

Abstract
While semiconductor quantum dots (QDs) have been used successfully in numerous single particle tracking (SPT) studies due to their high photoluminescence efficiency, photostability, and broad palette of emission colors, conventional QDs exhibit fluorescence intermittency or ‘blinking,’ which causes ambiguity in particle trajectory analysis and limits tracking duration. Here, non-blinking ‘giant’ quantum dots (gQDs) are exploited to study IgE-FcεRI receptor dynamics in live cells using a confocal-based 3D SPT microscope. There is a 7-fold increase in the probability of observing IgE-FcεRI for longer than 1 min using the gQDs compared to commercially available QDs. A time-gated photon-pair correlation analysis is implemented to verify that selected SPT trajectories are definitively from individual gQDs and not aggregates. The increase in tracking duration for the gQDs allows the observation of multiple changes in diffusion rates of individual IgE-FcεRI receptors occurring on long (>1 min) time scales, which are quantified using a time-dependent diffusion coefficient and hidden Markov modeling. Non-blinking gQDs should become an important tool in future live cell 2D and 3D SPT studies, especially in cases where changes in cellular dynamics are occurring on the time scale of several minutes.
1. Introduction
Over the past several decades, single particle tracking (SPT) has been used extensively to interrogate a variety of processes in live cells, ranging from membrane receptor dynamics[1] to motor protein kinetics.[2] For SPT, the molecules or proteins of interest can be labeled with large (>100 nm) fluorescent particles such as polystyrene beads[2a] or gold nanoparticles that scatter light,[1c,3] or smaller fluorophores such as fluorescent proteins[4] and organic dyes.[5] Unfortunately, large particles (>30 nm) used for scattering may hinder diffusion and fluorescent proteins and organic dyes are susceptible to photobleaching.
Alternatively, quantum dots (QDs), which are generally smaller (<10 nm in diameter) than large scattering particles and relatively resistant to photobleaching, have gained wide-spread use in SPT studies.[1d,e, 6] By a combination of composition and size-tuning, QDs can be produced to have a variety of emission wavelengths for multiplexed imaging using a single excitation source. Furthermore, they can be coated with biocompatible surface ligands and conjugated to a variety of biomolecules for specific targeting in vivo or in vitro.[7] However, QDs are characterized by fluorescence intermittency or blinking,[8] which limits tracking duration[9] and causes ambiguity in the interpretation of the particle’s trajectory as well as inaccurate quantification of the diffusion rate.[10] Recently, a new functional class of core/shell QD was developed[11] and extensively studied[12] that in contrast with conventional QDs provides substantially suppressed blinking and significantly enhanced photo- and chemical stability.[13] The novel properties were first realized in CdSe/CdS core/shell QDs comprised of a unusually thick CdS shell (8–20 monolayers CdS, where one shell monolayer is 0.3375 nm). These thick-shell[11b] or so-called ‘giant’ QDs (gQDs)[11a] have been further optimized to have essentially blinking-free behavior over extended continuous excitation.[14] Importantly, these non-blinking gQDs are not so large (<20 nm) as to hinder the diffusion of certain biomolecules, such as membrane proteins, and have been used successfully for SPT applications in viscous solvents[12d] and to resolve ~50 nm vertical displacements of microtubules in vitro.[15]
Here, we explore the application of these uniquely stable gQDs for three-dimensional tracking of single proteins in live cells. As a model system, we investigate the dynamics of the IgE-FcεRI receptor, which is involved in the allergic response.[16] The dynamics of the IgE-FcεRI system have been characterized previously using SPT and commercial QDs, with QD-conjugated IgE shown to behave essentially the same as IgE lacking the QD label.[1e,17] Specifically, using conventional QDs it was revealed that a range of different dynamic processes occur during different stages of the signal transduction cascade.[1e] On unstimulated mast cells, the IgE-FcεRI receptor diffuses on the membrane surface with a diffusion constant, D, of ~10−1 μm2/s.[1e,17,18] Upon crosslinking multiple IgE-FcεRI receptors with a multivalent antigen, the receptors become immobile and initiate a phosphorylation cascade leading to the release of histamine and cytokines.[19] To desensitize the cell or dampen the stimulatory signal, the IgE-FcεRI receptors are endocytosed,[20] having transport velocities of ~1 μm/s.[2c,6b]
Notably, the IgE-FcεRI receptor can transverse the entire range of the mast cell, ~10 μm in width (the x and y dimension) and ~5 μm in height (the z dimension), with a wide range of diffusion constants and velocities. Conventional 2-dimensional image-plane based SPT studies therefore fail to capture a significant portion of the IgE-FcεRI motion, particularly in the z dimension as the fluorophore goes out of focus after moving on the order of ±500 nm. Therefore, much of what is known about the IgE-FcεRI dynamics comes from combining information from many short SPT trajectories at different image planes. A SPT method and probe which interrogates individual receptor dynamics throughout the entire spatial extent of the cell during the entire lifecycle of the allergic response (receptor activation, stimulation, and downregulation) would be of great utility.
To overcome the limits of 2D SPT methods, we track the IgE-FcεRI receptor in three dimensions (3D) exploiting a fast (5 ms update) high resolution (~50 nm tracking accuracy in xy and ~100 nm in z) confocal-based 3D tracking microscope.[4c,6b,9,18,21] Our 3D tracking microscope is essentially a custom designed stage-scanning confocal microscope that employs a unique 4-element spatial filter geometry and active feedback 200 times per second to follow fast, 3D motion.[4c,6b,9,18,21] This 3D tracking microscope provides high temporal tracking resolution (5 ms) as well as a large tracking range (30 μm in x and y, and 10 μm in z) needed to follow IgE-FcεRI at high diffusion or transport rates throughout the entire spatial extent of the mast cell.
