

Abstract
The anticancer activity of n-3 fatty acids, especially those derived from fish, such as eicosapentaenoic acid (EPA) and docosahexaenoic acid) (DHA), has been studied for centuries. While there is a growing body of evidence that EPA and DHA may influence cancer initiation and development through targeting multiple events of tumor development, the underlying mechanisms responsible for these activities are still not fully understood. A number of studies have suggested that the anticancer activities of EPA and DHA are associated with their effects on eicosanoid metabolism by which they inhibit prostaglandin E2 (PGE2) production. In contrast to DHA, EPA can function as a substrate for cyclooxygenases (COXs) to synthesize unique 3-series prostaglandin compounds, especially PGE3. With advance technology in mass spectrometry, there is renewed interest in studying the role of PGE3 in EPA elicited anti-proliferative activity in various cancers, with some promising results. Here, we summarize the regulation of PGE3 synthesis in cancer cells and its role in EPA elicited anticancer activity. The development of PGE3 and its metabolites as potential biomarkers for future clinical evaluation of EPA and fish oil in cancer care is discussed.
Keywords: n-3 Fatty acids, PGE3, Metabolism, Cancer cells, Tumor tissues
Introduction
Cancer is a leading cause of death worldwide and a second in the United States, exceeded only by heart disease. One in every four deaths in the United States is due to cancer. Despite the advancement in various treatment strategies, such as combinations of surgical resection, radiation or chemotherapies and immune therapies, the 5-year survival rate for some cancers is still relatively low. Furthermore, the underlying cause of cancer remains unclear. Thus, there is an unmet need to develop an effective strategy for preventing the development of this devastating disease. While the results of large chemoprevention trials thus far are not encouraging, a 20-year follow up study with aspirin, a non-steriodal anti-inflammatory agent that acetylates cyclooxygenase 2 (COX-2), showed that the mortality rates from all solid cancers were 20% lower for those receiving aspirin, with adenocarcinoma being the most reduced (34%) [1], suggesting the role of anti-inflammatory agents such as COX inhibitors in cancer prevention. Further studies indicated that aspirin use has no impact on the risk of colon adenocarcinomas that do not overexpress COX-2 [2,3]. Overexpression of COX-2 has been observed in a number of malignant diseases, especially epithelial cancers. Prostaglandin E2 (PGE2), a metabolite of n-6 fatty acid (arachidonic acid, AA), produced by COX-2 catabolism inhibits cancer cell apoptosis, increases invasiveness and angiogenesis in the tumor through pathways such as NF-κB, MAPkinase/JNK/p38, PI3kinase/Akt [4,5] as well as epigenetic modifications [6]. Thus, there has continued interest in using selective COX-2 inhibitors, such as celecoxib and rofecoxib, in chemoprevention. However, the cardiotoxicity of these agents has become an impediment to their long-term chemopreventive usage. In contrast to synthetic COX inhibitors, n-3 fatty acids are natural modulators of COX-2, with the ability to alter COX-2 metabolites and regulate the activity of downstream receptors while reducing blood triglyceride levels [7–10].
Long chain n-3 polyunsaturated fatty acids (PUFA) or n-3 fatty acids including α-linolenic acid (C18:3, ALA), eicosapentaenoic acid (C20:5, EPA), docosapentaenoic acid (C22:5, DPA) and docosahexaenoic acid (C22:6, DHA) are a group of compounds possessing the first double bond after the third carbon atom from the methyl end of fatty acid chains. These fatty acids, especially EPA and DHA, have been shown to have anti-inflammatory and immunomodulatory properties and are believed to be beneficial to cardiac, musculoskeletal, gastrointestinal, and immune systems in humans [11]. Epidemiological and preclinical evidence support the notion that n-3 fatty acids, especially EPA and DHA, have anticancer activities. For example, n-3 fatty acids have been shown to reduce onset of different cancers and protect against late stage cancers in carcinogen induced mouse tumors, human tumor mouse xenografts and spontaneous mouse tumors induced by transgenes [12]. Additionally, human studies have demonstrated that a higher intake of n-3 fatty acid is linked to a reduced risk of skin, colorectal, lung, prostate and breast cancers [13–17]. A recent study demonstrated that intake of EPA and DHA was associated with approximate 25% reduced risk of additional breast cancer events and had a dose-dependent reduced risk of all-cause mortality [15]. These findings together provide compelling evidence that n-3 fatty acids, EPA and DHA, could not only prevent the initiation of cancer, but also delay further development of cancer possibly through different mechanisms (reviewed in [12]). Among a number of plausible mechanisms, the ability of n-3 fatty acids, especially EPA, to modulate eicosanoid metabolism, particularly reduction of cyclooxygenase derived prostaglandin metabolism, has been extensively studied. Additionally, EPA and DHA derived resolvins from acetylated COX-2 (by aspirin) or 15-lipoxygenase demonstrate anti-inflammatory, neuroprotective and anticarcinogenic activities which could be an additive benefit of EPA or DHA in cancer risk reduction [18–20]. Compelling evidence supports that EPA could function as a selective COX-2 inhibitor because it can essentially act as a competitive inhibitor of AA to COXs, resulting in reduction of the 2-series prostaglandin (PG), such as PGE2, and concomitant generation of the 3-series PGs, i.e., PGE3 [9,21,22]. Compared to PGE2, PGE3 and other 3-series prostaglandins tend to have antiproliferative and anti-inflammatory activities and could potentially antagonize the tumor promoting effect of PGE2 in tumorigenic cells [21,23– 25]. In normal mouse colonic organoid culture, a recently report demonstrated that PGE2 induced proliferation of Lgr5+ colonic stem cells and promoted growth of mouse colon organoids while PGE3 did not support the colonic stem cell expansion in the same system [25]. The pharmacokinetics of AA derived PGE2 and EPA derived PGE3 are differentially regulated in normal and cancer cells, however, due to the increased expression of COX-2 in cancer cells, PGE3 production in tumors can be much higher than the surrounding normal tissue. Given that, in general, cellular metabolism fundamentally differs in cancer cells and normal cells, an understanding of how EPA is metabolized in cancer cells becomes a critical component of research investigations focusing on anticancer activity of n-3 fatty acids.
In this review, focusing on PGE3 metabolism, we summarize the pharmacokinetics of EPA that produces PGE3 in normal and cancer cells, the comparisons of the 3-series to the 2-series PG receptors, the regulation of PGE3 metabolism in cancer cells, and the association between production of PGE3 and antitumor or chemopreventive effects of n-3 fatty acids. Additionally, we emphasize the significance of developing appropriate biomarkers for EPA, such as PGE3 and its metabolites, to further determine the anticancer function of n-3 in future clinical applications.
Overview of AA and EPA metabolism by cyclooxygenases
In humans, n-3 and n-6 series fatty acids are ingested in the physiologically active forms of EPA and AA or as these molecules' respective precursors ALA and linoleic acid (LA, 18:2 n-6) [26]. LA, the major dietary source of n-6 fatty acids, is efficiently converted to AA. Studies have reported that because of the high oxidation rate, only a small proportion of ALA is converted to EPA, DPA or DHA [27]. Omega 3 polyunsaturated fatty acid DPA is an elongated metabolite of EPA and is an intermediary product between EPA and DHA [28]. Burdge and Wootton discovered that young men possessed the capacity to synthesize EPA and DPA from ALA, but that DHA synthesis was limited [29]. The fractional conversion of ALA to EPA varies between 0.3% and 8%, and less than 4% to DHA in males [30,31]. In women, the conversion of ALA to long chain n-3 fatty acids appears to be more efficient (up to 20% is converted to EPA and up to 9% is converted to DHA) [31]. DHA itself also serves as a substrate for metabolic retro-conversion to EPA and DPA through a β-oxidation reaction [32]. The retro-conversion rate of DHA to EPA, which can be affected by hormonal therapies in women [33], is approximately 1.4% with normal intake of DHA from food sources, while increases to 10% (9.4–12%) with DHA supplementation [32,34,35]. Similarly, DPA can also be retro-converted to EPA [36]. High ALA diets seem to only increase the rate of ALA oxidation with little conversion to EPA or DHA because diets high in ALA limit ALA accumulation in plasma [37]. With considerable variability in the conversion rates of ALA, along with modest ALA intakes and high amounts of LA in the American diet, it is reasonable to believe that ALA cannot reliably replace EPA or DHA in the diet [32,38]. However, the combined intake of EPA and DHA is estimated at only about 100 mg/d, due to limited intake of marine foods that are the major dietary source of n-3 fatty acids in the United States [39].
Fatty acid incorporation in membrane phospholipid
After being absorbed from the GI track, both n-3 and n-6 fatty acids compete for esterification at the sn-2 position of membrane phospholipids (PLs) [32]. Dietary intake of EPA and DHA, not ALA metabolic conversion, is the major contributor of EPA and DHA in cellular membrane phospholipids, because ALA is not efficiently incorporated into membrane phospholipids. In humans, short term supplementation with DPA not only significantly increases the incorporation of DPA in both plasma PL and triglycerides (TGs), but also enhances the incorporation of EPA and DHA into plasma TGs and cholesterol esters (CEs). In contrast, EPA supplementation only results in increased incorporation of EPA in plasma PLs and CEs without altering the levels of DPA and DHA incorporation into plasma lipids, suggesting that DPA and EPA incorporate into plasma phospholipids in different patterns [40]. Additionally, rodent and most recently published human data suggested that dietary DPA and EPA may be metabolized differently even though both of them exert anti-inflammatory and anticarcinogeneic activities [28,41,42]. On the other hand, high dietary intake of LA, which is efficiently incorporated into the membrane, reduces the membrane EPA phospholipid pool. Upon incorporation into the membrane, AA and EPA can be released from cellular phospholipids by phospholipases following extracellular stimulation, while free DHA released from the membrane is undetectable [43]. Cytosolic free AA or EPA in cells can further be converted to downstream metabolites that differ both in the number of their double bonds for COX products (prostaglandins, PG) and lipoxygenase (LOX) products (HETEs and leukotrienes) and in their biological actions in a wide range of physiologic and pathologic processes [44], including inflammation and immunity [45], hemostasis [46,47], atherosclerosis [48,49] and cancer [50,51]. Further, the balance in tissues eicosanoids derived from AA and EPA is regulated by complex interactions/competitions between AA and EPA at multiple levels of the eicosanoid biosynthetic pathway [52].
