as a treatment in our mouse model of vitiligo. We found that simvastatin both prevented vitiligo and reversed depigmentation in mice with established, widespread disease. It had multiple effects on melanocyte-specific T cells, reducing their numbers in the skin, but also reducing their proliferation and production of IFN-γ, suggesting that multiple pleiotropic effects of simvastatin may contribute to its positive therapeutic response.
Results
Simvastatin treatment reduces the extent of depigmentation in vitiligo
In order to test simvastatin as a potential treatment for vitiligo, we induced vitiligo in mice with black skin and black hair as described previously (Harris et al., 2012). Briefly, KRT14-Kitl*4XTG2Bjl (Krt14-Kitl*) mice express a non-cleavable, membrane-bound form of Kit ligand under the keratin 14 promoter, and therefore expression is limited to the epidermis. As a consequence, these mice have black skin and black hair, without other sequelae (Kunisada et al., 1998). Sublethally irradiated (500 RAD) Krt14-Kitl* mice were used as hosts for the adoptive transfer of PMEL-specific TCR transgenic CD8+ T cells (PMELs), which recognize both mouse and human PMEL (also known as gp100), a melanocyte-specific differentiation antigen (Overwijk et al., 1998). PMELs were isolated from the spleen of donors, column-purified using negative selection, and 106 cells were transferred to hosts intravenously (i.v.). Mice were then infected intraperitoneally (i.p.) with 106 plaque-forming units of recombinant vaccinia virus that expresses human PMEL (rVV-hPMEL), a potent antigenic stimulus for PMELs that results in their activation and expansion in vivo (Overwijk et al., 2003). We have found that this treatment results in depigmentation of the skin 5–7 weeks later, and mice are assigned a Vitiligo Score based on the extent of depigmentation on their ears, nose, feet, and tail (Harris et al., 2012).
To identify the optimal treatment dose of simvastatin we performed a dose-response in our model, testing three increasing doses (0.2mg, 0.4mg, 0.8mg), with the highest dose consistent with treatment in a mouse model of rheumatoid arthritis (Leung et al., 2003). Beginning 1 day after vitiligo induction, we began treatment of hosts with simvastatin (i.p. 3x weekly) or vehicle control until scoring them 5 weeks later. We found that simvastatin treatment reduced depigmentation compared to vehicle-treated controls. There was a strong correlation between the dose and clinical response, with 0.8mg having the most significant response (Figure 1a). Flow cytometry analysis revealed a similar correlation between the dose and the reduction of total number of PMELs present in the ear skin (Figure 1b), and we selected 0.8mg as the optimal dose for repeat experiments (Figure 2a,b). Next, we analyzed the ear skin, tail skin, skin-draining lymph nodes, spleen, and blood by flow cytometry to quantify the total number of PMELs present at those locations in simvastatin-treated versus vehicle-treated controls. We found significantly reduced numbers of PMELs in the skin in simvastatin-treated hosts compared with controls, but no change in number of PMELs at other sites (Figure 2c,d). These observations are consistent with our previous studies blocking IFN-γ in this model (Harris et al., 2012), which suggested that simvastatin could be interfering with IFN-γ signaling.
Figure 1. Simvastatin dose correlates with clinical response and reduction of melanocyte-specific CD8+ T cell in the ear skin.

Vitiligo was induced and mice were treated with 0.2mg, 0.4mg, or 0.8 mg of simvastatin or vehicle control (No Tx) three times weekly for five weeks. (a) There is a strong correlation between the simvastatin dose and clinical response, with 0.8mg having the most significant effect. P-value calculated by ANOVA is shown. Post test for linear trend was also significant (p=0.0014). (b) A similar correlation was found between simvastatin dose and reduction of PMELs in the ear skin, but not skin draining lymph nodes. P-value calculated by ANOVA is shown. Post test for linear trend was also significant (p=0.0003). ns, not significant; *p<0.05; **p<0.01; ***p < 0.001.
Figure 2. Simvastatin prevents depigmentation and melanocyte-specific CD8+ T cell accumulation in the skin despite no global effect on T cell frequency.