In addition to advanced 3D SPT methods, here we introduce the use of non-blinking and photobleaching-suppressed gQDs for use as a stable molecular probe to study IgE-FcεRI receptor dynamics in live cells. The combination of these two novel technologies, non-blinking gQDs and confocal-based 3D tracking, enables tracking receptor dynamics for extended periods of time as compared to conventional 2D imaging using blinking and/or short-lived probes. This extended tracking duration in 3D allows for a unique characterization of changes in biophysical dynamics which occur on long time scales (minutes) and offers the potential to characterize IgE-FcεRI dynamics at the single molecule level throughout the lifecycle of receptor-mediated allergic response.
2. Results and Discussion
2.1. gQD Water Solubilization, Bioconjugation, and Mast Cell Labeling
Our optimized non-blinking gQD sample consisted of a 4-nm CdSe core with a shell of 16 CdS monolayers. To render the gQDs water soluble and biocompatible, ligand exchange was performed using a custom polyethylene glycol (PEG) ligand.[22] One end of the polymer contains a dihydrolipoic acid (DHLA) group for anchoring the PEG to the QD surface, while the other end contains a carboxyl group for subsequent bioconjugation. This custom-made ligand[22] afforded the gQDs adequate stability over a range of pH conditions and salt concentrations and within the harsh environment of the living cell. After ligand exchange, the hydrodynamic diameter of the gQDs was determined to be ~20 nm using particle sizing by SPT and atomic force microscopy (Figure S1). This size agrees with the hard diameter of ~15 nm obtained by transmission electron microscopy. The carboxyl-terminated gQDs were then conjugated to streptavidin (SA) using carbodiimide coupling chemistry (Figure 1a), followed by removal of excess SA with size-exclusion chromatography (Figure 1b). The conjugation of gQD to SA was verified by agarose gel electrophoresis (Figure 1c). To label the IgE, we added ~1 equivalent of biotin-IgE[1e] (with an antigen binding region specific to 2,4-dinitrophenol, DNP) to gQD-SA (Figure 1a). To selectively label FcεRI receptors, we incubated Rat Basophilic Leukemia (RBL-2H3) mast cells containing FcεRI with the resulting gQD-IgE conjugate. gQD-IgE specifically bound to RBL-2H3 cells when compared to a gQD-SA control (see Section 4). In addition, gQD-IgE competently binds multivalent antigen (bovine serum albumin containing ~23 DNP groups, DNP-BSA), as measured by an enzyme-linked immunosorbent assay (see Section 4). Labeling the IgE with the gQD enables the study of IgE-FcεRI dynamics in both unstimulated and stimulated mast cells upon addition of DNP-hapten bearing ligand. To monitor the dynamics of the antigen, we added ~10 equivalents of a custom made biotin-fibritin-DNP trimer (biotin-DNP) to gQD-SA (Figure 1a), which could then be added to the RBL-2H3 cells primed with unlabeled IgE. Details regarding the growth and labeling of the RBL-2H3 cells with gQD-IgE or gQD-DNP are found in Section S1. To compare measured gQD trajectrories in live cells with those of conventional blinking QDs, we performed similar experiments with Qdot 655 SA (QD655, Life Technologies) conjugated to biotin-IgE as described previously.[1e,6b]
Figure 1.
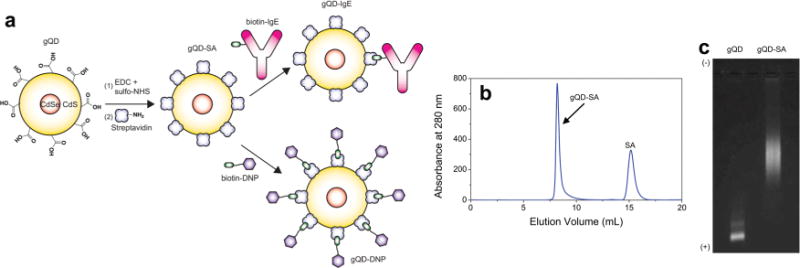
(a) Bioconjugation of gQDs to IgE or DNP. Carboxyl-terminated gQDs are functionalized with succinimidyl (NHS) ester groups by addition of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and sulfo-NHS, and subsequently conjugated to lysine amine groups within SA. gQD-SA could be coupled either to biotin-IgE or biotin-DNP. (b) Following coupling to SA, size exclusion chromatography showed two distinct peaks, corresponding to SA and gQD-SA. (c) 1% Agarose gel showing shift in gQD-SA relative to free gQD, verifying bioconjugation.
2.2. Extended Tracking Duration of Non-blinking gQDs Compared to QD655
Figure 2 directly compares 3D trajectories obtained during tracking for the commercially available blinking QD655-IgE versus the non-blinking gQD-IgE. We note the emission rate from the gQD-IgE (Figure 2b) is much steadier than that of commercial QD655 labeled IgE (Figure 2a), which often has intensity values dropping down to baseline levels. The distributions of intensity values for each example trajectory are shown in Figure 2c and d, along with the theoretical Poisson distribution centered at the average emission value. The blinking QD655-IgE intensity distribution (Figure 2c) clearly deviates from that of a Poisson distribution, as the ratio of the standard deviation over the square root of the mean, η, is ~10 (η = 1 for a pure Poisson distribution). These intensity fluctuations are what lead to a short tracking duration or ambiguity in the interpretation of the particle’s trajectory as our 3D tracking feedback loop benefits from a constant emission signal. In contrast, η ~ 2 for the stable gQD-IgE intensity distribution (Figure 2d) which allows for an extended tracking duration. The η value here is not quite 1 due to technical noise, background, and tracking error. We note the peak average photon detection rate for hundreds of gQD-IgE trajectories (~40 kHz) is consistent with the gQD brightness per particle (BPP) measured by fluorescence correlation spectroscopy (FCS) (Section S2).
Figure 2.