Eicosanoids metabolites
The term “eicosanoid” is used to denote twenty carbon PGs and lipoxins that are synthesized from n-3 and n-6 fatty acids in response to various hormones and physical stimuli [53]. The detailed prostaglandin biosynthesis including both 2 and 3 series PGs using AA and EPA has been comprehensively reviewed [54]. In this review, we will focus on PG synthesis, especially for 3-series prostaglandin, demonstrated in Fig. 1. Eicosanoids are synthesized and released from the cells rapidly (seconds) in response to extracellular stimuli [53]. Syntheses of the 2- and 3- series PG metabolites from AA or EPA, respectively, share three common steps in which particular synthases are involved (Fig. 1): firstly, AA or EPA is released from membrane phospholipids by cytosolic phospholipase A2 (cPLA2) or secretory sPLA2; secondly AA/EPA is converted to prostaglandin endoperoxide (PGH2 from AA and PGH3 from EPA) by COX-1 or COX-2; lastly, isomerization of PGH to “2-series” or “3-series” products– PGE2 or PGE3, PGD2 or PGD3, PGF2α or PGF3α, PGI2 or PGI3, or thromboxane A2 (TxA2) or TxA3 by specific synthases.
Fig. 1.
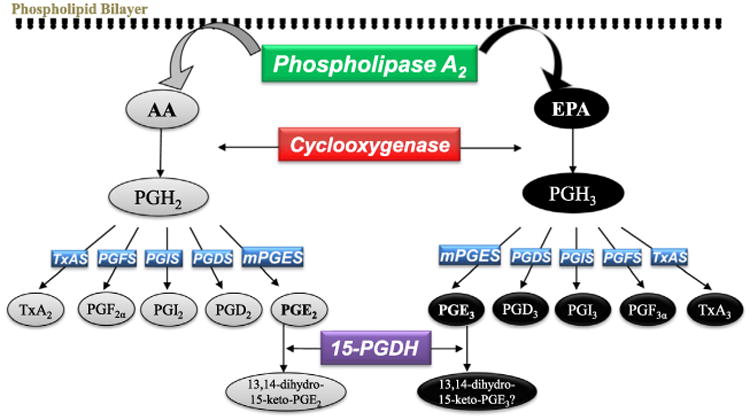
Comparison of EPA and AA cyclooxygenase metabolism. Modified from Pirman et al. [68], Wada et al. [22].
Although metabolisms of AA and EPA share the same set of enzymes, the catabolic activities of the enzymes in each of the metabolic step towards the two groups of substrates are somewhat different. cPLA2 is found in cytosol of cells at resting state. It translocates to the plasma membrane upon hormone-induced mobilization of intracellular Ca2+. While both EPA- and AA-containing phospholipids are substrates for cPLA2 with approximately equal activities [55,56], cPLA2 hydrolyzes DHA poorly [57]. COX-1 has been reported to preferentially catalyze AA with very low activity on EPA (10% compared to AA). EPA is a reasonably good inhibitor of AA oxygenation by COX-1; at equimolar concentrations EPA inhibits AA oxygenation by 50%. In contrast to COX-1, COX-2 converts EPA to PGH3 at about 30% of the rate of conversion of AA to PGH2. However, studies from our laboratory and other investigators demonstrated that EPA is a better substrate for human COX-2 enzyme than COX-1 enzyme [21,58]. EPA is a poor inhibitor of AA oxygenation by COX-2 [22,58,59]. Microsomal PGE synthase-1 (mPGES-1) and PGD synthases (PGDS) were reported to be more than 3-fold less active with PGH3 than with PGH2 [22], but the activity of cytosolic PGES on PGH3 has not been reported [60]. PGF synthase (PGFS) catalyzes the biosyntheses of PGF2α and PGF3α, however the substrate-enzyme binding/catabolic activities with PGH2 vs. PGH3 are unknown. PGI synthase (PGIS) and TxA synthase (TxAS) demonstrated similar activities with PGH2 and PGH3 [61,62]. Additionally, 15-hydroxyprostaglandin dehydrogenase (15-PGDH) catalyzes the rate-limiting step of PGE2 catabolism [63], however whether it is responsible for PGE3 degradation has not been reported. The ratios of the enzyme activities with EPA-vs. AA-derived reactants and products in vitro were documented [22], however the calculation needs to be tested in humans. Regardless, metabolite productivities of the entire enzymatic system closely depend on the substrate pools and levels of the various enzymes involved in the synthesis of the 2-series and the 3-series PGs.
EPA alters cellular PG metabolite profile
EPA competes with AA for incorporation into phospholipids in cell membranes, therefore partial substitution of AA by EPA in membrane phospholipids should lead to the replacement of AA-derived 2-series eicosanoid metabolites by EPA-derived 3-series metabolites. In several in vitro normal cell models, the effect of EPA on PG biosynthesis has been tested. Belury et al. first reported that PGE3 formation in EPA treated murine epidermal cells was more than PGE2 when these freeze–thawed cells were treated with equal molar of 14C-AA and 14C-EPA, while significant more PGF was produced in AA treated than in EPA treated cells [64]. Norris et al. showed that EPA treatment reduced the amount of AA-derived COX metabolites (20% lower) and increased EPA-derived COX metabolites PGD3 and PGE3 compared to control RAW264.7 macrophage cells [65]. In the same study, PGD3 was the predominant EPA derived metabolite produced in EPA-supplemented cells and was about 3-fold higher than in control cells, which was mainly due to PGD synthase being predominant subtype prostaglandin synthases in macrophages cells [65]. In another study using cultured human mast cells, EPA treatment decreased PGD2 generation by inhibiting the COX-2 pathway [66]. Limited in vivo data from animal studies have also shown that supplementation of dietary EPA reduces PGE2 and increases PGE3 in mouse colon [51,67], lung [68] or dog peripheral blood mononuclear cells (PBMCs) [69].
PGE3 and PG receptors
Due to rapid metabolism, PGs function at or near their sites of synthesis [22]. Newly formed PGs primarily function through G-protein coupled receptors (GPCRs) as autocrine or paracrine mediators [53]. Research studying the actions of PG receptors have reported the presence of PG receptors for the D, F, I, and E types PGs and TxA, named DP, FP, IP, EP, and TP receptor, respectively (Table 1) [22,70]. Originally interactions between E type PGs and EP receptors were discovered based on PGE2 actions [71]. There are four GPCR subtypes of EP receptors, EP1, EP2, EP3, and EP4, which exhibit differences in signal transduction, tissue localization, and regulation of expression [61]. This molecular and biochemical heterogeneity of PGE receptors leads to E type PGs being the most versatile PG [61]. Evidence for receptors for 3-series PGs is scant, with most research focused on downstream actions of PGE3. Limited literature suggests that PGE3 shares the same EP receptor system with PGE2 albeit with different binding affinities and potencies (Table 1).
Table 1.
3-Series PG related EP receptors.
| 3-Series PGs | Receptors | EC50 ratios (PGE3 vs. PGE2) | Tissue/Cell | References |
|---|---|---|---|---|
| PGE3 (partial agonists for EP1, EP2, and EP3) | EP1 | 2 | Human embryonic kidney cells | [22] |
| EP2 | 2.55 | Human pancreatic cancer cells, human embryonic kidney cells | [22,50] | |
| EP3 | 3.06 | Human embryonic kidney cells | [22] | |
| EP4 | 6.03 | Human CRC cells, human embryonic kidney cells | [22,24] | |
| PGD3 | DP1 | 0.46a | Human platelets | [22,119] |
| DP2 | 1.14a | Human eosinophil | [22,119] | |
| PGF3α (partial agonists) | FP | 4.79 | Human embryonic kidney cells | [22] |
| PGI3 | IP | 1.41 | Human embryonic kidney cells, human and rabbit platelets | [22,120] |
| TxA3 | TP | 1.31 | Human embryonic kidney cells | [22] |
Comparison of geometric means.
