

The incidence of invasive fungal infections poses a serious global health threat, killing nearly 1.4 million people a year—a number comparable to deaths from tuberculosis. Factors that contribute to this disease burden and high mortality include insufficient diagnostics and treatment, and the increasing numbers of individuals with immune system defects who are particularly susceptible to fungal pathogens [1].
Because of the high risk of fungal infections in immunocompromised individuals—e.g., low–birth-weight neonates or patients undergoing organ transplantation or chemotherapy—it has become common medical practice to utilize antifungal prophylaxis in these patients during immunosuppression [2–4]. However, the expanding use of antifungal drugs has been associated with increasing incidence of antifungal drug resistance resulting from inherently less sensitive species and/or acquisition of drug class–specific resistance mechanisms [5]. Most alarming in recent years, multidrug-resistant strains of certain Candida species have emerged that are resistant to azoles and echinocandins, the two most widely used classes of antifungal drugs [6,7].
In this article we explore the notion that frequent and prolonged exposures of fungal cells to antifungal drugs activate fungal stress responses, which both support the short-term cellular adaptation to the drugs [8,9] and promote genetic instability to facilitate the emergence of stable drug resistant mutants refractory to therapy [9,10], including multidrug-resistant (MDR) strains (Fig. 1). Antifungal drug-induced stress has been associated with genetic instability in such distantly related fungi as Candida and Cryptococcus [11,12], suggesting that it is a broadly conserved phenomenon. In this article, we focus on drug resistance in Candida spp. because of its clinical significance, especially with the development of emerging multidrug resistance, and because mechanisms of drug resistance and stress-associated genetic instability are best studied in Candida, both in the clinic and in the lab.
Fig 1. A model for the formation of multidrug-resistant strains of C. glabrata.
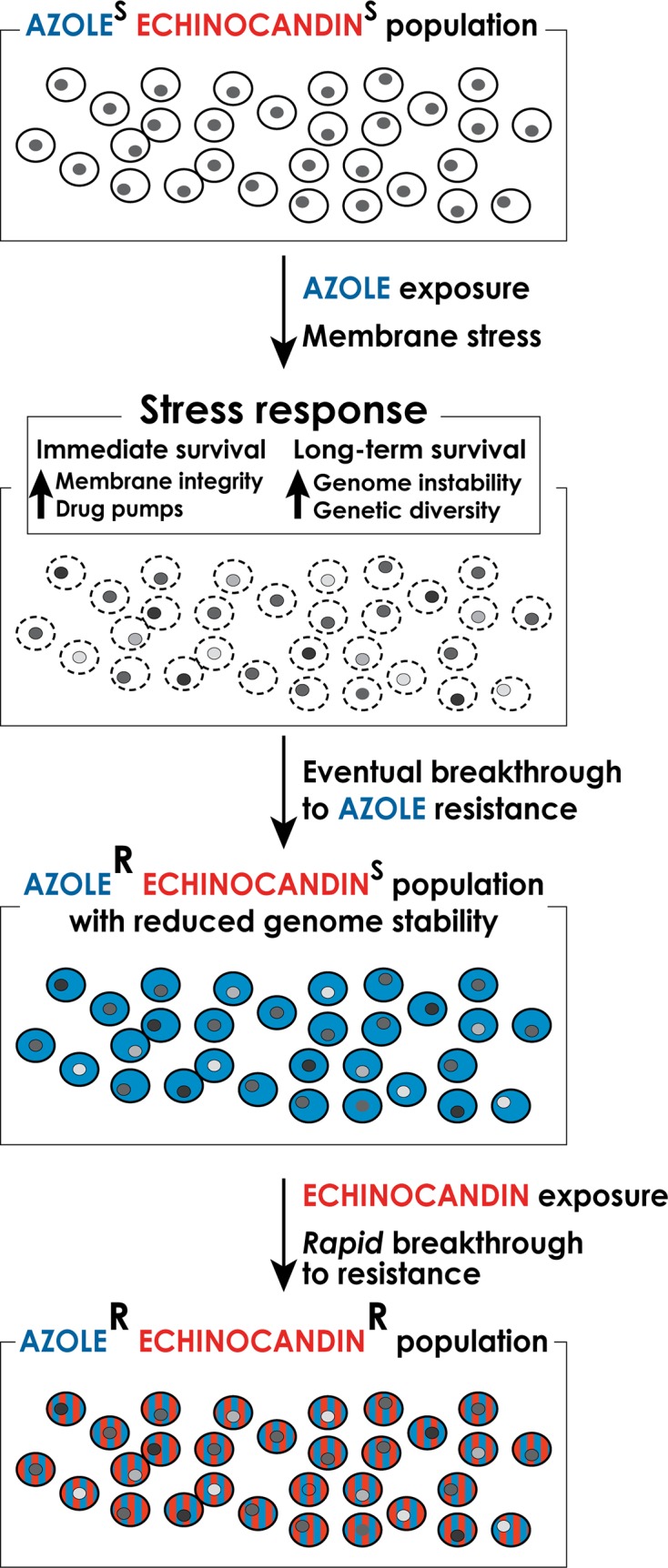
Long-term exposure to antifungal drugs triggers stress responses that both help maintain cellular integrity in the face of acute stress and promote genomic instability and genetic diversity to facilitate the emergence of genetic variants with enhanced intrinsic resistance. In the case of long-term prophylaxis with azole class drugs in Candida glabrata, this process results in formation of azole-resistant mutants with reduced genome stability. These mutants, when challenged with echinocandin class drugs, rapidly develop echinocandin resistance as well, resulting in formation of multidrug-resistant C. glabrata.