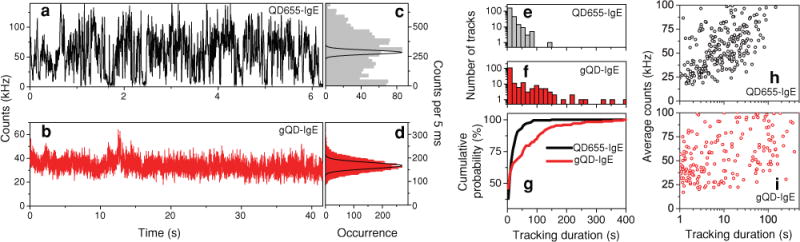
Representative intensity trajectories for (a) QD655-IgE and (b) gQD-IgE during 3D tracking in live cells and corresponding intensity distributions for (c) QD655-IgE and (d) gQD-IgE. Both intensity distributions are overlayed with the theoretical Poisson distribution centered at the average count rate. Distribution of tracking durations for (e) 239 QD655-IgE tracks and (f) 177 gQD-IgE tracks and corresponding cumulative distributions (g) for particles exhibiting average count rates less than 100 kHz and a tracking duration greater than 1 s. Average count rate vs. tracking duration for (h) QD655-IgE and (i) gQD-IgE. All data were acquired using pulsed, 10 MHz, 470-nm laser excitation at a power of 20 μW.
To compare tracking durations between QD655-IgE and gQD-IgE in live cells, we analyzed tracks with an average count rate below 100 kHz to minimize the effect of having aggregates contributing to the analysis. In addition, we only included trajectories with tracking durations greater than 1 s to discard a large number of false positives or aborted tracks that result from our method of searching for particles in the 3D tracking microscope. As shown in Figure 2e and f, there is a much greater probability of tracking the IgE-FcεRI receptor for longer periods of time when using gQDs as compared to commercial QDs. The average tracking duration for QD655-IgE (Figure 2 e) is 16 ± 1 s (± standard error of the mean), with a standard deviation of 19 s. The average tracking duration for gQD-IgE (Figure 2 f) is 40 ± 5 s, with a standard deviation of 67 s. It should be noted that the average tracking duration varied from day to day depending on microscope alignment, and was as much as 111 ± 26 s for gQD-IgE during optimal configuration. While a factor of 2.5 increase in overall average tracking duration might seem a modest improvement, the gQD-IgE tracking data contains significantly more tracks in the longer time regime. For example, the cumulative probability distribution of tracking durations (Figure 2g) shows that the probability of tracking IgE-FcεRI for greater than 1 min is 20% for gQD-IgE as opposed to only 3% for QD655-IgE. The 7-fold increase in probability for this longer time regime is important for studying cellular processes or changes between processes occurring on this time scale. For example, unstimulated diffusion, antigen-induced crosslinking, and down-regulation mediated by endocytosis could all in principle be followed for an individual IgE-FcεRI more readily using non-blinking gQDs.
We find that there is an increase in tracking duration with an increase in count rate for QD655-IgE (Figure 2h, Pearson correlation coefficient, ρ = 0.47). Interestingly, the tracking duration seems to be less dependent on the count rate for the non-blinking gQD-IgE (Figure 2i, ρ = 0.31). As shown in Figure 2i, gQD particles with count rates as low as ~25 kHz can still be tracked with our 3D microscope for extended periods of time due to their stable emission signal.
The upper limit of tracking duration for the gQDs using 3D confocal feedback-based SPT can be limited by several factors. First, the gQDs do occasionally blink and eventually photobleach in an aqueous environment over the course of minutes. Additionally, due to shot noise, background of the cells, and tracking error, our correction to the stage position during 3D tracking can occasionally be in the wrong direction resulting in the loss of the particle. The latter limitation should not be of concern in performing conventional 2D SPT.
2.3. Distinguishing Individual gQD Particles Using Time-gated Photon Pair Correlation
Although the relative count rate of the emission signal can be used to estimate whether single or multiple quantum dots are being tracked, one can more definitively determine the number of particles by using photon-pair correlation (PPC) analysis.[23] In addition to recording the count rate and 3D position, we can simultaneously record the arrival time of every photon detected via time-correlated single photon counting (TCSPC) (Section S3). This TCSPC data includes both the macro time, as determined by a laser pulse every 100 ns, and the micro time, the photon arrival time with respect to the laser pulse. The absolute arrival time of the photon is the sum of the macro and micro times. The PPC method histograms the time delay between successive photons measured by different detectors. A single fluorophore can on average emit only one photon per laser pulse, assuming the average fluorescence lifetime is much shorter than the time between laser pulses. If multiple photons are detected simultaneously within the same laser pulse by multiple detectors, this suggests there is more than one fluorophore. We have previously implemented PPC to distinguish single particles from aggregates while tracking in 3D and to verify we are tracking individual fluorescent proteins.[4c,6b,9]
However, it is well documented that non-blinking gQDs can emit multiple photons simultaneously,[12c,e] which would cause ambiguity in using PPC to determine the number of particles. Mangum et al.[12j] developed a technique to overcome this caveat that takes advantage of the fact that the multi-exciton emission occurs on a fast time scale whereas the single-exciton emission occurs on a longer time scale. If one applies a time-gate to only analyze photons which arrive later than a fixed time following the excitation pulse, the multi-exciton contributions are minimized and the PPC histogram can be used to determine if the fluorescence is coming from a single quantum emitter.