Wada et al. studied the receptor binding specificities of PGE2/PGE3 to EP and PGF2α/PGF3α to FP receptors using cell membrane from human kidney cells [22]. They determined that the relative affinities of the 2-series PGs were significantly greater than the 3-series PGs except for the EP4 receptor. The difference between PGF2α and PGF3α with the FP receptor can be as high as 78-fold. In the same study, the authors reported the potencies of the 2-vs. the 3-series PGs in stimulating second messenger formation via the EP and FP receptors and found that the 2-series PGs exhibited significantly higher potency than the 3-series PGs with the exception for EP4. Interestingly, the data further suggested that PGE3 is a partial agonist of the EP receptors. Whether there are functional differences in downstream signaling between PGE3 and PGE2 was not detailed in the same publication. One recent study reported PGE3 acts as an antagonist to EP3 to promote the inhibitory effects of PGE3 on platelet function, by which the cardiovascular benefits of dietary n-3 fatty acids may be conferred [72]. Another study illustrated that PGE3 bound to EP4 with reduced affinity and efficacy compared with PGE2 in human colorectal cancer (CRC) cells, but in the presence of PGE2, PGE3 acted as an antagonist of EP4 in cyclic AMP production [24]. A different research group found that both PGE2 and PGE3 induced cyclic AMP in RAW 264.7 cells, however, accumulation of intracellular cyclic AMP in PGE3 treated cells was only half as high as PGE2 treated cells [73]. These data suggest that although PGE2 and PGE3 both activate the EP receptor system, the substrate-receptor binding actions vary dramatically depending on the circumstance.
cPLA2, COX-2, and PGE3 metabolism in cancer
EPA functions as a substrate for COXs and results in synthesis of the 3-series PG compounds, such as PGE3, PGD3 and PGI3 [9]. Even though the theory of formation of the 3-series PGs by EPA has been studied for decades, understanding of the synthetic capability of the 3-series PGs, especially PGE3 from EPA in cancer cells is still inconclusive. This is due, in part, to the lack of specific and sensitive analytical techniques that could be used to determine the endogenous levels of PGE3 in various biological matrices. Additionally, the amount of EPA derived PGE3 is much lower than AA derived PGE2 because of the low COX-2 protein expression in normal tissue [9]. Published studies reveal that COX-2 protein is overexpressed in various cancers, such as lung, colon, breast, and pancreatic tumors as well as in their relevant in vitro cancer cells [5,74]. As a result, the PGE3 metabolism in cancer cells can be markedly different from that in the normal epithelial and smooth muscle cells mainly due to the different levels of expression and activities of the enzymes, such as cPLA2, COX-2, and mPGES-1 regulating the release of EPA and biosynthesis of PGE3 in the cancer cells or tumor tissues. Given that studies on the role of mPGES in PGE3 synthesis are limited, here we will only elaborate on EPA availability and COXs regulation of the biosynthesis of PGE3 in cancer cells or tumor tissues.
Membrane phospholipid pool
In contrast to the general consensus of PGE3 biosynthesis being relatively low compared to that of PGE2 in the normal cells or tissues as described earlier, the formation of PGE3 appears to be similar to that of PGE2 in cancer cells or tumor tissues when similar levels of EPA and AA are incorporated in the membrane phospholipid. We have found that the formation of PGE3 was similar in the A549 non-small cell lung cancer (NSCLC) treated with EPA alone compared to that of the same cells treated with a combination of equal concentration of EPA and AA [21]. In line with this, the formation of PGE3 and PGE2 was generated at equivalent levels (ratio of PGE3/PGE2 > 1.0) when a similar amount of EPA and AA were incorporated into the membrane phospholipids of MC-26 mouse CRC liver tumors [75]. However, the actual kinetics of the formation of PGE3 in cancer cells or tumor tissues when both AA and EPA are present has yet to be fully understood, although Hawcroft et al. have indicated that biosynthesis of PGE3 in tumor tissues might also be associated with the ratio of EPA/AA, i.e., the ratio needs to be higher than 0.1 in order to allow biosynthesis of PGE3 [75]. The formation of PGE3 is relatively proportional to the amount of EPA incorporated in cell membrane of tumor tissues, but certainly the relationship is non-linear. For example, the amount of EPA incorporated in phospholipids of MC-26 mouse CRC cells liver tumor in mice fed with 5% EPA free fatty acids, was almost 3-fold higher than that from mice fed with 2.5% EPA free fatty acids. The formation of PGE3 was only about 2-fold higher in livers from mice fed with 5% EPA free fatty acids compared to that in livers from the 2.5% group [75]. This could potentially be affected by the amount cytosolic fatty acid release from the membrane phospholipids and the status of the COXs or mPGES-2 proteins as n-3 fatty acids have the ability to inhibit the expression of COX-2 [76] and mPGES-2 [51].
cPLA2
The release of AA or EPA from the phospholipids of cell membrane is catalyzed by phospholipases and cPLA2 plays a critical role in regulation of eicosanoid metabolism [77,78]. For example, macrophages from mice release much less AA than macrophages from WT mice upon inflammatory stimuli, such as phorbol mysristate acetate and lipopolysaccharides (LPS) [77]. Consequently less PGE2 is produced in cPLA2 deficient macrophages compared to the controls [77]. By profiling of the whole cell metabolomics using a combination of a matrix-assisted laser desorption (MALDI) mass spectrometry and LC/MS-MS, our recent study suggested that cPLA2 status could be critical for regulation of EPA derived PG metabolism in cancer cells or tumor tissues because of its ability to affect the hydrolysis of these fatty acids from the membrane phospholipids [68]. While both A549 and H596 NSCLC cells express similar levels of COX-2 and mPGES-1, the formation of PGE3 was substantially lower in H596 cells than in A549 cells. This appears to be due to the lower expression and activity of cPLA2 in H596 cells than that of A549 cells. As a result, even though the abilities of EPA incorporation into membrane phospholipids were similar in both cell lines, the release of EPA from the phospholipids in H596 cells was far less than that of A549 cells because low cPLA2 expression and activity limited the amount of free EPA presented to COXs. As described earlier, cPLA2 hydrolyzes EPA and AA in the membrane phospholipids at similar rate, but it is less active in releasing DHA from the membrane phospholipids [9]. The differential activities on the release of EPA and DHA could potentially influence the biosynthesis of PGE3 when both these n-3 fatty acids are incorporated into the membrane phospholipids. Based on our unpublished data, it is possible that the presence of DHA might reduce the formation of PGE3 from EPA. This is based on the fact that the levels of PGE3 in lung tumor tissues from mice treated with Lovaza, a fish oil supplement containing equal amounts of EPA and DHA, were only about 50% of that in the tumors from mice treated with same concentration of EPA alone. This may be caused by DHA competing with EPA for incorporation into phospholipids (Yang P unpublished data). Studies delineating the impact of DHA on EPA derived PGs and on EPA elicited anti-inflammatory and anti-proliferative activities will be interesting as the two n-3 fatty acids in general are administered simultaneously. Taken together, these studies suggested that the bioavailability of EPA, which could be regulated through the incorporation into phospholipids as well as the release from phospholipids, is important in PGE3 metabolism in cancer cells.
Cox-2
The critical role of COX2 in PGE3 metabolism in cancer cells and tumors has been documented in a number of studies [21,24]. By applying a sensitive and specific LC/MS-MS method in COX-2 expressing A549 cells, Yang et al. have demonstrated that the formation of PGE3 in these cells appears to be mainly mediated through COX-2 pathway because the level of PGE3 was reduced by 50% when the cells were exposed to a combination of EPA (25 µM) and celecoxib, a selective COX-2 inhibitor. Compared to EPA alone, a combination of EPA and COX-1 inhibitor did not reduce the formation of PGE3 in this particular cell line [21]. Similarly, Xia et al. and Hawcroft et al. reported that the formation of PGE3 in EPA treated mouse melanoma and human HCA-7 CRC cells was blocked by indomethacin [79] or a selective COX-2 inhibitor, SC-236, respectively [24]. The important role of COX-2 in regulating PGE3 metabolism was further supported by the finding that a substantially lower amount of PGE3 was detected in the H1299 cells (COX-2 null NSCLC cells) and its xenograft tumor tissues compared to that in A549 cells (COX-2 constitutively expressed) and its xenograft [80]. The formation of PGE3 was partially blocked in the COX-2 knockdown A549 cells compared with the control siRNA administrated A549 cells. Furthermore, while the level of PGE2 was doubled in the colon mucosa from irradiated azoxymethane (AOM) treated rats compared to that in the colon mucosa from non-irradiated controls, there was 3-fold increase in colonic mucosal PGE3 from irradiated AOM-rats than that of non-irradiated AOM-rats which can be due to the higher mucosal COX-2 expression in the irradiated rats than non-irradiated rats [51]. The increased PGE3 in these studies might be linked to the anticancer activity of EPA or fish oil containing EPA. Literature on studies of different n-3 fatty acids, either EPA alone or menhaden oil in various tumors, including NSCLC, colon, breast and pancreatic cancer, consistently demonstrated that tumor cells have the ability to notably increased the ratio of PGE3/PGE2 in the tumor tissues of interest. Intriguingly, the ratio of PGE3/PGE2 was 3-fold higher in EPA free fatty acid (5%) treated MC-26 mouse CRC cell liver tumors than that in the normal liver tissues of this mouse model [75], suggesting that there should be relatively higher PGE3 production in cancer cells as oppose to that of normal cells that potentially is due to reduced expression of COX-2 enzyme in normal tissues. Thus, these studies together provide strong support that PGE3 metabolism is not only dependent on COX-2, but is also highly associated with cPLA2 expression and activity. In light of COX-2 expression and activity being an important regulator of PGE3 synthesis, it certainly challenges the concept of using combination of the n-3 fatty acids, especially EPA, and selective COX-2 inhibitor simultaneously in cancer therapy.
EPA induced changes in 2 and 3-series PGs in cancer
When EPA instead of AA is incorporated into cell membranes, not only would less AA-derived products be available but the EPA-derived substrates and products are typically less active than AA-derived substrates and products with potentially different biological activities [64]. Studies have shown that the 3-series eicosanoid metabolites are generally less pro-inflammatory than the homologous 2-series [21,22,45]. Thus, the role of n-3 fatty acids on modulation of eicosanoids has been one of the molecular mechanisms that is currently heavily studied in the context of n-3 fatty acid elicited anticancer activities [12]. The early studies conducted on n-3 fatty acids and cancer prior to the year of 2000 were mainly focused on the reduction of PGE2 in n-3 fatty acid treated cancer cells or tissues. For examples, consumption of a diet enriched with menhaden oil significantly reduced PGE2 metabolites in both plasma and tumor tissues in mice bearing the PG-producing HSDM1 fibrosarcoma [81]. The reduction of PGE2 in either tumor cells or tissues has been documented in EPA, DHA or menhaden oil treated AOM-induced rat colon tissues [82–84], fish oil treated human prostate cancer DU145 xenograft model [85], 7,12-dimethylenz[a]anthracene induced rat mammary carcinoma [86], and EPA or DHA treated human lung mucoepidermoid carcinoma [87]. The reduction of PGE2 appeared to be associated with the inhibitory effect of either fish oil (both EPA and DHA) or one of the n-3 fatty acids alone in all the aforementioned tumor models.