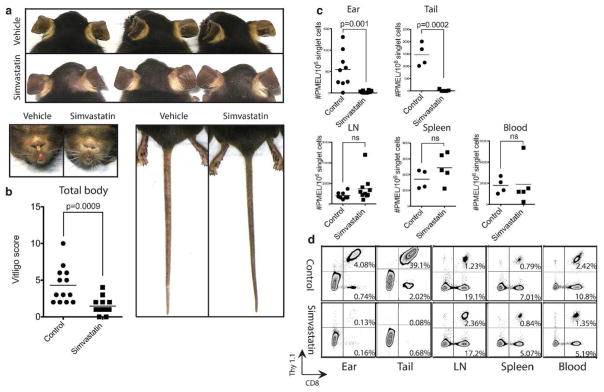
Vitiligo was induced and mice were treated with simvastatin (0.8mg) or vehicle control three times weekly for five weeks. (a) Representative mouse ears, noses, footpads and tails from each group are shown. (b) The effects of simvastatin on vitiligo score and (c) the total numbers of PMELs in the ear skin, tail skin, lymph nodes (LN), spleen and blood. (d) Representative flow cytometry plots are shown.
Acute treatment with simvastatin reduces autoreactive T cells in the skin, but does not affect CXCL10 expression
Long-term, preventative treatment with simvastatin as above could affect PMELs in multiple ways, including reducing the activation or proliferation of PMELs early during priming, migration of PMELs into the skin, or altering their function after their recruitment to the skin. In order to determine whether simvastatin affects T cells directly within the skin, we induced vitiligo in hosts as before and allowed them to develop depigmentation for five weeks. Next, we treated the hosts with 1 dose of simvastatin, 3 daily doses of simvastatin, or vehicle control and analyzed the numbers of PMELs within the skin by flow cytometry. We found that 3 doses of simvastatin significantly reduced the number of PMELs within the ear epidermis, and even a single dose suggested a trend toward reduction, however PMEL numbers at other sites were not affected, including the dermis (Figure 3a,b). Antibody neutralization of IFN-γ had a similar effect (Figure 3c). These results suggest that simvastatin acutely affects PMEL homing or retention directly within the skin, and may inhibit IFN-γ signaling.
Figure 3. Acute treatment with simvastatin reduces T cell numbers in the epidermis.

Vitiligo was induced and mice were treated with 1 dose or 3 daily doses of simvastatin (0.8mg) or vehicle control five weeks after vitiligo induction. (a) Representative flow plots show a reduced number of PMELs in the ear epidermis of the mice that received 1 or 3 daily doses of simvastatin when compared to vehicle. (b) The effects of acute treatment with simvastatin on the total number of PMELs on ear skin, tail skin, lymph nodes, and spleen. (c) Neutralization of IFN-γ has a similar effect on ear skin. (d) The relative expression of CXCL10 was unaffected in the ear skin of mice treated with 1 or 3 daily doses of simvastatin.
Our previous observations revealed that IFN-γ is required for PMEL migration to the skin and progression of disease, as well as the maintenance of depigmentation. In addition, it appeared that IFN-γ-induced CXCL10 may specifically promote PMEL migration within the skin to the epidermis (Rashighi et al., 2014). In order to determine whether CXCL10 expression is directly inhibited by simvastatin, we analyzed CXCL10 expression in the skin by RT-PCR in mice treated with vehicle, 1 dose of simvastatin or 3 daily doses of simvastatin. We found that despite a reduction of PMELs in the epidermis following treatment with simvastatin, CXCL10 expression was unchanged (Figure 3d).
Simvastatin reduces T cell proliferation and effector function in vitro
Because simvastatin did not affect CXCL10 expression in the skin, we tested whether it altered the activation or effector function of T cells directly. PMELs were isolated from the spleen of donors and column-purified for CD8 T cells using negative selection. In order to track proliferation after activation, purified PMELs were labeled with CFSE. PMELs were then activated for 3 days in vitro using anti-CD3 and anti-CD8 antibodies, and treated with either 1, 5, 10 or 100 μM simvastatin or vehicle control. We found that treatment with simvastatin reduced both the proliferation and IFN-γ production by PMELs (Figure 4a,b), suggesting that simvastatin may directly affect T cell function in vitiligo, rather than indirectly by decreasing CXCL10.
Figure 4. Simvastatin inhibits both the proliferation and IFN-γ production of melanocyte-specific CD8+ T cells in vitro through inhibition of the HMG-CoA reductase pathway.