Genetic Basis of Antifungal Drug Resistance
The most prevalent fungal pathogens belong to the Candida genus, with C. albicans and C. glabrata being the most common and second or third most commonly isolated bloodstream fungal pathogens in the United States [13,14], respectively. For more than three decades, Candida infections have been treated with azole antifungal agents, which target fungal membranes by inhibiting the biosynthesis of ergosterol. However, nearly 20% of C. glabrata strains exhibit intrinsic resistance to azoles, and even susceptible strains rapidly acquire resistance. This inherent resistant property has contributed to C. glabrata being the most frequently isolated Candida species in some high-risk centers [15,16]. To address this issue, echinocandin drugs are now recommended as first-line therapy to treat a range of candidiasis [17], since they do not exhibit cross-resistance to azole-resistant yeasts. A relatively recent addition to the antifungal drug repertoire, echinocandin class compounds target the fungal cell wall by inhibiting the biosynthesis of the key cell wall polymer β-(1,3)D-glucan. Troublingly, however, C. glabrata resistance to echinocandins is on the rise [18], with >10% of isolates now showing resistance in some hospital settings [7]. Many of the echinocandin-resistant C. glabrata isolates are also resistant to azoles, leaving extremely few options to treat patients infected with MDR strains [7].
Clinical resistance to echinocandins resulting in treatment failures is nearly always due to limited mutations in two highly conserved “hot spot regions” of genes encoding subunits of β-glucan synthase: FKS1 for most Candida species and FKS1 and/or FKS2 in C. glabrata [19,20]. Echinocandin resistance is always acquired during therapy, and the resulting amino acid changes in glucan synthase significantly decrease the sensitivity of the enzyme to the drug, resulting in higher MIC values and reduced pharmacodynamic responses [20].
In contrast to the echinocandins, azole drug resistance resulting in clinical failure may be caused by a variety of genetic changes, most of which affect the expression of fungal drug transporters or the structure and/or expression of fungal drug targets [9]. For instance, resistance to azoles can be caused by overexpression of transporters that mediate drug efflux out of the cell [21]. This overexpression can be achieved by amplification of the transporter genes, by large scale chromosomal rearrangement or chromosome gain that result in higher transporter gene copy number, or by point mutations in transcription factors that regulate transporter gene expression [22]. Resistance to azoles can also be mediated by mutations in the gene encoding their cellular target—the Erg11 protein, or by mutations in transcriptional regulators of ERG11 that result in Erg1 overexpression [23,24]. In C. albicans, clinical resistance to azoles frequently occurs in a stepwise manner, with a gradual increase in resistance associated with a sequential accumulation of mutations and genome rearrangements that together result in high-level azole resistance [25]. In contrast, in C. glabrata a single gain-of-function mutation in a transcriptional activator of drug efflux pumps is often sufficient to confer high-level azole resistance [26].