It was only after the year 2000 when we and other investigators established a sensitive and specific LC/MS-MS method which allowed determination of the 3-series PGs, especially PGE3, in various biological matrices [51,67,88,89] that the role of PGE3 in influencing the growth of cancer cells or tumor tissues began to be documented (Table 2). The anti-proliferative effect of EPA might be mediated not only through reduction of PGE2, but also through concomitant increase of PGE3 in A549 cells, because the COX-2 selective inhibitor, celecoxib, but not the COX-1 inhibitor SC-560, blocked formation of PGE3 as well as antiproliferative effect of EPA in the A549 cells [21]. EPA induced inhibition of cell proliferation of A549 cells constitutively expressing COX-2 was almost 10-times stronger than that of H1299 cells (COX-2 null), and 2-fold stronger than that of COX-2 knockdown A549 cells. Additionally, dietary menhaden oil significantly inhibited the growth of A549 tumors but showed no tumor inhibitory effect in H1299 xenografts, which could be linked to the relatively higher ratio of PGE3/PGE2 in A549 xenografts fed with menhaden oil compared to that of H1299 tumors [80]. In line with this, Vanamala et al. reported that fish oil enriched diet protected against radiation enhanced colon cancer by reduction of PGE2 and increased formation of PGE3 in the colonocytes in an AOM rat colon cancer model [51]. Similar results, i.e., reduction of PGE2 and increased PGE3 were identified in menhaden oil treated human pancreatic cancer BxPC3 xenografts [50] and in EPA treated mouse MC-26 mouse CRC cells liver tumors [75]. These results could contribute to the mechanisms of anticancer activities of n-3 fatty acids. Furthermore, two studies using mice carrying the Fat-1 transgene, which restores the synthesis of n-3 fatty acids (EPA and DHA), inoculated with either mouse melanoma B16 cells or mouse HER2 positive breast cancer E0771 cells, showed that the development of melanoma and HER2 positive mammary gland tumors were substantially reduced after 15 days of cell inoculation while the tumors in the Fat-1 wild type (WT) mice continued to grow [79,90]. Interestingly, Xia et al. demonstrated that the reduction of PGE2 was moderate in the stromal tissues and stronger in the tumors in the Fat-1 WT mice, while PGE3 was markedly increased in both the stromal and the tumor tissues in the Fat-1 transgenic mice with melanoma [79]. Similarly, there was notably increase in PGE3 production in the mammary tumors of the Fat-1 transgenic mice compared to that in the Fat-1 WT mice [90]. The significant contribution of COX-2 to the chemopreventive activity of fish oil is further strengthened by recent findings revealing that higher consumption of n-3 PUFA (EPA and DHA) lowered the risk of aggressive prostate cancer with this effect being more pronounced in men carrying a particular COX-2 variant [14]. Therefore, these studies suggested that the antiproliferative activity of n-3 fatty acids, especially EPA, could be enhanced by the formation of PGE3 primarily through COX-2, and COX-2 could be a pivotal target for EPA or fish oil mediated anti-proliferative or chemopreventive activities.
Table 2.
Dietary n-3 fatty acids targeting 3-series PGs in cancer prevention.
| Cancer type | Models | Interventionsa | Cancer outcome | PGE2 | PGE3 | References |
|---|---|---|---|---|---|---|
| Breast | Fat-1 mice injected with E0771 murine breast cancer cells | The diet contained 10% safflower oil with high LA and less than 0.1% n-3 fatty acids. | No palpable tumors in the fat-1 mice but 600 mm tumors in WT mice | No difference in the tumor | ↑ From undetectable to 7.65 of ng/mg in tumor | [90] |
| Colon | AOM induced Sprague– Dawley rats | 15% fat in the diets: a FO diet (EPA, 18.2% + DHA, 11.3%); a CO diet (LA 55.4%) | Not reported | ↓ 78% in mucosa | ↑ From undetectable to 1.43 pg/µg protein | [51] |
| Colon cancer liver metastasis | BALB/c AnN mice with intrasplenic injection of MC-26 cells | AIN-93G with 7% CO (control) vs. 4.5% CO + 2.5%EPA-FFA; 2% CO + 5%EPA-FAA | Liver metastasis was reduced; liver weight was significantly lower in 5% EPA-FFA group compared to controls | ↓ 60% in the tumor in EPA-FFA fed mice (5% but not 2.5%) | ↑ Dose dependently reaching 321 pg/mg in the tumors in 5% EPA-FFA treated animals | [75] |
| Lung | BALB/c athymic (Nu/Nu) mice injected with A549 cells | AIN-76 based: a SO diet with 15% SO; a FO diet with 10% of menhaden oil + 5% SO | Reduced tumor growth by 50–60% in A549 xenograft | ↓ 52% in the tumor in A549 xenograft | ↑ (0 to 0.89 ng/mg protein) in A549 xenograft | [80] |
| Melanoma | Fat-1 mice injected with B16 cells | A diet with 10% safflower oil | Reduced incidence of tumor formation and tumor growth rate | ↓ In the tumor and surrounding tissues | ↑ In the tumor | [79] |
| Pancreas | Nude mice injected with BxPC-3 cells | AIN-93G based diets: a CO diet (8.3% CO) vs. a FO diet (5.7% n-3 + 2.6% CO) | Tumor size reduced by 60% | ↓ 35% in the tumor | ↑ From undetectable to 0.5 ng/mg tumor | [50] |
| Colon | HCA-7 human CRC cells; MC-26 cells | 0.05–0.8 µM EPA-FFA | Apoptosis induced | ↓ 90% in HCA-7 cell conditioned medium (maximal synthesis at 24 h) | ↑ In MC-26 cells | [24] |
| Lung | A549 human NSCLC cells | 100 µM EPA, DHA | Not reported | ↓ 64% in cells treated with EPA ↓ 48% in cells treated with DHA |
↑ From 0.43 ng/5 million cells in control cells to 5.12 ng/5 million cells in treated cells | [89] |
| Lung | A549 human NSCLC cells | 10–50 µM EPA | Reduced proliferation and induced cell death | Not reported | ↑ Intracellular and extracellular production | [21] |
| Lung | H1299 and A549 human NSCLC cells | 10 and 50 µM EPA | Reduced cell growth | Not reported | ↑ Dose dependently ↑ PGE3/PGE2 ratio | [80] |
| Pancreas | Human pancreatic cancer cells BxPC-3 | 10 µM EPA | Decrease cell growth | ↓ to below baseline | ↑ Extracellular production to 209 pg/106 cells | [50] |
CO: corn oil; FO: fish oil; SO: soybean oil; FFA: free fatty acid.
Potential mechanisms in PGE3 anticancer activities
In comparison to our understanding of the biosynthesis of the n-3 series of PGs, knowledge of their biologic functions is limited. To understand the mechanism(s) underlying the effects of PGE3 on cancer progression, researchers have been investigating how PGE3 regulates tumor growth and its signaling pathways. It appears that the multiple signaling pathways including the three major ones AKT, ERK1/2 and PKA are involved in PGE3 elicited anticancer activity (Fig. 2). Several reports have shown that PGE3 suppressed tumor growth by inhibiting angiogenesis, cell invasion, cell growth and survival, which could be mediated through the EP receptors. Studies from our laboratory indicate that PGE3 inhibits proliferation of A549 cells, whereas PGE2 slightly stimulates the growth of these cells [21]. PGE3 appears to inhibit the proliferation of mouse melanoma B16 cells by induction of apoptosis [79]. A similar effect was observed in human pancreatic cancer BxPC3 cells, when the cells were exposed to PGE2 and PGE3 [50]. In the same study, the anti-proliferative activity of PGE3 was shown to be mediated through both the EP2 and EP4 receptor using EP2 and EP4 transfected MiaPaca cells (EP receptor null). Interestingly, the anti-proliferative effect of PGE3 does not appear being mediated through PKA/cyclic AMP pathways. Others have shown that PGE3 is less effective in stimulating cell proliferation and IL-6 production [45,91]. Additionally, PGE2 eliminated the growth inhibitory effect of fish oil in hepatoma cells, while PGE3 reduced the invasiveness of the same cells pretreated with safflower oil [92]. Moreover, PGE3 suppressed the induction of angiopoietin-2 and resulted in inhibition of angiogenesis in human umbilical vein endothelial cells [93]. PGE3 also modulated COX-2–mediated invasion and angiogenesis in brain-metastatic melanoma [94]. Furthermore, our studies demonstrated that PGE3 inhibits proliferation of A549 cells and acts as an antagonist to PGE2-mediated increases in cell proliferation [21]. Hawcroft et al. observed that PGE3 antagonizes the protumorigenic activity of PGE2 in EP4 transfected HT-29 CRC cells that lack expression of COX-2 and other EP receptors, but not in HCA-7 CRC cells with higher endogenous levels of PGE2 [24]. What was also interesting is that PGE3 acts as an agonist when exposed alone to LoVo human CRC cells, while it becomes antagonist in the presence of natural ligand PGE2 in these particular cells. Nevertheless, the evidence is limited and more research is necessary to address the question whether PGE3 activates one or more of the EP receptor axis during tumor development.
Fig. 2.