(a,b) PMEL CD8+ T cells were isolated, labeled with CFSE, and activated for 3 days in vitro using anti-CD3 and anti-CD8 antibodies in the presence of either 1, 5, 10 or 100 μM simvastatin or vehicle control. PMEL CD8+ T cells were then stained for intracellular IFN-γ and analyzed for (a) proliferation or (b) IFN-γ production by flow cytometry. (c,d) Simvastatin-treated PMEL cultures were supplemented with 1mM mevalonate or vehicle control and CD8+ T cell (c) proliferation and (d) IFN-γ production were analyzed by flow cytometry. Representative (e) histograms for CFSE dilution and (f) dot plots for IFN-γ. Blue represents control unstimulated samples.
We next determined whether these direct effects of simvastatin on T cells were through inhibition of the HMG-CoA reductase pathway, or through an off-target effect. HMG-CoA reductase catalyzes the formation of mevalonate, an intermediate in the cholesterol synthesis pathway (Zhao et al., 2011). In order to test whether mevalonate supplementation could rescue proliferation and IFN-γ production by PMELs in the presence of simvastatin, we supplemented simvastatin-treated PMEL cultures with 1 mM mevalonate or vehicle control. We found that mevalonate rescued both proliferation and IFN-γ production by PMELs, suggesting that direct inhibition of the HMG-CoA reductase pathway, rather than an off-target effect, was responsible for reduced proliferation and function of PMELs in the presence of simvastatin (Figure 4c–f).
Simvastatin reverses established vitiligo
Because simvastatin prevented depigmentation in our mouse model and directly reduced the numbers of PMELs in the skin in vivo as well as the function of PMELs in vitro, we tested whether simvastatin could also reverse established depigmentation in our mouse model of vitiligo. Mice were induced with vitiligo as above, allowed to depigment, and then stabilize for ten to twelve weeks. Those with >50% depigmentation on their tails were randomized into two treatment groups, either with simvastatin 0.8mg three times weekly or vehicle control for a total of four to six weeks. Photographs of the tails before and after treatment were analyzed using ImageJ to calculate the percent pigmentation of the tail, and therefore the degree of repigmentation, after treatment. Despite no effect on the total number of melanocyte-specific CD8+ T cells present in the skin, we found that there was a significant increase in pigmentation in the group treated with simvastatin compared to the control group, as evidenced by the newly appearing macules of pigment around the follicles (Figure 5a–d), providing a rationale for clinical studies in humans to test simvastatin as a potential treatment for vitiligo.
Figure 5. Simvastatin reverses established vitiligo.

Mice with extensive depigmentation on the tail (>50%) were treated with simvastatin (0.8mg) or vehicle control three times weekly beginning twelve weeks post vitiligo induction for a total of four to six weeks. Photographs of each tail before and after treatment were analyzed using ImageJ software to calculate the amount of repigmentation. (a) Simvastatin treatment in established vitiligo did not significantly affect total number of PMELs in skin. (b) The mean percent change in pigmentation from baseline was −3.1% and 8.4% for the mice treated with PBS or simvastatin, respectively. (c) Paired t-test showed a significant increase in percent tail pigmentation only in mice treated with simvastatin. (d) A representative tail from each group before and after treatment is shown.
Discussion
Here we report the ability of simvastatin to both prevent and reverse established depigmentation in a mouse model of vitiligo. This is consistent with previous observations that the IFN-γ-signaling pathway is critical for both the progression and maintenance of depigmentation in our mouse model (Harris et al., 2012)), that STAT1 activation is required for IFN-γ signaling (Aaronson and Horvath, 2002), that simvastatin inhibits IFN-γ-induced STAT1 activation in vitro (Li et al., 2011; Zhao et al., 2011), and that simvastatin reversed depigmentation in a patient with vitiligo (Noel et al., 2004). We hypothesized that the mechanism of action of simvastatin in vitiligo was to inhibit IFN-γ-induced CXCL10 production in the skin, and therefore interfere with proper homing of autoreactive T cells to the epidermis. However despite its clear efficacy in our mouse model of vitiligo, its mechanism of action does not appear to be straightforward. This is consistent with multiple previous studies showing pleiotropic effects of statins on various T-cell mediated autoimmune diseases (Greenwood et al., 2006).