Theoretical and Experimental Support for Stress-Induced Genetic Instability
How do genetic changes that cause drug resistance arise and become fixed in fungal populations? The classical theory of evolution and natural selection posits that rare, random mutations arise in a population (e.g., due to occasional spontaneous errors in DNA replication and repair processes) and that a change in conditions, such as appearance of antifungal drug, would favor mutants that happen to be more fit under the new conditions (i.e., resistant to the drug). However, it seems clear that in thriving populations well adapted to their stable environments, genetic diversity is far less valuable than in maladapted populations, or during times of environmental change. From this point of view, a winning evolutionary strategy for an organism would contain the capacity to enhance its genetic diversity specifically during the times of stress. Indeed, mathematical modeling shows that the ability to generate increased genetic diversity by mutation or recombination specifically during the times of decreased fitness, i.e., stress, is itself a beneficial trait that would be favored by natural selection [27,28]. In other words, an organism with the capacity to modulate its spontaneous mutation and recombination rates—keeping them low during conditions of low stress and increasing them during conditions of high stress—would have a selective advantage over organisms with constant (either constitutively low or high) mutation and recombination rates.
The modeling studies are supported by empirical observations of increased genetic instability in fungi exposed to stresses such as high temperature, starvation, proteotoxic stress, and antifungal drugs. Starving laboratory strains of baker’s yeast (Saccharomyces cerevisiae) induces gross chromosomal rearrangements at rates several-fold higher than non-starving cells [29]. Interestingly, only a fraction of these rearrangements shows increased fitness relative to the parent strain under the starvation conditions, suggesting that rearrangements arise not by the selection of pre-existing genetic variants but by random starvation-induced genetic variability [29]. Furthermore, growth of laboratory yeast strains at high temperature or in the presence of antifungal drugs strongly induces gross chromosomal rearrangements and chromosomal gain or loss [30]. Studies of clinical C. albicans isolates from patients treated with azoles show that these strains frequently exhibit gross chromosomal rearrangements and/or aneuploidy [11]. Likewise, in C. glabrata isolated from patients, chromosomes are frequently reshuffled resulting in new genetic configurations, including appearance of small chromosomes [31,32]. Moreover, passaging C. albicans through a mouse in the absence of drug treatment increases chromosomal rearrangements relative to C. albicans passaged in regular laboratory medium, indicating that the stresses encountered in vivo, such as oxidative stress and elevated temperature promote genetic instability [33].
Roles of Cellular Stress Responses in Genetic Instability and Drug Resistance
Cellular stress responses are generally considered in terms of their role in helping cells tolerate and survive acute stress. For instance, echinocandin exposure compromises the cell wall and induces a number of stress signaling cascades, including those dependent on protein kinase C (PKC), Ca2+/calcineurin, HSP90 and high osmolarity glycerol (HOG) kinase, and up-regulation of chitin production to enhance cell wall stability [8,20]. These compensatory mechanisms produce a drug tolerant state where cells appear to resist the fungicidal properties of echinocandin drugs. The enhanced drug tolerance may explain why significant levels of surviving persister C. albicans cells were observed in mice after echinocandin treatment [34]. It is likely that stress response–mediated enhanced drug tolerance allows some cells sufficient time to develop a resistance-conferring mutation in FKS1 or FKS2.
However, the observations of chromosomal instability during stress strongly suggest that another role of cellular stress responses is to help increase genetic diversity during stress, presumably by altering the machinery responsible for maintaining genome integrity [10]. Indeed, several fungal stress response factors have been implicated in promoting genetic instability during stress. For instance, in S. cerevisiae, general stress response transcription factors Msn2 and Msn4 help promote certain types of mutagenesis in response to proteotoxic stress [35]. Furthermore, in both Saccharomyces and Candida, stress-induced aneuploidy depends on the function of a stress-inducible protein chaperone HSP90, which regulates the folding of a number of client proteins that function in chromosome segregation and cell cycle progression [30,36]. Another mechanism of stress-induced aneuploidy has been observed in several Candida species, including C. albicans and C. glabrata, involving mis-regulation of the fungal cell cycle shortly upon exposure to an antifungal agent. Within a few hours of exposure to azoles or echinocandins, Candida cells were observed to uncouple spindle formation and nuclear division from cell division, ultimately leading to the formation of tetraploid cells that underwent unequal division to produce aneuploid progeny [37].
While induction of aneuploidy appears to be an early event following stress, aneuploidy can promote other types of genomic rearrangements and mutagenic lesions [38]. This effect of aneuploidy is attributed to the fact that it alters gene dosage of a subset of the genome, thus disrupting the stoichiometry of complexes involved in chromosome maintenance and DNA repair whose components are encoded by different chromosomes.