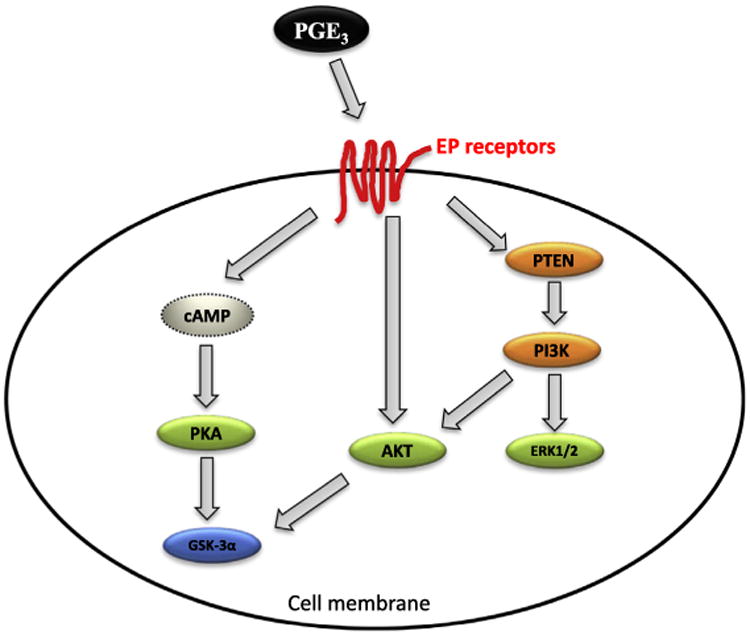
Proposed schematic signaling pathways for PGE3 induced EP receptor activation [80,121].
Recently the impact of downstream signaling by PGE3 has been studied more rigorously regardless of how the EP receptors are involved. PGE3 appears to downregulate PI3kinase signaling by either increasing PTEN expression [79] or directly suppressing the phopsphorylation of Akt, as opposed to increased pAkt by PGE2 in these particular cells [21]. PGE3 also inhibits the expression of HER3 and cMYC proteins which might contribute to the antitumor effect of n-3 fatty acids derived from de novo synthesis in Fat-1 transgenic mice. It is not clear whether or not the influence of PGE3 on cell signaling proteins is mediated through specific EP receptors, which needs to be further investigated. Overall, these studies suggest that the 3-series metabolite of EPA, PGE3, is associated with EPA elicited anti-proliferative activity in various types of tumors and could serve as a biomarker for EPA intake and biological response.
Prostaglandin 3-series as biomarkers for EPA status
Blood PGEs
The levels of lipids in cellular membranes reflect the net outcome of dietary intake, absorption, transport and metabolism of fats. Plasma EPA concentration increases in response to dietary EPA or DHA [32]. Plasma n-3 fatty acid levels have been used in clinical trials in testing the beneficial effects of n-3 fatty acids on a variety of debilitating conditions, including cancer [95]. However, plasma n-3 levels only provide evidence of n-3 fatty acid uptake, but not the utility of these n-3 fatty acids in relevant tissues, such as tumors, because the biosynthesis of PGE3 in the tumor tissues or cells could be mediated by the expression and activity of other enzymes mentioned earlier that are involved in de novo prostaglandin synthesis. In terms of measuring eicosanoids in the blood, there is not sufficient evidence to extract meaningful data. Kearns et al. studied the effect of dietary n-6 and n-3 fatty acid ratios on eicosanoid production in peripheral blood mononuclear cells (PBMCs) in young and aged dogs [69]. The authors isolated PBMCs from dogs that were treated with different ratios of n-6/n-3 diets and stimulated with LPS before measuring prostaglandins. Interestingly, they found that PGE2 production was not affected by dietary fatty acid ratios but there was a significant higher PGE3 production in PBMCs from dogs fed with high n-3 diet. In humans, a few studies have reported blood prostaglandin production after dietary n-3 fatty acid interventions. Most data were limited to PGE2 in ex vivo lipopolysaccharide stimulated PBMCs [96,97] and whole blood [98,99]. Dosing with 1 g/d EPA plus DHA caused the biggest decrease in PGE2 in stimulated PBMCs [100]. One recent randomized controlled trial conducted by Dawczynski et al. showed that higher consumption of PUFAs increase EPA and PGE3 in both plasma and red blood cells in mildly hypertriacylglycerolemic patients [101]. The available data suggested that dietary EPA consumption is correlated with reduction of PGE2 production and increase of PGE3 in the blood.
Tissue PGs
Compared to blood PG levels, the amount of PGE2 and PGE3 in tissue can be easily assayed by ELISA or LC/MS-MS, which are commonly used in research laboratories. In human studies, dietary EPA reduced PGE2 and ornithine decarboxylase activity [83,102,103]. Several rodent studies have found that dietary EPA reduces tissue PGE2 by 50–90% depending on models used in the studies and doses of EPA given to the animals [84,104–106]. In a human study, fish oil (MaxEPA) with 5.4 g EPA plus DHA decreased colonic PGE2 and TxB2 within three weeks in patients with inflammatory bowel disease [107]. One piece of information lacking in these studies was PGE3 levels because reliable internal standard was not available for the analysis.
In carcinogenic conditions, COX-2 is commonly overexpressed which leads to induced PGE3 formation. Several in vivo studies using rodent cancer models have reported increased PGE3 concentrations in tumors or tumor bearing animals upon dietary EPA consumption (Table 2). Interestingly, a few studies have indicated that an increased dose of EPA in both cancer animals [75] or cell lines [68] can dose dependently increase PGE3 production which further supports the theory that PGE3 can be used as a biomarker for EPA intake in vivo. Our recent study also suggested that the ratio of PGE3/PGE2 in mouse lung tumor tissues was significantly increased by 40-fold in mice fed with EPA enriched diet compared to that in soybean-fed mice [68]. Therefore, both in vitro and in vivo data appear to support that tissue PGE3 production is responding to dietary EPA, indicating that PGE3 can be a useful biomarker for evaluation of EPA associated anti-proliferative activity. Although tissue production of PGE3 may be quantifiable as a relevant biomarker linked to EPA elicited anticancer activity, obtaining tissue from human subjects would be inconvenient and prohibitory in some cases. For these reasons, development of blood or urine biomarkers is a preferable strategy.
Urinary PG metabolites
Quantification of urinary PGE metabolites [108] and the major urinary metabolite of PGI [109] can potentially be another biomarker for fatty acid status. In 1988, Kivits et al. reported that in rats, an AA diet increased urinary tetranor-prostaglandin E1 (PGE-M) which is the major urinary metabolite of PGE2 [110]. The authors also detected much higher conversion of PGE3 to Δ17-tetranor-prostaglandin E1, the major metabolite of PGE3, upon dietary EPA feeding. There are some human studies that reported changes in PGE-M in the urine from subjects who consumed n-3 fatty acids. Ferretti and colleagues conducted a clinical trial involving a group of healthy male volunteers. They found that a low-fat diet or a high-fat diet supplemented with fish oil decreased the urinary PGE-M by 14% [111,112]. Interestingly, body weight had a significant effect on the difference in the urinary PGE-M levels in that lighter weight individuals showed a bigger reduction in urinary PGE-M levels. Murff et al. demonstrated that the dietary intake of n-3 fatty acids in women but not in men was negatively correlated to urinary PGE-M concentrations [113]. However, Young et al. performed a similar study with n-3 fatty acids in healthy postmenopausal women and did not observe changes in PGE-M in urine samples from people who consumed EPA and DHA for 8 weeks [114]. These data indicate that PGE-M as a marker for estimation of consumption of dietary fish oil may be appropriate in certain populations. More studies with larger numbers and more diverse participants are necessary to verify this observation. However, one would think that measuring urinary PGE3 or its metabolites will provide a better approach than detecting reduction of the metabolites from the 2-series PGs upon EPA intake.
Fischer et al. reported that 10-fold increase of urinary PGE3 was detected when the cod liver oil was ingested [115]. Additionally, two urinary PGE3 metabolites, 7α, 11α-dihydroxy-5-ketotetranorprosta-9,13-dienoic acid and 11α-hydroxy-5-ketotetranoprosta-4(8),9, 13-trienoic acids were identified in the rat urine after the PGE3 was administered to rat [116]. Recently data showed that two related 2,3-dinor metabolites were the major urinary metabolites of 6-keto-PGF2α from prostacyclin (PGI3) in mice [117], which may facilitate the development of methods to measure urinary metabolites of the 3-series PGs. Whether these metabolites could serve as urinary biomarkers for EPA status and whether this correlates to its biological responses need to be explored in cancer settings. Another important issue is that urinary metabolite levels are markers of the total body production of PGs, which may be altered in a different manner than specific tissue levels of PGs [118]. Therefore, the questions are whether systemic changes will mirror changes in a particular tissue and whether the function of these eicosanoid metabolites will correlate with changes in absolute amounts of PGs. We anticipate that systemic levels of PGs should represent dietary fatty acid consumption. Whether fatty acid derived PGs are biologically active will depend on not only the substrate availability but accessibility of the receptors that receive the signals as well.
Conclusion and challenge
Even though researchers have been studying n-3 fatty acids and cancer for decades, there is still growing interest in defining the effects of n-3 fatty acids, EPA or DHA, in preventing the development of malignant diseases. Preclinical studies continually support the notion of n-3 fatty acids being effective in preventing the initiation and progression of various cancers, however, results from human clinical evaluation on the efficacies of n-3 fatty acids in cancer management are still controversial. This certainly could be due to several factors, such as the type and dose of n-3 fatty acids used in the studies, nutritional status of patient, and lack of biomarkers to monitor bioavailability in in situ tissues. Thus, it becomes imperative to concomitantly analyze both plasma levels of EPA and DHA as well as their metabolites, such as PGE3, in the tumor tissues. Compared to other 3-series prostaglandins, accumulating in vivo evidence supports the findings that tumor PGE3 derived from EPA or fish oil appears to be associated with EPA elicited antiproliferative activity. However, most studies have focused thus far on determination of EPA derived eicosanoids, especially PGE3 in cancer cells or tumor tissues. We contend that more effort needs to be taken to identify the regulation of PGE3 de novo synthesis and its influence on downstream signaling, which will ultimately improve our understanding on the role of EPA in cancer prevention and treatment. Identifying the key factors mediating the ability of cancer cells to produce PGE3, such as substrate availability when other n-3 (DPA and DHA) and n-6 fatty acids (AA) are present and expression of other enzymes involved in PGE synthesis, such as mPGES, are critical for understanding the biosynthesis of PGE3 in the tumor tissues and its biological consequences. An advanced technology, such as MALDI mass spectrometry coupled with LC/MS/MS, makes it possible to measure PGE3 metabolites in different surrogate tissues, such as erythrocytes, tissues and urine and further validate PGE3 metabolites as potential biomarkers for the COX-2 mediated anticancer activity of EPA. This information is pivotal for clinical studies of n-3 fatty acids in cancer management. Additionally, it is critical important for delineating the toxicity of the n-3 fatty acids and for optimizing the use of the n-3 fatty acids in the prevention and treatment of cancer, which has not been fully addressed.