Simvastatin did indeed prevent the migration of PMEL T cells to the skin during vitiligo progression while not decreasing their viability, as their numbers were unaffected in skin-draining lymph nodes, spleen, and blood. These effects are similar to what we observed in our model when we neutralized IFN-γ (Harris et al., 2012). Short-term treatment with simvastatin reduced the number of PMELs within the ear epidermis but not tail, which may be due to the difference in their thickness. The epidermis of the tail is multilayered, while the epidermis of the ear is only one cell thick. CD8+ T cells, which drive melanocyte destruction in humans and our mouse model, have been reported to express adhesion molecules upon entry into the epidermis, which helps to retain them there. One possibility is that the thicker epidermis of the tail with more adhesion molecules is better able to retain the PMELs than that of the thinner ear epidermis. Alternatively, simvastatin may have better penetration of the thinner epidermis of the ear compared to the tail, although high lipophilicity of simvastatin makes this less likely.
Despite the ability of simvastatin to reduce the number of PMELs in the epidermis, we found that CXCL10 expression was not reduced in the skin after short-term treatment, suggesting that this effect of simvastatin was independent of CXCL10. We then found that simvastatin reduced T cell proliferation and IFN-γ production in vitro. This is consistent with a study that reported decreased superantigen-induced IFN-γ production by human CD4+ T cells ex vivo isolated from subjects treated with simvastatin compared to their responses before treatment (Fehr et al., 2004), suggesting that it has additional effects on T cells. STAT1 is also phosphorylated by IL-12 (Schroder et al., 2004), a cytokine that induces IFN-γ production, and certain STAT1 mutations in humans result in decreased IFN-γ production after IL-12 stimulation (van de Veerdonk et al., 2011), suggesting that STAT1 inhibition by simvastatin may have effects upstream of IFN-γ production as well. Alternatively, inhibition of HMG-CoA reductase may affect signaling mediators other than STAT1 that are required for T cell proliferation and/or IFN-γ production. Interestingly, despite reversing vitiligo, simvastatin did not affect the number of PMELs cells in the skin in established disease, suggesting that simvastatin applies this effect primarily by mechanisms other than recruitment of autoreactive T cells. Previous studies have shown that statins could have multiple additional effects on T cells, in addition to their ability to influence T cell recruitment and proliferation. Consistent with this, we found a direct effect on IFN-γ production after stimulation in vitro, and thus hypothesize that simvastatin inhibits the activation or effector status of PMELs that remain in the skin to promote repigmentation. Further experiments are needed to elucidate the exact mechanisms by which simvastatin acts to reverse vitiligo in our mouse model.
The dose of simvastatin used for our in vivo studies in mice (up to 40 mg/kg) is much higher than what is used in humans (up to 80 mg/day, ~1 mg/kg). However high doses are required for treatment of rodents because of their rapid upregulation of HMG-CoA reductase in response to treatment with statins (Kita et al., 1980). The optimal dose we identified in our mouse model is consistent with established active doses in rodents (Dimmeler et al., 2001; Mundy et al., 1999; Ni et al., 2001), as well as doses tested in a mouse model of rheumatoid arthritis (Leung et al., 2003). Notably, the one vitiligo patient who reportedly responded to simvastatin clinically was on the highest FDA-approved dose of simvastatin, 80mg per day (Noel et al., 2004).
Statins are metabolized and concentrate in the liver, resulting in low levels circulating in the plasma and through tissues (Stamm and Ornstein, 2005). Therefore, high doses may be required for efficacy in humans if the target tissue is the skin or blood. However the 80mg dose of simvastatin increases the risk of myopathy and rhabdomyolysis, which led to removal of this dose by the FDA (Desai et al., 2014). Whether simvastatin will be required for successful treatment of vitiligo patients, or whether other statins with fewer side effects could be effective, is unknown. Simvastatin is highly lipophilic (Stamm and Ornstein, 2005), which may promote its accumulation in multiple tissues other than the liver, including the skin. This may contribute to its therapeutic effects in vitiligo, but may also be why it has more side effects. Future studies in humans may consider testing simvastatin in a topical formulation, as this should result in fewer side effects, and topical simvastatin was reported to reduce inflammation in a mouse model of irritant dermatitis (Otuki et al., 2006).
Simvastatin is an inexpensive, relatively safe FDA-approved medication that prevents and reverses depigmentation in our vitiligo mouse model, and reversed depigmentation in one reported vitiligo patient (Noel et al., 2004). Its use may be repurposed to provide the first systemic treatment option for patients with vitiligo, and could also be reformulated as a topical treatment option. Based on these observations, a clinical trial in a small number of patients with high-dose simvastatin would be useful to determine whether larger studies to prove efficacy would be a worthwhile investment.