In Figure 3 we implement this time-gated PPC analysis to verify that we are readily tracking individual gQD-IgE (Figure 3 a–e) and individual gQD-DNP (Figure 3 f–j) particles, rather than aggregates. In addition to the intensity (Figure 3a,f), 3D coordinates (Figure 3b,g) and white-light image of the cell to show relative position of the gQD (Figure 3c,h), we can also record TCSPC data that give us the fluorescence lifetime decay (Figure 3d, i). After applying a time gate to the photons with micro times between 4 and 20 ns, we can analyze the PPC distributions with negligible multi-exciton contributions (Figure 3e, j). To determine the number of independent quantum emitters present in the particle being tracked, we compute the ratio, R, between the center peak area, AC, and the average lateral peak area, AL, surrounding the t = 0 peak.[9,23c] In an ideal background free case, R = AC/AL = 1 – 1/m, where m is the number of independent quantum emitters. Because of background contributions and multi-exciton emission that make it through the time-gate, individual fluorophores exhibit R values above 0, but still less than 0.5. The time gate between 4 and 20 ns was empirically chosen to maximize the total number of fluorescence photons used for generating the PPC histogram while minimizing the contribution from multiexciton emission and background counts. For the examples shown in Figure 3, R = 0.48 for gQD-IgE and R = 0.41 for gQDDNP, which are both indicative of single gQD particles. It is worth noting that this analysis demonstrates that single gQDs can have count rates as high as 100 kHz, which supports the use of this value as a threshold in our tracking duration analysis (Figure 2).
Figure 3.
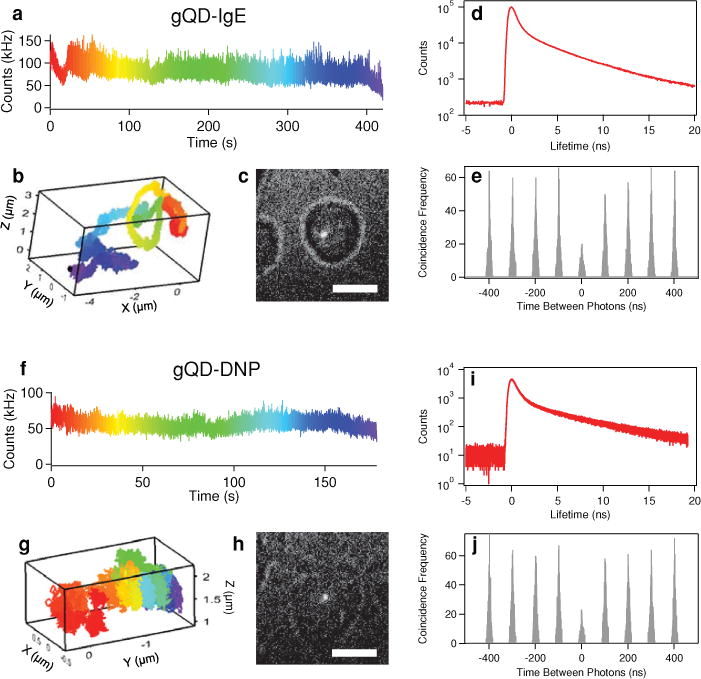
Live cell tracking of gQD-IgE: (a) Intensity as a function of time, with a rainbow color code used to denote passage of time, (b) correlated 3D trajectory, and (c) white-light image displaying respective location of gQD-IgE within the cell (scale bar is 10 μm). (d) Fluorescence lifetime histogram and (e) PPC histogram using a time gate between 4 and 20 ns to minimize multi-exciton contributions. Here, R = 0.48, which is indicative of a single gQD-IgE particle. Live cell tracking of gQD-DNP: (f) Color-coded intensity, (g) correlated 3D trajectory, and (h) white-light image displaying respective location of gQD-DNP within the cell (scale bar is 10 μm). (i) Fluorescence lifetime histogram and (j) PPC histogram using a time gate between 4 and 20 ns to minimize multi-exciton contributions. Here, R = 0.41, which is indicative of a single gQD particle.
2.4. Extended Tracking Duration Allows for Observation of Heterogeneous Diffusion
Using non-blinking gQDs to track individual IgE-FcεRI receptors in 3D for extended periods of time in live cells can offer unique insight into long time scale dynamics or changes between such dynamics. Figure 4 a shows the intensity and x, y z coordinates of a gQD-IgE particle being tracked for ~6.5 min after stimulation with 1 μg/mL DNP-BSA. Movie S1 shows the evolution of the 3D trajectory through time (played at 10× real time). While TCSPC data were not acquired for this particular trajectory, the count rate is consistent with an individual gQD. White light images showing the relative location of the gQD-IgE within the cell at different time points (Figure 4b) and the color-coded 3D trajectory (Figure 4c) are also shown for perspective.
Figure 4.
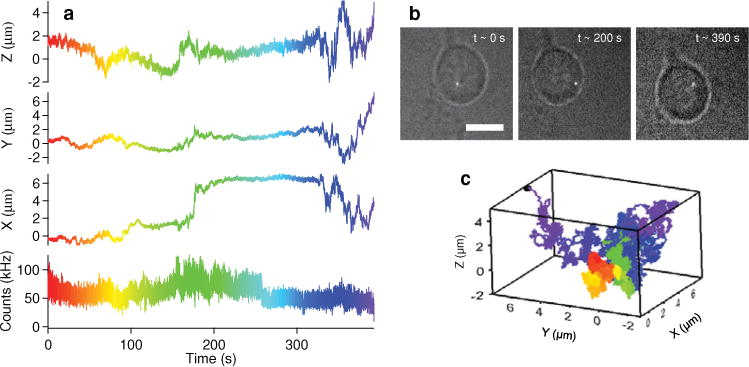
(a) Intensity and corresponding x, y, and z position of gQD-IgE monitored for ~6.5 min. (b) White light images to show relative cellular location of gQD-IgE at different time points (scale bar is 10 μm). (c) 3-dimensional trajectory of gQD-IgE (color-coded to correlate with trajectory displayed in a).
For this trajectory, it is clear that the IgE-FcεRI receptor diffusion is highly heterogeneous. After about ~175 s of slow movement, the IgE-FcεRI receptor undergoes a sudden large displacement in x, followed by a period of being fairly immobile. Then, at ~325 s, the IgE-FcεRI receptor undergoes rapid displacement in x, y and z until the end of the trajectory. Because the probability of tracking a single blinking QD655-IgE beyond 100 s is very rare (<1%, Figure 2e,g), these transitions between diffusion rates would unlikely be observed using a commercially available QD. The higher probability of observing longer gQD-IgE trajectories (Figure 2f,g), however, increases the likelihood of observing such heterogeneities in diffusion.