Acknowledgments
This work was partially supported by the National Cancer Institute through grant R01-CA144053 to P.Y. We thank Dr. Ekem Efuet for his help with manuscript preparation.
Footnotes
Conflict of Interest: Authors have no conflict of interest to declare that is relevant to this review.
References
- 1.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 2.Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. New Engl J Med. 2007;356:2131–2142. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- 3.Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009;302:649–658. doi: 10.1001/jama.2009.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCarty MF. Minimizing the cancer-promotional activity of cox-2 as a central strategy in cancer prevention. Med Hypotheses. 2012;78:45–57. doi: 10.1016/j.mehy.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 5.Wang D, Dubois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2010;29:781–788. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia D, Wang D, Kim SH, Katoh H, DuBois RN. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat Med. 2012;18:224–226. doi: 10.1038/nm.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park Y, Harris WS. Omega-3 fatty acid supplementation accelerates chylomicron triglyceride clearance. J Lipid Res. 2003;44:455–463. doi: 10.1194/jlr.M200282-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Qi K, Fan C, Jiang J, Zhu H, Jiao H, Meng Q, Deckelbaum RJ. Omega-3 fatty acid containing diets decrease plasma triglyceride concentrations in mice by reducing endogenous triglyceride synthesis and enhancing the blood clearance of triglyceride-rich particles. Clin Nutr. 2008;27:424–430. doi: 10.1016/j.clnu.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Smith W. Cyclooxygenases, peroxide tone and the allure of fish oil. Curr Opin Cell Biol. 2005;17:174–182. doi: 10.1016/j.ceb.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Spencer L, Mann C, Metcalfe M, Webb M, Pollard C, Spencer D, Berry D, Steward W, Dennison A. The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. Eur J Cancer. 2009;45:2077–2086. doi: 10.1016/j.ejca.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 11.Riediger ND, Othman RA, Suh M, Moghadasian MH. A systemic review of the roles of n-3 fatty acids in health and disease. J Am Diet Assoc. 2009;109:668–679. doi: 10.1016/j.jada.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 12.Jing K, Wu T, Lim K. Omega-3 polyunsaturated fatty acids and cancer, Anticancer Agents Med. Chem. 2013;13:1162–1177. doi: 10.2174/18715206113139990319. [DOI] [PubMed] [Google Scholar]
- 13.Cockbain AJ, Toogood GJ, Hull MA. Omega-3 polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut. 2011;61:135–149. doi: 10.1136/gut.2010.233718. [DOI] [PubMed] [Google Scholar]
- 14.Fradet V, Cheng I, Casey G, Witte JS. Dietary omega-3 fatty acids, cyclooxygenase-2 genetic variation, and aggressive prostate cancer risk. Clin Cancer Res. 2009;15:2559–2566. doi: 10.1158/1078-0432.CCR-08-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patterson RE, Flatt SW, Newman VA, Natarajan L, Rock CL, Thomson CA, Caan BJ, Parker BA, Pierce JP. Marine fatty acid intake is associated with breast cancer prognosis. J Nutr. 2011;141:201–206. doi: 10.3945/jn.110.128777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takezaki T, Hirose K, Inoue M, Hamajima N, Yatabe Y, Mitsudomi T, Sugiura T, Kuroishi T, Tajima K. Dietary factors and lung cancer risk in Japanese: with special reference to fish consumption and adenocarcinomas. Br J Cancer. 2001;84:1199–1206. doi: 10.1054/bjoc.2001.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodes LE, Shahbakhti H, Azurdia RM, Moison RM, Steenwinkel MJ, Homburg MI, Dean MP, McArdle F, Beijersbergen van Henegouwen GM, Epe B, Vink AA. Effect of eicosapentaenoic acid, an omega-3 polyunsaturated fatty acid, on UVR-related cancer risk in humans. An assessment of early genotoxic markers. Carcinogenesis. 2003;24:919–925. doi: 10.1093/carcin/bgg038. [DOI] [PubMed] [Google Scholar]
- 18.Serhan CN. Novel eicosanoid and docosanoid mediators: resolvins, docosatrienes, and neuroprotectins. Curr Opin Clin Nutr Metab Care. 2005;8:115–121. doi: 10.1097/00075197-200503000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Janakiram NB, Mohammed A, Rao CV. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metast Rev. 2011;30:507–523. doi: 10.1007/s10555-011-9311-2. [DOI] [PubMed] [Google Scholar]
- 20.Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96:27–36. doi: 10.1016/j.prostaglandins.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang P, Chan D, Felix E, Cartwright C, Menter DG, Madden T, Klein RD, Fischer SM, Newman RA. Formation and antiproliferative effect of prostaglandin E(3) from eicosapentaenoic acid in human lung cancer cells. J Lipid Res. 2004;45:1030–1039. doi: 10.1194/jlr.M300455-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Wada M, DeLong CJ, Hong YH, Rieke CJ, Song I, Sidhu RS, Yuan C, Warnock M, Schmaier AH, Yokoyama C, Smyth EM, Wilson SJ, FitzGerald GA, Garavito RM, Sui de X, Regan JW, Smith WL. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J Biol Chem. 2007;282:22254–22266. doi: 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- 23.Hegde S, Kaushal N, Ravindra KC, Chiaro C, Hafer KT, Gandhi UH, Thompson JT, van den Heuvel JP, Kennett MJ, Hankey P, Paulson RF, Prabhu KS. Delta12-prostaglandin J3, an omega-3 fatty acid-derived metabolite, selectively ablates leukemia stem cells in mice. Blood. 2011;118:6909–6919. doi: 10.1182/blood-2010-11-317750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawcroft G, Loadman PM, Belluzzi A, Hull MA. Effect of eicosapentaenoic acid on E-type prostaglandin synthesis and EP4 receptor signaling in human colorectal cancer cells. Neoplasia. 2010;12:618–627. doi: 10.1593/neo.10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan YY, Davidson LA, Callaway ES, Goldsby JS, Chapkin RS. Differential effects of 2 and 3-series E-prostaglandins on in vitro expansion of Lgr5+ intestinal stem cells. Carcinogenesis. 2014 doi: 10.1093/carcin/bgt412. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marszalek JR, Lodish HF. Docosahexaenoic acid, fatty acid-interacting proteins, and neuronal function: breastmilk and fish are good for you. Annu Rev Cell Dev Biol. 2005;21:633–657. doi: 10.1146/annurev.cellbio.21.122303.120624. [DOI] [PubMed] [Google Scholar]
- 27.Goyens PL, Spilker ME, Zock PL, Katan MB, Mensink RP. Compartmental modeling to quantify alpha-linolenic acid conversion after longer term intake of multiple tracer boluses. J Lipid Res. 2005;46:1474–1483. doi: 10.1194/jlr.M400514-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Kaur G, Cameron-Smith D, Garg M, Sinclair AJ. Docosapentaenoic acid (22:5n–3): a review of its biological effects. Prog Lipid Res. 2011;50:28–34. doi: 10.1016/j.plipres.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Burdge GC, Wootton SA. Conversion of alpha-linolenic acid to eicosapentaenoic, docosapentaenoic and docosahexaenoic acids in young women. Br J Nutr. 2002;88:411–420. doi: 10.1079/BJN2002689. [DOI] [PubMed] [Google Scholar]
- 30.Burdge GC, Finnegan YE, Minihane AM, Williams CM, Wootton SA. Effect of altered dietary n-3 fatty acid intake upon plasma lipid fatty acid composition, conversion of [13C] alpha-linolenic acid to longer-chain fatty acids and partitioning towards beta-oxidation in older men. Br J Nutr. 2003;90:311–321. doi: 10.1079/bjn2003901. [DOI] [PubMed] [Google Scholar]
- 31.Burdge GC, Jones AE, Wootton SA. Eicosapentaenoic and docosapentaenoic acids are the principal products of alpha-linolenic acid metabolism in young men*. Br J Nutr. 2002;88:355–363. doi: 10.1079/BJN2002662. [DOI] [PubMed] [Google Scholar]
- 32.Arterburn LM, Hall EB, Oken H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am J Clin Nutr. 2006;83:1467S–1476S. doi: 10.1093/ajcn/83.6.1467S. [DOI] [PubMed] [Google Scholar]
- 33.Stark KD, Holub BJ. Differential eicosapentaenoic acid elevations and altered cardiovascular disease risk factor responses after supplementation with docosahexaenoic acid in postmenopausal women receiving and not receiving hormone replacement therapy. Am J Clin Nutr. 2004;79:765–773. doi: 10.1093/ajcn/79.5.765. [DOI] [PubMed] [Google Scholar]