Clinical Implications of Stress-Induced Genetic Instability
The potential for stress-induced genetic instability is especially troubling for drugs such as azoles, which are fungistatic—i.e., they do not kill fungal cells but only arrest their growth. As outlined above, arrested cells activate stress responses that promote genome destabilization and genetic diversity. Indeed, data from the clinic indicating rapid development of drug resistance following initiation of therapy [39] support the notion that exposure of C. glabrata to an azole may create fungal cells with highly variable, “evolvable” genomes capable of quick breakthrough of resistance to any other types of drug, such as echinocandins (Fig. 1). Thus, patients encountering fluconazole prophylaxis may show infections with azole resistant C. glabrata followed by rapid breakthrough on echinocandin therapy, resulting in multidrug-resistant strains. In light of this phenomenon, it may be necessary to review the current practice of prophylaxis in some settings, which promotes such genetic instability. Furthermore, there is an urgent need for new fungicidal drugs, for safe combinations of antifungal drugs that would require mutagenesis of more than one target to create resistance (a much more rare event), and for a deeper understanding of mechanisms underlying stress-induced genetic instability so that they could potentially be pharmacologically mitigated during antifungal therapy.
Funding Statement
This work was supported by NIH grants AI109025 and AI069397 to DSP and grants from Pfizer and Astellas. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, et al. (2012) Hidden killers: human fungal infections. Sci Transl Med 4: 165rv113 10.1126/scitranslmed.3004404 [DOI] [PubMed] [Google Scholar]
- 2. Ziakas PD, Kourbeti IS, Mylonakis E (2014) Systemic antifungal prophylaxis after hematopoietic stem cell transplantation: a meta-analysis. Clin Ther 36: 292–306 e291. 10.1016/j.clinthera.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 3. Greenberg RG, Benjamin DK Jr. (2014) Neonatal candidiasis: Diagnosis, prevention, and treatment. J Infect 69 Suppl 1: S19–22. 10.1016/j.jinf.2014.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robenshtok E, Gafter-Gvili A, Goldberg E, Weinberger M, Yeshurun M, et al. (2007) Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol 25: 5471–5489. [DOI] [PubMed] [Google Scholar]
- 5. Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M (2011) Geographic variations in species distribution and echinocandin and azole antifungal resistance rates among Candida bloodstream infection isolates: report from the SENTRY Antimicrobial Surveillance Program (2008 to 2009). J Clin Microbiol 49: 396–399. 10.1128/JCM.01398-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfaller MA, Messer SA, Woosley LN, Jones RN, Castanheira M (2013) Echinocandin and triazole antifungal susceptibility profiles for clinical opportunistic yeast and mold isolates collected from 2010 to 2011: application of new CLSI clinical breakpoints and epidemiological cutoff values for characterization of geographic and temporal trends of antifungal resistance. J Clin Microbiol 51: 2571–2581. 10.1128/JCM.00308-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, et al. (2013) Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis 56: 1724–1732. 10.1093/cid/cit136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walker LA, Gow NA, Munro CA (2010) Fungal echinocandin resistance. Fungal Genet Biol 47: 117–126. 10.1016/j.fgb.2009.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cowen LE, Steinbach WJ (2008) Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot Cell 7: 747–764. 10.1128/EC.00041-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galhardo RS, Hastings PJ, Rosenberg SM (2007) Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol 42: 399–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Selmecki A, Forche A, Berman J (2006) Aneuploidy and isochromosome formation in drug-resistant Candida albicans . Science 313: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sionov E, Lee H, Chang YC, Kwon-Chung KJ (2010) Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog 6: e1000848 10.1371/journal.ppat.1000848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wisplinghoff H, Ebbers J, Geurtz L, Stefanik D, Major Y, et al. (2014) Nosocomial bloodstream infections due to Candida spp. in the USA: species distribution, clinical features and antifungal susceptibilities. Int J Antimicrob Agents 43: 78–81. 10.1016/j.ijantimicag.2013.09.005 [DOI] [PubMed] [Google Scholar]