2.5. Quantification of Heterogeneous Diffusion by Hidden Markov Modeling
The standard method to quantify a diffusion coefficient (D) in SPT is to plot the mean squared displacement, MSD = 〈Δx2〉+ 〈Δy2〉+ 〈Δz2〉, as a function of lag time (Δt). For Brownian motion, the slope of this line is 6D for 3-dimensional diffusion and 4D for 2-dimensional diffusion. However, as just discussed, the trajectory shown in Figure 4 contains multiple diffusion rates. For clear transitions between diffusive regimes, one can simply divide the trajectory by visual inspection, plot MSD vs. Δt for each region, and determine the corresponding diffusion coefficient. We have implemented this approach for the trajectory in Figure 4 in Section S4.
However, where and how to divide a trajectory into different sections for analysis can be ambiguous and subject to interpretation. For example, the slight motion in z around ~60 s seen in Figure 4 may be interpreted by some as a separate diffusion regime, while others may classify all of the motion up to ~175 s as one regime. Several methods have been developed to eliminate this ambiguity in quantifying heterogeneous diffusion, including hidden Markov models,[24] change-point analysis,[25] and computing an average diffusion coefficient through time.[6a] Here we implement a Variational Bayesian Hidden Markov Modeling (vbHMM) approach developed by Persson et al.[24b] along with an average diffusion coefficient, , within a sliding window throughout the trajectory.[6a,25]
| (1) |
In the Equation 1, N is the number of data points (each 5 ms time point) within the sliding window, Δt is the lag time, and MSDi is given by Δx2 + Δy2 + Δz2 for a fixed Δt. Our analysis here also assumes Brownian motion throughout, which is supported by the linearity of the MSD vs Δt (Section S4). Figure 5a shows as a function of time for the trajectory from Figure 4 with an averaging time window of 1 s (N = 200) and a Δt of 250 ms. Justifications for choosing this particular N and Δt are found in Section S5. Details regarding the vbHMM analysis are also found in Section S6. The most probable state trajectory from the vbHMM analysis is shown in red to illustrate changes in diffusion through time. The corresponding distribution of is shown in Figure 5b along with the relative occupancy of each state found by the vbHMM analysis. The vbHMM analysis suggests three states which have diffusion coefficients of ~0.05, ~0.2, and ~0.4 μm2/s with average dwell times of ~8, ~2, and ~15 s respectively. We apply this analysis to additional example trajectories with long tracking durations exhibiting heterogeneous diffusion (Section S7), which showed a broad distribution of dwell times (10−1–10−1 s) for states found by the vbHMM. This broad distribution of dwell times further illustrates the need for high temporal tracking resolution (afforded by our confocal-based tracking system) and extended tracking duration (afforded by our non-blinking gQDs) needed to quantify multiple transitions between each state. The range of diffusive states found by vbHMM, 10−2–100 μm2/s, are consistent with that observed in previous 2D SPT studies using commercial QDs,[1e,17] and likely correspond to antigen-induced receptor cross-linking (10−2 μm2/s), membrane diffusion (10−1 μm2/s), and fast endocytotic transport (100 μm/s). In addition, transient confinement zones encountered by the receptor may also explain the observed heterogeneity. Moreover, vast heterogeneity exists in the cluster size of the antigen-induced IgE-FcεRI aggregation state.[17,26] The observed stochastic interconversion between these receptor aggregation states may allow the cell to modulate the allergic signaling response.
Figure 5.
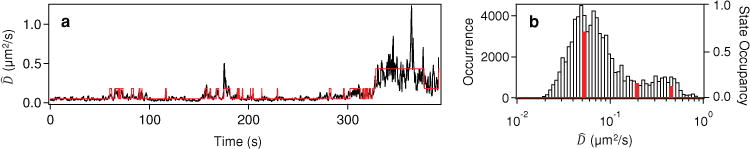
(a) Average diffusion coefficient over time, , from the trajectory shown in Figure 4 using a lag time of 250 ms and an averaging window size of 1 s is shown in black. The most probable trajectory from vbHMM analysis is shown in red. (b) The projected distribution of (black bars) along with the relative occupancy of the diffusion states obtained from vbHMM analysis (red bars). The vbHMM analysis yields three diffusion states at ~0.05, ~0.2, and ~0.4 μm2 /s with dwell times of 8, 2, and 15 s, respectively.
3. Conclusion
We have shown that the unique non-blinking behavior and overall enhanced photostability of gQDs significantly improves live cell SPT in three dimensions, enabling observation times of single receptors for several minutes. This increased observation time is enabled by the highly stable emission from a gQD and the enhanced tracking range enabled by 3D SPT via confocal feedback. Here, we found the use of the gQD-IgE compared to commercial QD655-IgE increases the probability of observing a single QD-IgE for a minute or greater duration by 7-fold. We emphasize that this “one minute” threshold is not an arbitrary temporal period, but rather is a critical time scale for many biological processes (such as IgE stimulation and downregulation). We point out that stable non-blinking gQD emission signal should also improve conventional image-plane based 2D SPT studies, by minimizing ambiguity in particle trajectory analysis caused by blinking. In addition, using time-gated PPC we differentiated single from multiple gQDs, a technique that could potentially be used to correlate the aggregation state of the IgE-FcεRI receptor with dynamics. Lastly, the increased probability of observing gQD-IgE for long time periods allows us to quantify multiple changes in receptor diffusion that occur on ~101 s timescales, as given by vbHMM analysis. We expect the use of non-blinking gQDs to be widely applicable for future SPT studies in both 2 and 3 dimensions where long time-scale changes in dynamics (on the order of minutes) are of interest.