- 34.Conquer JA, Holub BJ. Supplementation with an algae source of docosahexaenoic acid increases (n-3) fatty acid status and alters selected risk factors for heart disease in vegetarian subjects. J Nutr. 1996;126:3032–3039. doi: 10.1093/jn/126.12.3032. [DOI] [PubMed] [Google Scholar]
- 35.Conquer JA, Holub BJ. Dietary docosahexaenoic acid as a source of eicosapentaenoic acid in vegetarians and omnivores. Lipids. 1997;32:341–345. doi: 10.1007/s11745-997-0043-y. [DOI] [PubMed] [Google Scholar]
- 36.Mozaffarian D, Wu JH. (n-3) fatty acids and cardiovascular health: are effects of EPA and DHA shared or complementary? J Nutr. 2012;142:614S–625S. doi: 10.3945/jn.111.149633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vermunt SH, Mensink RP, Simonis MM, Hornstra G. Effects of dietary alpha-linolenic acid on the conversion and oxidation of 13C-alpha-linolenic acid. Lipids. 2000;35:137–142. doi: 10.1007/BF02664762. [DOI] [PubMed] [Google Scholar]
- 38.Emken EA, Adlof RO, Gulley RM. Dietary linoleic acid influences desaturation and acylation of deuterium-labeled linoleic and linolenic acids in young adult males. Biochim Biophys Acta. 1994;1213:277–288. doi: 10.1016/0005-2760(94)00054-9. [DOI] [PubMed] [Google Scholar]
- 39.Ervin RB, Wright JD, Wang CY, Kennedy-Stephenson J. Dietary intake of fats and fatty acids for the United States population: 1999–2000. Adv Data. 2004:1–6. [PubMed] [Google Scholar]
- 40.Miller E, Kaur G, Larsen A, Loh SP, Linderborg K, Weisinger HS, Turchini GM, Cameron-Smith D, Sinclair AJ. A short-term n-3 DPA supplementation study in humans. Eur J Nutr. 2013;52:895–904. doi: 10.1007/s00394-012-0396-3. [DOI] [PubMed] [Google Scholar]
- 41.Ghasemi Fard S, Linderborg KM, Turchini GM, Sinclair AJ. Comparison of the bioavailability of docosapentaenoic acid (DPA, 22:5n–3) and eicosapentaenoic acid (EPA, 20:5n–3) in the rat. Prostaglandins Leukot Essent Fatty Acids. 2014;90:23–26. doi: 10.1016/j.plefa.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Linderborg KM, Kaur G, Miller E, Meikle PJ, Larsen AE, Weir JM, Nuora A, Barlow CK, Kallio HP, Cameron-Smith D, Sinclair AJ. Postprandial metabolism of docosapentaenoic acid (DPA, 22:5n–3) and eicosapentaenoic acid (EPA, 20:5n–3) in humans. Prostaglandins Leukot Essent Fatty Acids. 2013;88:313–319. doi: 10.1016/j.plefa.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 43.Fischer S, von Schacky C, Siess W, Strasser T, Weber PC. Uptake, release and metabolism of docosahexaenoic acid (DHA, c22:6 omega 3) in human platelets and neutrophils. Biochem Biophys Res Commun. 1984;120:907–918. doi: 10.1016/s0006-291x(84)80193-x. [DOI] [PubMed] [Google Scholar]
- 44.Samuelsson B. Prostaglandins, thromboxanes, and leukotrienes: formation and biological roles. Harvey Lect. 1979;75:1–40. [PubMed] [Google Scholar]
- 45.Bagga D, Wang L, Farias-Eisner R, Glaspy JA, Reddy ST. Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc Natl Acad Sci USA. 2003;100:1751–1756. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorenz R, Spengler U, Fischer S, Duhm J, Weber PC. Platelet function, thromboxane formation and blood pressure control during supplementation of the Western diet with cod liver oil. Circulation. 1983;67:504–511. doi: 10.1161/01.cir.67.3.504. [DOI] [PubMed] [Google Scholar]
- 47.Phang M, Lincz LF, Garg ML. Eicosapentaenoic and docosahexaenoic acid supplementations reduce platelet aggregation and hemostatic markers differentially in men and women. J Nutr. 2013;143:457–463. doi: 10.3945/jn.112.171249. [DOI] [PubMed] [Google Scholar]
- 48.Dehmer GJ, Popma JJ, van den Berg EK, Eichhorn EJ, Prewitt JB, Campbell WB, Jennings L, Willerson JT, Schmitz JM. Reduction in the rate of early restenosis after coronary angioplasty by a diet supplemented with n-3 fatty acids. New Engl J Med. 1988;319:733–740. doi: 10.1056/NEJM198809223191201. [DOI] [PubMed] [Google Scholar]
- 49.Dessi M, Noce A, Bertucci P, Manca di Villahermosa S, Zenobi R, Castagnola V, Addessi E, Di Daniele N. Atherosclerosis, Dyslipidemia, and Inflammation: The Significant Role of Polyunsaturated Fatty Acids. ISRN Inflammat. 2013;2013:191823. doi: 10.1155/2013/191823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Funahashi H, Satake M, Hasan S, Sawai H, Newman RA, Reber HA, Hines OJ, Eibl G. Opposing effects of n-6 and n-3 polyunsaturated fatty acids on pancreatic cancer growth. Pancreas. 2008;36:353–362. doi: 10.1097/MPA.0b013e31815ccc44. [DOI] [PubMed] [Google Scholar]
- 51.Vanamala J, Glagolenko A, Yang P, Carroll RJ, Murphy ME, Newman RA, Ford JR, Braby LA, Chapkin RS, Turner ND, Lupton JR. Dietary fish oil and pectin enhance colonocyte apoptosis in part through suppression of PPARdelta/PGE2 and elevation of PGE3. Carcinogenesis. 2008;29:790–796. doi: 10.1093/carcin/bgm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rubin D, Laposata M. Cellular interactions between n-6 and n-3 fatty acids: a mass analysis of fatty acid elongation/desaturation, distribution among complex lipids, and conversion to eicosanoids. J Lipid Res. 1992;33:1431–1440. [PubMed] [Google Scholar]
- 53.Smith W, Murphy R. The eicosanoids: cyclooxygenase, lipoxygenase, and epoxygenase pathways. In: Vance D, Vance J, editors. Biochemistry of Lipids, Lipoproteins and Membranes. Elsevier; Amsterdam: 2002. pp. 341–372. [Google Scholar]
- 54.Smith WL. Nutritionally essential fatty acids and biologically indispensable cyclooxygenases. Trends Biochem Sci. 2008;33:27–37. doi: 10.1016/j.tibs.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 55.Mahadevappa VG, Holub BJ. Quantitative loss of individual eicosapentaenoyl-relative to arachidonoyl-containing phospholipids in thrombin-stimulated human-platelets. J Lipid Res. 1987;28:1275–1280. [PubMed] [Google Scholar]
- 56.Clark JD, Schievella AR, Nalefski EA, Lin LL. Cytosolic Phospholipase a(2) J Lipid Mediat Cell. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- 57.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 58.Laneuville O, Breuer DK, Xu N, Huang ZH, Gage DA, Watson JT, Lagarde M, DeWitt DL, Smith WL. Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2. Formation of 12-hydroxy-(9Z, 13E/Z, 15Z)- octadecatrienoic acids from alpha-linolenic acid. J Biol Chem. 1995;270:19330–19336. doi: 10.1074/jbc.270.33.19330. [DOI] [PubMed] [Google Scholar]
- 59.Liu W, Cao D, Oh SF, Serhan CN, Kulmacz RJ. Divergent cyclooxygenase responses to fatty acid structure and peroxide level in fish and mammalian prostaglandin H synthases. Faseb J. 2006;20:1097–1108. doi: 10.1096/fj.05-5273com. [DOI] [PubMed] [Google Scholar]
- 60.Tanikawa N, Ohmiya Y, Ohkubo H, Hashimoto K, Kangawa K, Kojima M, Ito S, Watanabe K. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem Biophys Res Commun. 2002;291:884–889. doi: 10.1006/bbrc.2002.6531. [DOI] [PubMed] [Google Scholar]
- 61.Wada M, Yokoyama C, Hatae T, Shimonishi M, Nakamura M, Imai Y, Ullrich V, Tanabe T. Purification and characterization of recombinant human prostacyclin synthase. J Biochem. 2004;135:455–463. doi: 10.1093/jb/mvh059. [DOI] [PubMed] [Google Scholar]
- 62.Hecker M, Ullrich V. On the mechanism of prostacyclin and thromboxane-A2 biosynthesis. J Biol Chem. 1989;264:141–150. [PubMed] [Google Scholar]
- 63.Kung-Chao DT, Tai HH. NAD+-dependent 15-hydroxyprostaglandin dehydrogenase from porcine kidney. I. Purification and partial characterization. Biochim Biophys Acta. 1980;614:1–13. doi: 10.1016/0005-2744(80)90161-8. [DOI] [PubMed] [Google Scholar]
- 64.Belury MA, Patrick KE, Locniskar M, Fischer SM. Eicosapentaenoic and arachidonic acid: comparison of metabolism and activity in murine epidermal cells. Lipids. 1989;24:423–429. doi: 10.1007/BF02535150. [DOI] [PubMed] [Google Scholar]
- 65.Norris PC, Dennis EA. Omega-3 fatty acids cause dramatic changes in TLR4 and purinergic eicosanoid signaling. Proc Natl Acad Sci USA. 2012;109:8517–8522. doi: 10.1073/pnas.1200189109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Obata T, Nagakura T, Masaki T, Maekawa K, Yamashita K. Eicosapentaenoic acid inhibits prostaglandin D-2 generation by inhibiting cyclo-oxygenase-2 in cultured human mast cells. Clin Exp Allergy. 1999;29:1129–1135. doi: 10.1046/j.1365-2222.1999.00604.x. [DOI] [PubMed] [Google Scholar]