Materials & Methods
Mice
KRT14-Kitl*4XTG2Bjl (Krt14-Kitl*) mice were a gift from BJ Longley, University of Wisconsin. Thy1.1+ PMEL TCR transgenic mice were obtained from The Jackson Laboratory (Bar Harbor, ME, stock no. 005023, B6.Cg Thy1a/CyTg(TcraTcrb)8Rest/J). All mice were on a C57BL/6J background, were maintained in pathogen-free facilities at the University of Massachusetts Medical School, and procedures were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee.
Simvastatin preparation
Simvastatin (Sigma-Aldrich, St Louis, MO) was activated prior to use as described previously (Sadeghi et al, 2000). Briefly, 12 mg of simvastatin was dissolved in 300 μl of ethanol and then 450 μl of 0.1 N NaOH was added to the solution and subsequently incubated at 50 °C for 2 hours. The pH was then brought to 7.0 by adding HCl, the final concentration of the stock solution was adjusted to 4 mg/ml and kept at 4°C.
Induction of vitiligo and treatment of mice
Vitiligo was induced through adoptive transfer of PMEL CD8+ T cells as described previously (Harris et al, 2012). Briefly, PMEL CD8+ T cells were isolated from the spleens of PMEL TCR transgenic mice through negative selection on microbeads (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions. Purified CD8+ T cells (1×106) were injected intravenously into sublethally irradiated (500 rads 1 day before transfer) Krt14-Kitl* hosts (12 to 16 weeks of age). Recipient mice also received intraperitoneal injection of 1 × 106 plaque-forming units of rVV-hPMEL (N. Restifo, National Cancer Institute, NIH) on the same day of transfer. Treatment with simvastatin to prevent vitiligo was performed by i.p. injection of 0.8 mg of the drug three times weekly for the duration of the 5 weeks. Control mice received either no treatment or were treated with an equal volume of PBS. Vitiligo score was objectively quantified by an observer blinded to the experimental groups, using a point scale based on the extent of depigmentation at four easily visible locations, including the ears, nose, rear footpads, and tails as described previously (Harris et al, 2012). Each location was examined, and the extent of depigmentation was estimated as a percentage of the anatomic site; both left and right ears and left and right rear footpads were estimated together and therefore evaluated as single sites. Points were awarded as follows: no evidence of depigmentation (0%) received a score of 0, >0 to 10% = 1 point, >10 to 25% = 2 points, >25 to 75% = 3 points, >75 to <100% = 4 points, and 100% = 5 points. The “vitiligo score” was the sum of the scores at all four sites, with a maximum score of 20 points. Acute treatment with simvastatin was performed by i.p. injection of either 1 dose or 3 daily doses of simvastatin (0.8mg) five weeks after vitiligo induction, or vehicle control. Acute IFN-γ blockade was through i.p. injection of 3 daily doses of IFN-γ–neutralizing antibody (500 μg, XMG-6) or vehicle control.
To induce repigmentation, 10 to 12 weeks after induction of vitiligo mice with at least 50% tail depigmentation were randomly assigned to receive either 0.8 mg of simvastatin as before or vehicle control for a total of 4 to 6 weeks. Treatment efficacy was objectively quantified by comparison of the tail photographs before and after treatment with ImageJ software (NIH). Briefly, all images were converted to 8-bit black and white, and the brightness threshold adjusted to one hundred, converting all pigmented areas to black and all depigmented areas to white. The outline of the tail was selected, and the mean area over threshold calculated, representing the fraction of pigmented tail skin. This was multiplied by one hundred, converting the fraction to percent pigmentation of the tail. Percent change over baseline reflects the percent pigmentation after treatment minus the percent pigmentation before treatment, and therefore, a positive number reflects percent repigmentation while a negative number reflects further depigmentation.