4. Experimental Section
gQD Ligand Exchange and Characterization
The gQDs were synthesized using successive ion layer adsorption and reaction (SILAR) as described previously.[14] Prior to ligand-exchange, gQDs dissolved in toluene were washed by several precipitation-centrifugation cycles with ethanol. After dissolving ~2 mg of gQDs in 3 mL of toluene (~1.6 μM), 30 mg of the custom DHLA-PEG ligand (7.7 mM) was added with stirring. After stirring for ~2 h at room temperature, a 2 mL aqueous solution of ~30 mM tetramethyl ammonium hydroxide (phase transfer catalyst) was added drop-wise. The gQDs immediately transferred to the water phase, which was stored overnight at 4 °C. The aqueous layer containing the gQDs was extracted and the excess ligand was removed via ultracentrifugation using a 15 mL 50 kDa molecular weight cut-off (MWCO) centrifugal unit (EMD Millipore). After ligand exchange, the hydrodynamic diameter was determined to be ~20 nm via 2D single-particle tracking (LM-12 Nanosight Ltd., Figure S1a) and atomic force microscopy (AFM, Veeco Instruments Bioscope SZ, Figure S1b). The broad ultraviolet absorption and fluorescence emission at 640 nm are shown in Figure S1c. The average BPP and approximate concentration of the gQDs were determined using FCS as described in Section S2.
Bioconjugation of gQD to SA
The carboxyl-terminated gQDs were first functionalized with NHS ester groups by adding a solid mixture of 4.4 mg (5.7 mM) EDC (Pierce) and 9 mg (10 mM) sulfo-NHS (Pierce) to 4 mL of 40 nM gQDs dissolved in 100 mM MES, pH 6 buffer. After incubating for 30 min at room temperature, the excess EDC and sulfo-NHS were quickly removed via ultracentrifugation using a 15 mL 50 kDa MWCO centrifugal unit (EMD Millipore) with a 100 mM phosphate, 150 mM NaCl, pH 7.4 (PBS) buffer. To ~300 μL of ~1 μM gQD-NHS ester, 1 mL of 94 μM SA (Pierce) in 50 mM sodium bicarbonate, pH 8.3 buffer was added. The conjugation reaction was incubated overnight at 4 °C. After concentrating the reaction to ~200 μL using a 100 kDa MWCO centrifugal unit (EMD Millipore), the excess SA was removed the following day using size-exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare, Figure 1b). The conjugation of the gQD to SA was verified using agarose gel electrophoresis (Figure 1c) and the diameter, ~23 nm, was measured again using 2D particle tracking (Figure S1a) to insure that aggregation had not occurred. The gQD-IgE or gQD-DNP conjugates were then prepared by adding either ~1 equivalent of biotin-IgE or ~10 equivalents of biotin-DNP to 20 nM gQD-SA in PBS buffer containing 1% BSA (Fisher Scientific).
Cell Binding Specificity
The RBL-2H3 mast cells were incubated with either ~1 nM gQD-SA or gQD-IgE in Hanks’ balanced salt solution (HBSS) buffer for 15 min at 37 °C. The cells were then rinsed 3 times with 200 μL aliquots of HBSS buffer and imaged with an EMCCD camera (Photon Max, Princeton Instruments) using a combination of red-light (>700 nm) to observe the cell outline and a wide-field 488 nm laser to excite the gQDs. A 593 nm long-pass filter (FF01–593/LP, Semrock) was used to filter the emission. Representative images of the cells incubated with gQD-IgE (Figure S2a) vs. gQD-SA (Figure S2b) clearly show that binding to the FcεRI receptor is specific for the gQD-IgE conjugate. The distribution of the number of gQDs observed on the top surface of the RBL-2H3 cells for both gQD-IgE and gQD-SA is shown in Figure S2c, which allows us to estimate a gQD-IgE binding specificity of ~96%.
Antigen Binding Assay
To assess the gQD-IgE’s ability to bind the multivalent antigen, we performed an Enzyme-Linked Immunosorbent Assay (ELISA). Each well within a NUNC MaxiSorp multi-well plate was incubated overnight at 4 °C with 100 μL of 10 μg/mL DNP-BSA in PBS buffer. After rinsing with PBS, the wells were passivated with 250 μL of PBS containing 5% BSA for 1 h at room temperature. After removal of the passivating solution, the wells were incubated for 1 hr with 100 μL of 20 nM gQD-IgE and corresponding controls (Figure S3). The wells were then rinsed thoroughly with PBS containing 0.1% Tween (PBST) followed by PBS. Then, 100 μL of 1000-fold diluted Goat anti-Mouse IgE – Horse Radish Peroxidase (HRP) conjugate (Pierce) in PBS with 1% BSA was added to each well, protected from light, and incubated for 30 min. After thoroughly rinsing the wells with PBST and PBS, 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Pierce) was simultaneously added to each well. After several seconds, 50 μL of 1 M H2SO4 was added simultaneously to stop the catalytic reaction of HRP. The absorbance at 405 nm of the oxidized TMB was measured using a multi-well plate reader (Multiskan Ascent, Thermo Electron Coorporation). The results (Figure S3) indicate that the gQD-IgE conjugate retains its ability to bind the multivalent DNP-BSA antigen.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (5R01AI097154–02) JHW. JAH also acknowledges partial support by NIH-NIGMS Grant 1R01GM084702–01. YG was supported in part by the Los Alamos National Laboratory Directed Research and Development Program. This work was performed at the Center for Integrated Nanotechnologies, a US Department of Energy, Office of Basic Energy Sciences user facility. Los Alamos National Laboratory is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the US Department of Energy under contract DE-AC52–06NA25396. Collaborative efforts were also supported by P50 GM065794 and R01AI051575 (BSW), the New Mexico Spatiotemporal Modeling Center (P50GM0852673) and NIH R01GM100114 (DSL). We thank Jennifer Martinez for advice on bioconjugation and general use of laboratory resources, Peter Goodwin for assistance with AFM measurements, Patrick Cutler and Cédric Cleyrat for advice on handling and labeling the RBL-2H3 mast cells, Darrick Williams for advice on QD water solubilization via ligand exchange, Han Htoon for advice on assessing the number of gQD particles using time-gated PPC, and Pengfei Zhang for assistance with the PPC analysis.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Aaron M. Keller, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
Dr. Yagnaseni Ghosh, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
Dr. Matthew S. DeVore, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
Mary E. Phipps, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
Dr. Michael H. Stewart, Optical Sciences Division, Code 5600, US Naval Research Laboratory, Washington, D.C. 20375, USA
Prof. Bridget S. Wilson, Department of Pathology and Cancer Research and Treatment Center, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 97131, USA
Prof. Diane S. Lidke, Department of Pathology and Cancer Research and Treatment Center, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 97131, USA
Dr. Jennifer A. Hollingsworth, Email: jenn@lanl.gov, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA.