- 14. Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M (2011) Candida bloodstream infections: comparison of species distributions and antifungal resistance patterns in community-onset and nosocomial isolates in the SENTRY Antimicrobial Surveillance Program, 2008–2009. Antimicrob Agents Chemother 55: 561–566. 10.1128/AAC.01079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farmakiotis D, Tarrand JJ, Kontoyiannis DP (2014) Drug-resistant Candida glabrata infection in cancer patients. Emerg Infect Dis 20: 1833–1840. 10.3201/eid2011.140685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lortholary O, Desnos-Ollivier M, Sitbon K, Fontanet A, Bretagne S, et al. (2011) Recent exposure to caspofungin or fluconazole influences the epidemiology of candidemia: a prospective multicenter study involving 2,441 patients. Antimicrob Agents Chemother 55: 532–538. 10.1128/AAC.01128-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pappas PG, Kauffman CA, Andes D, Benjamin DK Jr., Calandra TF, et al. (2009) Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 48: 503–535. 10.1086/596757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pfaller MA, Castanheira M, Lockhart SR, Ahlquist AM, Messer SA, et al. (2012) Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata . J Clin Microbiol 50: 1199–1203. 10.1128/JCM.06112-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perlin DS (2007) Resistance to echinocandin-class antifungal drugs. Drug Resist Updat 10: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perlin DS (2014) Echinocandin resistance, susceptibility testing and prophylaxis: implications for patient management. Drugs 74: 1573–1585. 10.1007/s40265-014-0286-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. White TC, Holleman S, Dy F, Mirels LF, Stevens DA (2002) Resistance mechanisms in clinical isolates of Candida albicans . Antimicrobial Agents and Chemotherapy 46: 1704–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schubert S, Barker KS, Znaidi S, Schneider S, Dierolf F, et al. (2011) Regulation of efflux pump expression and drug resistance by the transcription factors Mrr1, Upc2, and Cap1 in Candida albicans . Antimicrob Agents Chemother 55: 2212–2223. 10.1128/AAC.01343-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Flowers SA, Barker KS, Berkow EL, Toner G, Chadwick SG, et al. (2012) Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans . Eukaryot Cell 11: 1289–1299. 10.1128/EC.00215-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flowers SA, Colon B, Whaley SG, Schuler MA, Rogers PD (2014) The contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans . Antimicrob Agents Chemother 59: 450–60. 10.1128/AAC.03470-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J (2009) Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5: e1000705 10.1371/journal.pgen.1000705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vermitsky J-P, Earhart KD, Smith WL, Homayouni R, Edlind TD, et al. (2006) Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61: 704–722. [DOI] [PubMed] [Google Scholar]
- 27. Ram Y, Hadany L (2014) Stress-induced mutagenesis and complex adaptation. Proc Biol Sci 281. pii: 20141025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hadany L, Beker T (2003) On the evolutionary advantage of fitness-associated recombination. Genetics 165: 2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coyle S, Kroll E (2008) Starvation induces genomic rearrangements and starvation-resilient phenotypes in yeast. Mol Biol Evol 25: 310–318. [DOI] [PubMed] [Google Scholar]
- 30. Chen G, Bradford WD, Seidel CW, Li R (2012) Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482: 246–250. 10.1038/nature10795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin JH, Chae MJ, Song JW, Jung SI, Cho D, et al. (2007) Changes in karyotype and azole susceptibility of sequential bloodstream isolates from patients with Candida glabrata candidemia. J Clin Microbiol 45: 2385–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poláková S, Blume C, Zárate JA, Mentel M, Jørck-Ramberg D, et al. (2009) Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata . Proc Natl Acad Sci USA 106: 2688–2693. 10.1073/pnas.0809793106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Forche A, Magee PT, Selmecki A, Berman J, May G (2009) Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 182: 799–811. 10.1534/genetics.109.103325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Slater JL, Howard SJ, Sharp A, Goodwin J, Gregson LM, et al. (2011) Disseminated candidiasis caused by Candida albicans with amino acid substitutions in Fks1 at position Ser645 cannot be successfully treated with micafungin. Antimicrob Agents Chemother 55: 3075–3083. 10.1128/AAC.01686-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shor E, Fox CA, Broach JR (2013) The yeast environmental stress response regulates mutagenesis induced by proteotoxic stress. PLoS Genet 9: e1003680 10.1371/journal.pgen.1003680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cowen LE (2009) Hsp90 orchestrates stress response signaling governing fungal drug resistance. PLoS Pathog 5: e1000471 10.1371/journal.ppat.1000471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harrison BD, Hashemi J, Bibi M, Pulver R, Bavli D, et al. (2014) A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol 12: e1001815 10.1371/journal.pbio.1001815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, et al. (2011) Aneuploidy drives genomic instability in yeast. Science 333: 1026–1030. 10.1126/science.1206412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lewis JS 2nd, Wiederhold NP, Wickes BL, Patterson TF, Jorgensen JH (2013) Rapid emergence of echinocandin resistance in Candida glabrata resulting in clinical and microbiologic failure. Antimicrob Agents Chemother 57: 4559–4561. 10.1128/AAC.01144-13 [DOI] [PMC free article] [PubMed] [Google Scholar]