In vitro T cell proliferation and cytokine production assays
TCR transgenic CD8+ T cells that recognize gp100 (PMELs) were isolated from the spleens of transgenic mice using a MACS CD8 negative isolation kit as mentioned above. Isolated CD8+ T cells were suspended at 1.0 × 107 cells/mL in 2mM CFSE (Invitrogen, Carlsbad, CA) in PBS with 0.1% Fetal Bovine Serum and incubated for 10 minutes at 37°C. Subsequently cold FBS was added at an equal volume, the cells were centrifuged at 350g, and re-suspended in T-cell media (RPMI 1640 gibco, 10% FBS, 2mM Glutamax, 1mM Sodium Pyruvate, 10mM HEPES, 0.5X Non-Essential Amino Acids, 50μM β-Mercaptoethanol). 5.0 × 104 cells per well were incubated in a 96 well plate for 72 hours at 37°C. Wells in a 96 well plate were previously coated overnight with 3μg/ml CD3 antibody in PBS at 4°. Stimulated cells were incubated in the presence of 2μg/mL of soluble CD28 antibody and unstimulated cells were incubated in uncoated wells. Cells were also incubated with simvastatin or both simvastatin and 1mM (S)- Mevalonic Acid Lithium Salt (Sigma-Aldrich, St Louis, MO). Surface staining for flow cytometry was then performed for CD45 (Biolegend clone 30-F11), CD8β (Biolegend clone: YTS1560707), Thy1.1 (Biolegend clone: OX-7), and intracellular staining was performed for IFN-γ (ebioscience clone: XMG1.2). Data were pooled from three separate experiments, and the average numbers from untreated groups (neither simvastatin nor mevalonate) were used for normalization.
Flow cytometry
Ears, tails, spleens, and skin-draining lymph nodes were harvested at the indicated times. Spleens were disrupted, and the red blood cells were lysed with RBC lysis buffer. Ear and tail skin were incubated in skin digest medium [RPMI containing 0.5% deoxyribonuclease (DNase) I (Sigma-Aldrich, St Louis, MO) and liberase TL enzyme blend (0.5 mg/ml) (Roche, Indianapolis, IN)] and processed with a medimachine (BD Biosciences, San Jose, CA) as described previously (Harris et al, 2012). For separation of the dermis and epidermis, tail skin samples were incubated with dispase (2.4 U/ml) (Roche, Indianapolis, IN) for 1 hour at 37°C. Epidermis was removed and mechanically disrupted with 70-μm cell strainers, and dermis samples were incubated with collagenase IV (1 mg/ml) with DNase I (0.5 mg/ml) (Sigma-Aldrich, St Louis, MO) for 1 hour at 37°C on a shaker. Cells were filtered through a 70-μm mesh before analysis. The following antibodies were obtained from BioLegend (San Diego, CA): mouse (CD8β, CD45.2, CD90.1, and Fc block). The data were collected and analyzed with a BD LSR II flow cytometer (BD Biosciences, San Jose, CA) and FlowJo (Tree Star Inc., Ashland, OR).
Quantitative real-time reverse transcriptase–PCR
The expression of CXCL10 in mouse tissues was performed using quantitative real-time reverse transcriptase-PCR. Ear and tail skin from 3 control and 3 simvastatin treated mice were analyzed in each experiment. The tissues obtained were stored at −80°C until RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Complimentary DNA (cDNA) was generated with iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Real-time PCR was conducted with cDNA and iQ SYBR Green (Bio-Rad, Hercules, CA) in a Bio-Rad iCycler iQ, according to manufacturer recommendations. Mouse primer sequences are as follows: Cxcl10 5′-AGGGGAGTGATGGAGAGAGG-3′ (sense), 5′-TGAAAGCGTTTAGCCAAAAAAGG-3′ (antisense); actin-beta (Actb): 5′-GGCTGTATTCCCCTCCATCG-3′ (sense), 5′-CCAGTTGGTAACAATGCCATGT-3′ (antisense). CXCL10 expression is reported after normalization to expression of ACTB. Data were pooled from three separate experiments and gene expression is reported relative to the lowest expression in untreated mice within each experiment after normalization to expression of β-actin.
Statistical analysis
Statistical analysis was performed using Prism software (GraphPad Software, La Jolla, CA). Dual comparisons were made using the paired or unpaired Student’s t-test when applicable. Groups of three or more were analyzed by ANOVA with Dunnett’s post-tests. P values < 0.05 were considered significant.
Acknowledgments
We would like to thank B. J. Longley for Krt14-Kitl* mice and N. Restifo for recombinant vaccinia virus. This project was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the NIH, under Award Number AR061437, and research grants from the Vitiligo Research Foundation, Kawaja Family Vitiligo Research Initiative Award, and Dermatology Foundation Stiefel Scholar Award (to J.E.H.).