Dr. James H. Werner, Email: jwerner@lanl.gov, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA.
References
- 1.a) Saxton MJ, Jacobson K. Annu Rev Biophys Biomol Struct. 1997;26:373. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]; b) Barak LS, Webb WW. J Cell Biol. 1981;90:595. doi: 10.1083/jcb.90.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kusumi A, Sako Y, Yamamoto M. Biophys J. 1993;65:2021. doi: 10.1016/S0006-3495(93)81253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dahan M, Levi S, Luccardini C, Rostaing P, Riveau B, Triller A. Science. 2003;302:442. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]; e) Andrews NL, Lidke KA, Pfeiffer JR, Burns AR, Wilson BS, Oliver JM, Lidke DS. Nat Cell Biol. 2008;10:955. doi: 10.1038/ncb1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Gelles J, Schnapp BJ, Sheetz MP. Nature. 1988;331:450. doi: 10.1038/331450a0. [DOI] [PubMed] [Google Scholar]; b) Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Science. 2005;308:1469. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]; c) Nan XL, Sims PA, Chen P, Xie XS. J Phys Chem B. 2005;109:24220. doi: 10.1021/jp056360w. [DOI] [PubMed] [Google Scholar]
- 3.Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, Kusumi A. J Cell Biol. 2002;157:1071. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Tsien RY. Annu Rev Biochem. 1998;67:509. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]; b) Yu J, Xiao J, Ren XJ, Lao KQ, Xie XS. Science. 2006;311:1600. doi: 10.1126/science.1119623. [DOI] [PubMed] [Google Scholar]; c) Han JJ, Kiss C, Bradbury ARM, Werner JH. ACS Nano. 2012;6:8922. doi: 10.1021/nn302912j. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt T, Schutz GJ, Baumgartner W, Gruber HJ, Schindler H. Proc Natl Acad Sci USA. 1996;93:2926. doi: 10.1073/pnas.93.7.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Pinaud F, Michalet X, Iyer G, Margeat E, Moore HP, Weiss S. Traffic. 2009;10:691. doi: 10.1111/j.1600-0854.2009.00902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wells NP, Lessard GA, Goodwin PM, Phipps ME, Cutler PJ, Lidke DS, Wilson BS, Werner JH. Nano Lett. 2010;10:4732. doi: 10.1021/nl103247v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat Mater. 2005;4:435. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]; b) Sperling RA, Parak WJ. Philos Transact A Math Phys Eng Sci. 2010;368:1333. doi: 10.1098/rsta.2009.0273. [DOI] [PubMed] [Google Scholar]
- 8.Nirmal M, Dabbousi BO, Bawendi MG, Macklin JJ, Trautman JK, Harris TD, Brus LE. Nature. 1996;383:802. [Google Scholar]
- 9.Wells NP, Lessard GA, Werner JH. Anal Chem. 2008;80:9830. doi: 10.1021/ac8021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Saxton MJ. Biophys J. 1997;72:1744. doi: 10.1016/S0006-3495(97)78820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Woll D, Kolbl C, Stempfle B, Karrenbauer A. Phys Chem Chem Phys. 2013;15:6196. doi: 10.1039/c3cp44693j. [DOI] [PubMed] [Google Scholar]
- 11.a) Chen Y, Vela J, Htoon H, Casson JL, Werder DJ, Bussian DA, Klimov VI, Hollingsworth JA. J Am Chem Soc. 2008;130:5026. doi: 10.1021/ja711379k. [DOI] [PubMed] [Google Scholar]; b) Mahler B, Spinicelli P, Buil S, Quelin X, Hermier JP, Dubertret B. Nat Mater. 2008;7:659. doi: 10.1038/nmat2222. [DOI] [PubMed] [Google Scholar]; c) Dennis AM, Mangum BD, Piryatinski A, Park YS, Hannah DC, Casson JL, Williams DJ, Schaller RD, Htoon H, Hollingsworth JA. Nano Lett. 2012;12:5545. doi: 10.1021/nl302453x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Garcia-Santamaria F, Chen YF, Vela J, Schaller RD, Hollingsworth JA, Klimov VI. Nano Lett. 2009;9:3482. doi: 10.1021/nl901681d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Spinicelli P, Buil S, Quelin X, Mahler B, Dubertret B, Hermier JP. Phys Rev Lett. 2009;102:136801. doi: 10.1103/PhysRevLett.102.136801. [DOI] [PubMed] [Google Scholar]; c) Htoon H, Malko AV, Bussian D, Vela J, Chen Y, Hollingsworth JA, Klimov VI. Nano Lett. 2010;10:2401. doi: 10.1021/nl1004652. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Vela J, Htoon H, Chen YF, Park YS, Ghosh Y, Goodwin PM, Werner JH, Wells NP, Casson JL, Hollingsworth JA. J Biophotonics. 