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–5. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Alghamdi KM, Khurrum H, Rikabi A. Worsening of vitiligo and onset of new psoriasiform dermatitis following treatment with infliximab. Journal of cutaneous medicine and surgery. 2011;15:280–4. doi: 10.2310/7750.2011.10068. [DOI] [PubMed] [Google Scholar]
- Alghamdi KM, Khurrum H, Taieb A, et al. Treatment of generalized vitiligo with anti-TNF-alpha Agents. J Drugs Dermatol. 2012;11:534–9. [PubMed] [Google Scholar]
- Alikhan A, Felsten LM, Daly M, et al. Vitiligo: a comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol. 2011;65:473–91. doi: 10.1016/j.jaad.2010.11.061. [DOI] [PubMed] [Google Scholar]
- Baker EL, Coleman CI, Reinhart KM, et al. Effect of Biologic Agents on Non-PASI Outcomes in Moderate-to-Severe Plaque Psoriasis: Systematic Review and Meta-Analyses. Dermatology and therapy. 2012;2:9. doi: 10.1007/s13555-012-0009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickers DR, Lim HW, Margolis D, et al. The burden of skin diseases: 2004 a joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol. 2006;55:490–500. doi: 10.1016/j.jaad.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Chapgier A, Boisson-Dupuis S, Jouanguy E, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS genetics. 2006a;2:e131. doi: 10.1371/journal.pgen.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapgier A, Wynn RF, Jouanguy E, et al. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol. 2006b;176:5078–83. doi: 10.4049/jimmunol.176.8.5078. [DOI] [PubMed] [Google Scholar]
- Dayel SB, Alghamdi K. Failure of alefacept in the treatment of vitiligo. J Drugs Dermatol. 2013;12:159–61. [PubMed] [Google Scholar]
- Desai CS, Martin SS, Blumenthal RS. Non-cardiovascular effects associated with statins. BMJ. 2014;349:g3743. doi: 10.1136/bmj.g3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–7. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis S, Dargemont C, Fieschi C, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–3. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- Dupuis S, Jouanguy E, Al-Hajjar S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- Fehr T, Kahlert C, Fierz W, et al. Statin-induced immunomodulatory effects on human T cells in vivo. Atherosclerosis. 2004;175:83–90. doi: 10.1016/j.atherosclerosis.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond) 2011;120:99–120. doi: 10.1042/CS20090603. [DOI] [PubMed] [Google Scholar]
- Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nature reviews Immunology. 2006;6:358–70. doi: 10.1038/nri1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JE, Harris TH, Weninger W, et al. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol. 2012;132:1869–76. doi: 10.1038/jid.2011.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T, Brown MS, Goldstein JL. Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of the reductase. J Clin Invest. 1980;66:1094–100. doi: 10.1172/JCI109938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisada T, Lu SZ, Yoshida H, et al. Murine cutaneous mastocytosis and epidermal melanocytosis induced by keratinocyte expression of transgenic stem cell factor. J Exp Med. 1998;187:1565–73. doi: 10.1084/jem.187.10.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laddha NC, Dwivedi M, Mansuri MS, et al. Vitiligo: interplay between oxidative stress and immune system. Exp Dermatol. 2013;22:245–50. doi: 10.1111/exd.12103. [DOI] [PubMed] [Google Scholar]
- Leung BP, Sattar N, Crilly A, et al. A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J Immunol. 2003;170:1524–30. doi: 10.4049/jimmunol.170.3.1524. [DOI] [PubMed] [Google Scholar]
- Li N, Salter RC, Ramji DP. Molecular mechanisms underlying the inhibition of IFN-gamma- induced, STAT1-mediated gene transcription in human macrophages by simvastatin and agonists of PPARs and LXRs. Journal of cellular biochemistry. 2011;112:675–83. doi: 10.1002/jcb.22976. [DOI] [PubMed] [Google Scholar]
- Linthorst Homan MW, Spuls PI, de Korte J, et al. The burden of vitiligo: patient characteristics associated with quality of life. J Am Acad Dermatol. 2009;61:411–20. doi: 10.1016/j.jaad.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–42. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Mosenson JA, Zloza A, Klarquist J, et al. HSP70i is a critical component of the immune response leading to vitiligo. Pigment Cell Melanoma Res. 2012;25:88–98. doi: 10.1111/j.1755-148X.2011.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosenson JA, Zloza A, Nieland JD, et al. Mutant HSP70 reverses autoimmune depigmentation in vitiligo. Science translational medicine. 2013;5:174ra28. doi: 10.1126/scitranslmed.3005127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy G, Garrett R, Harris S, et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286:1946–9. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- Ni W, Egashira K, Kataoka C, et al. Antiinflammatory and antiarteriosclerotic actions of HMG-CoA reductase inhibitors in a rat model of chronic inhibition of nitric oxide synthesis. Circulation research. 2001;89:415–21. doi: 10.1161/hh1701.096614. [DOI] [PubMed] [Google Scholar]
- Noel M, Gagne C, Bergeron J, et al. Positive pleiotropic effects of HMG-CoA reductase inhibitor on vitiligo. Lipids Health Dis. 2004;3:7. doi: 10.1186/1476-511X-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg GS, Rod Dunbar P, Romero P, et al. High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo. The Journal of experimental medicine. 1998;188:1203–8. doi: 10.1084/jem.188.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongenae K, Beelaert L, van Geel N, et al. Psychosocial effects of vitiligo. J Eur Acad Dermatol Venereol. 2006;20:1–8. doi: 10.1111/j.1468-3083.2005.01369.x. [DOI] [PubMed] [Google Scholar]
- Otuki MF, Pietrovski EF, Cabrini DA. Topical simvastatin: preclinical evidence for a treatment of skin inflammatory conditions. J Dermatol Sci. 2006;44:45–7. doi: 10.1016/j.jdermsci.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. The Journal of experimental medicine. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–86. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passeron T, Ortonne JP. Activation of the unfolded protein response in vitiligo: the missing link? J Invest Dermatol. 2012;132:2502–4. doi: 10.1038/jid.2012.328. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Han Y, et al. Stat1-independent regulation of gene expression in response to IFN-gamma. Proc Natl Acad Sci U S A. 2001;98:6674–9. doi: 10.1073/pnas.111164198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashighi M, Agarwal P, Richmond JM, et al. CXCL10 Is Critical for the Progression and Maintenance of Depigmentation in a Mouse Model of Vitiligo. Science translational medicine. 2014;6:223ra23. doi: 10.1126/scitranslmed.3007811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi MM, Collinge M, Pardi R, et al. Simvastatin modulates cytokine-mediated endothelial cell adhesion molecule induction: involvement of an inhibitory G protein. J Immunol. 2000;165:2712–8. doi: 10.4049/jimmunol.165.5.2712. [DOI] [PubMed] [Google Scholar]
- Schallreuter KU, Bahadoran P, Picardo M, et al. Vitiligo pathogenesis: autoimmune disease, genetic defect, excessive reactive oxygen species, calcium imbalance, or what else? Exp Dermatol. 2008;17:139–40. doi: 10.1111/j.1600-0625.2007.00666_1.x. discussion 41–60. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, et al. Interferon-gamma: an overview of signals, mechanisms and functions. Journal of leukocyte biology. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Shah AA, Sinha AA. Oxidative stress and autoimmune skin disease. Eur J Dermatol. 2013;23:5–13. doi: 10.1684/ejd.2012.1884. [DOI] [PubMed] [Google Scholar]
- Spritz RA. Six decades of vitiligo genetics: genome-wide studies provide insights into autoimmune pathogenesis. J Invest Dermatol. 2012;132:268–73. doi: 10.1038/jid.2011.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm JA, Ornstein DL. The role of statins in cancer prevention and treatment. Oncology (Williston Park) 2005;19:739–50. discussion 53–4. [PubMed] [Google Scholar]
- Toosi S, Orlow SJ, Manga P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J Invest Dermatol. 2012;132:2601–9. doi: 10.1038/jid.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Plantinga TS, Hoischen A, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011;365:54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- van den Boorn JG, Konijnenberg D, Dellemijn TA, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. 2009;129:2220–32. doi: 10.1038/jid.2009.32. [DOI] [PubMed] [Google Scholar]
- van den Boorn JG, Melief CJ, Luiten RM. Monobenzone-induced depigmentation: from enzymatic blockade to autoimmunity. Pigment Cell Melanoma Res. 2011;24:673–9. doi: 10.1111/j.1755-148X.2011.00878.x. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Gartner U, Smith FJ, et al. Statins downregulate K6a promoter activity: a possible therapeutic avenue for pachyonychia congenita. J Invest Dermatol. 2011;131:1045–52. doi: 10.1038/jid.2011.41. [DOI] [PubMed] [Google Scholar]