2010;3:706. doi: 10.1002/jbio.201000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Park YS, Malko AV, Vela J, Chen Y, Ghosh Y, Garcia-Santamaria F, Hollingsworth JA, Klimov VI, Htoon H. Phys Rev Lett. 2011;106:1874011. doi: 10.1103/PhysRevLett.106.187401. [DOI] [PubMed] [Google Scholar]; f) Galland C, Ghosh Y, Steinbruck A, Sykora M, Hollingsworth JA, Klimov VI, Htoon H. Nature. 2011;479:203. doi: 10.1038/nature10569. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Malko AV, Park YS, Sampat S, Galland C, Vela J, Chen YF, Hollingsworth JA, Klimov VI, Htoon H. Nano Lett. 2011;11:5213. doi: 10.1021/nl2025272. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Pal BN, Ghosh Y, Brovelli S, Laocharoensuk R, Klimov VI, Hollingsworth JA, Htoon H. Nano Lett. 2012;12:331. doi: 10.1021/nl203620f. [DOI] [PubMed] [Google Scholar]; i) Galland C, Ghosh Y, Steinbruck A, Hollingsworth JA, Htoon H, Klimov VI. Nat Commun. 2012;3 doi: 10.1038/ncomms1916. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Mangum BD, Ghosh Y, Hollingsworth JA, Htoon H. Opt Express. 2013;21:7419. doi: 10.1364/OE.21.007419. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Kundu J, Ghosh Y, Dennis AM, Htoon H, Hollingsworth JA. Nano Lett. 2012;12:3031. doi: 10.1021/nl3008659. [DOI] [PubMed] [Google Scholar]
- 13.Hollingsworth JA. Chem Mater. 2013;25:1318. doi: 10.1021/cm304161d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh Y, Mangum BD, Casson JL, Williams DJ, Htoon H, Hollingsworth JA. J Am Chem Soc. 2012;134:9634. doi: 10.1021/ja212032q. [DOI] [PubMed] [Google Scholar]
- 15.Marchuk K, Guo YJ, Sun W, Vela J, Fang N. J Am Chem Soc. 2012;134:6108. doi: 10.1021/ja301332t. [DOI] [PubMed] [Google Scholar]
- 16.Gould HJ, Sutton BJ. Nat Rev Immunol. 2008;8:205. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 17.Andrews NL, Pfeiffer JR, Martinez AM, Haaland DM, Davis RW, Kawakami T, Oliver JM, Wilson BS, Lidke DS. Immunity. 2009;31:469. doi: 10.1016/j.immuni.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wells NP, Lessard GA, Phipps ME, Goodwin PM, Lidke DS, Wilson BS, Werner JH. Proc SPIE. 2009;7185:71850Z. doi: 10.1117/12.809412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Dvorak AM, Galli SJ, Schulman ES, Lichtenstein LM, Dvorak HF. Fed Proc. 1983;42:2510. [PubMed] [Google Scholar]; b) Pfeiffer JR, Seagrave JC, Davis BH, Deanin GG, Oliver JM. J Cell Biol. 1985;101:2145. doi: 10.1083/jcb.101.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blank U, Rivera J. Trends Immunol. 2004;25:266. doi: 10.1016/j.it.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 20.a) Santini F, Keen JH. J Cell Biol. 1996;132:1025. doi: 10.1083/jcb.132.6.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wilson BS, Pfeiffer JR, Oliver JM. J Cell Biol. 2000;149:1131. doi: 10.1083/jcb.149.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Molfetta R, Belleudi F, Peruzzi G, Morrone S, Leone L, Dikic I, Piccoli M, Frati L, Torrisi MR, Santoni A, Paolini R. J Immunol. 2005;175:4208. doi: 10.4049/jimmunol.175.7.4208. [DOI] [PubMed] [Google Scholar]
- 21.a) Lessard G, Goodwin PM, Werner JH. Proc SPIE. 2006;6092:6092051. [Google Scholar]; b) Lessard GA, Goodwin PM, Werner JH. Appl Phys Lett. 2007;91:224106. [Google Scholar]
- 22.a) Susumu K, Uyeda HT, Medintz IL, Pons T, Delehanty JB, Mattoussi H. J Am Chem Soc. 2007;129:13987. doi: 10.1021/ja0749744. [DOI] [PubMed] [Google Scholar]; b) Susumu K, Mei BC, Mattoussi H. Nat Protoc. 2009;4:424. doi: 10.1038/nprot.2008.247. [DOI] [PubMed] [Google Scholar]
- 23.a) Ambrose WP, Goodwin PM, Enderlein J, Semin DJ, Martin JC, Keller RA. Chem Phys Lett. 1997;269:365. [Google Scholar]; b) Tinnefeld P, Muller C, Sauer M. Chem Phys Lett. 2001;345:252. [Google Scholar]; c) Weston KD, Dyck M, Tinnefeld P, Muller C, Herten DP, Sauer M. Anal Chem. 2002;74:5342. doi: 10.1021/ac025730z. [DOI] [PubMed] [Google Scholar]
- 24.a) Low-Nam ST, Lidke KA, Cutler PJ, Roovers RC, Henegouwen PMPVE, Wilson BS, Lidke DS. Nat Struct Mol Biol. 2011;18:1244. doi: 10.1038/nsmb.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Persson F, Linden M, Unoson C, Elf J. Nat Methods. 2013;10:265. doi: 10.1038/nmeth.2367. [DOI] [PubMed] [Google Scholar]
- 25.Montiel D, Cang H, Yang H. J Phys Chem B. 2006;110:19763. doi: 10.1021/jp062024j. [DOI] [PubMed] [Google Scholar]
- 26.Shelby SA, Holowka D, Baird B, Veatch SL. Biophys J. 2013;105:2343. doi: 10.1016/j.bpj.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.