- 67.Neilson AP, Djuric Z, Ren JW, Hong YH, Sen A, Lager C, Jiang Y, Reuven S, Smith WL, Brenner DE. Effect of cyclooxygenase genotype and dietary fish oil on colonic eicosanoids in mice. J Nutr Biochem. 2012;23:966–976. doi: 10.1016/j.jnutbio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pirman DA, Efuet E, Ding XP, Pan Y, Tan L, Fischer SM, DuBois RN, Yang P. Changes in cancer cell metabolism revealed by direct sample analysis with MALDI mass spectrometry. PLoS ONE. 2013;8:e61379. doi: 10.1371/journal.pone.0061379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kearns RJ, Hayek MG, Turek JJ, Meydani M, Burr JR, Greene RJ, Marshall CA, Adams SM, Borgert RC, Reinhart GA. Effect of age, breed and dietary omega-6 (n-6): omega-3 (n-3) fatty acid ratio on immune function, eicosanoid production, and lipid peroxidation in young and aged dogs. Vet Immunol Immunopathol. 1999;69:165–183. doi: 10.1016/s0165-2427(99)00052-5. [DOI] [PubMed] [Google Scholar]
- 70.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Clin Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- 72.Iyu D, Glenn JR, White AE, Johnson A, Heptinstall S, Fox SC. The role of prostanoid receptors in mediating the effects of PGE3 on human platelet function. Thromb Haemost. 2012;107:797–799. doi: 10.1160/TH11-11-0794. [DOI] [PubMed] [Google Scholar]
- 73.Bansal V, Syres KM, Makarenkova V, Brannon R, Matta B, Harbrecht BG, Ochoa JB. Interactions between fatty acids and arginine metabolism: implications for the design of immune-enhancing diets. J Parenter Enteral Nutr. 2005;29:S75. doi: 10.1177/01486071050290S1S75. [DOI] [PubMed] [Google Scholar]
- 74.Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 75.Hawcroft G, Volpato M, Marston G, Ingram N, Perry SL, Cockbain AJ, Race AD, Munarini A, Belluzzi A, Loadman PM, Coletta PL, Hull MA. The omega-3 polyunsaturated fatty acid eicosapentaenoic acid inhibits mouse MC-26 colorectal cancer cell liver metastasis via inhibition of PGE2-dependent cell motility. Br J Pharmacol. 2012;166:1724–1737. doi: 10.1111/j.1476-5381.2012.01882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Calviello G, Di Nicuolo F, Gragnoli S, Piccioni E, Serini S, Maggiano N, Tringali G, Navarra P, Ranelletti FO, Palozza P. N-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE2 induced ERK-1 and -2 and HIF-1alpha induction pathway. Carcinogenesis. 2004;25:2303–2310. doi: 10.1093/carcin/bgh265. [DOI] [PubMed] [Google Scholar]
- 77.Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 78.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 79.Xia SH, Wang JD, He CW, Hong S, Serhan CN, Kang JX. Melanoma growth is reduced in fat-1 transgenic mice: impact of omega-6/omega-3 essential fatty acids. Proc Natl Acad Sci USA. 2006;103:12499–12504. doi: 10.1073/pnas.0605394103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang P, Cartwright C, Chan D, Ding J, Felix E, Pan Y, Pang J, Rhea P, Block K, Fischer SM, Newman RA. Anticancer activity of fish oils against human lung cancer is associated with changes in formation of PGE(2) and PGE(3) and Alteration of Akt Phosphorylation. Mol Carcinogen. 2013 doi: 10.1002/mc.22008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tashjian AH, Jr, Voelkel EF, Robinson DR, Levine L. Dietary menhaden oil lowers plasma prostaglandins and calcium in mice bearing the prostaglandin-producing HSDM1 fibrosarcoma. J Clin Invest. 1984;74:2042–2048. doi: 10.1172/JCI111627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takahashi M, Fukutake M, Isoi T, Fukuda K, Sato H, Yazawa K, Sugimura T, Wakabayashi K. Suppression of azoxymethane-induced rat colon carcinoma development by a fish oil component, docosahexaenoic acid (DHA) Carcinogenesis. 1997;18:1337–1342. doi: 10.1093/carcin/18.7.1337. [DOI] [PubMed] [Google Scholar]
- 83.Bartram HP, Gostner A, Scheppach W, Reddy BS, Rao CV, Dusel G, Richter F, Richter A, Kasper H. Effects of fish oil on rectal cell proliferation, mucosal fatty acids, and prostaglandin E2 release in healthy subjects. Gastroenterology. 1993;105:1317–1322. doi: 10.1016/0016-5085(93)90135-y. [DOI] [PubMed] [Google Scholar]
- 84.Minoura T, Takata T, Sakaguchi M, Takada H, Yamamura M, Hioki K, Yamamoto M. Effect of dietary eicosapentaenoic acid on azoxymethane-induced colon carcinogenesis in rats. Cancer Res. 1988;48:4790–4794. [PubMed] [Google Scholar]
- 85.Karmali RA, Reichel P, Cohen LA, Terano T, Hirai A, Tamura Y, Yoshida S. The effects of dietary omega-3 fatty acids on the DU-145 transplantable human prostatic tumor. Anticancer Res. 1987;7:1173–1179. [PubMed] [Google Scholar]
- 86.Sasaki T, Kobayashi Y, Shimizu J, Wada M, In'nami S, Kanke Y, Takita T. Effects of dietary n-3-to-n-6 polyunsaturated fatty acid ratio on mammary carcinogenesis in rats. Nutr Cancer. 1998;30:137–143. doi: 10.1080/01635589809514653. [DOI] [PubMed] [Google Scholar]
- 87.de Bravo MG, de Antueno RJ, Toledo J, De Tomas ME, Mercuri OF, Quintans C. Effects of an eicosapentaenoic and docosahexaenoic acid concentrate on a human lung carcinoma grown in nude mice. Lipids. 1991;26:866–870. doi: 10.1007/BF02535969. [DOI] [PubMed] [Google Scholar]
- 88.Yang P, Chan D, Felix E, Madden T, Klein RD, Shureiqi I, Chen X, Dannenberg AJ, Newman RA. Determination of endogenous tissue inflammation profiles by LC/MS/MS: COX- and LOX-derived bioactive lipids. Prostaglandins Leukot Essent Fatty Acids. 2006;75:385–395. doi: 10.1016/j.plefa.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 89.Yang P, Felix E, Madden T, Fischer SM, Newman RA. Quantitative high-performance liquid chromatography/electrospray ionization tandem mass spectrometric analysis of 2- and 3-series prostaglandins in cultured tumor cells. Anal Biochem. 2002;308:168–177. doi: 10.1016/s0003-2697(02)00218-x. [DOI] [PubMed] [Google Scholar]
- 90.Zou Z, Bellenger S, Massey KA, Nicolaou A, Geissler A, Bidu C, Bonnotte B, Pierre AS, Minville-Walz M, Rialland M, Seubert J, Kang JX, Lagrost L, Narce M, Bellenger J. Inhibition of the HER2 pathway by n-3 polyunsaturated fatty acids prevents breast cancer in fat-1 transgenic mice. J Lipid Res. 2013;54:3453–3463. doi: 10.1194/jlr.M042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeng L, An S, Goetzl EJ. EP4/EP2 receptor-specific prostaglandin E2 regulation of interleukin-6 generation by human HSB.2 early T cells. J Pharmacol Exp Ther. 1998;286:1420–1426. [PubMed] [Google Scholar]
- 92.Hagi A, Nakayama M, Miura Y, Yagasaki K. Effects of a fish oil-based emulsion on rat hepatoma cell invasion in culture. Nutrition. 2007;23:871–877. doi: 10.1016/j.nut.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 93.Szymczak M, Murray M, Petrovic N. Modulation of angiogenesis by omega-3 polyunsaturated fatty acids is mediated by cyclooxygenases. Blood. 2008;111:3514–3521. doi: 10.1182/blood-2007-08-109934. [DOI] [PubMed] [Google Scholar]
- 94.Denkins Y, Kempf D, Ferniz M, Nileshwar S, Marchetti D. Role of omega-3 polyunsaturated fatty acids on cyclooxygenase-2 metabolism in brain-metastatic melanoma. J Lipid Res. 2005;46:1278–1284. doi: 10.1194/jlr.M400474-JLR200. [DOI] [PubMed] [Google Scholar]
- 95.Bartoli GM, Palozza P, Marra G, Armelao F, Franceschelli P, Luberto C, Sgarlata E, Piccioni E, Anti M. N-3 PUFA and alpha-tocopherol control of tumor cell proliferation. Mol Aspects Med. 1993;14:247–252. doi: 10.1016/0098-2997(93)90011-2. [DOI] [PubMed] [Google Scholar]
- 96.Gallai V, Sarchielli P, Trequattrini A, Franceschini M, Floridi A, Firenze C, Alberti A, Di Benedetto D, Stragliotto E. Cytokine secretion and eicosanoid production in the peripheral blood mononuclear cells of MS patients undergoing dietary supplementation with n-3 polyunsaturated fatty acids. J Neuroimmunol. 1995;56:143–153. doi: 10.1016/0165-5728(94)00140-j. [DOI] [PubMed] [Google Scholar]
- 97.Swails WS, Kenler AS, Driscoll DF, DeMichele SJ, Babineau TJ, Utsunamiya T, Chavali S, Forse RA, Bistrian BR. Effect of a fish oil structured lipid-based diet on prostaglandin release from mononuclear cells in cancer patients after surgery. JPEN J Parenter Enteral Nutr. 1997;21:266–274. doi: 10.1177/0148607197021005266. [DOI] [PubMed] [Google Scholar]
- 98.Faber J, Berkhout M, Fiedler U, Avlar M, Witteman BJ, Vos AP, Henke M, Garssen J, van Helvoort A, Otten MH, Arends J. Rapid EPA and DHA incorporation and reduced PGE2 levels after one week intervention with a medical food in cancer patients receiving radiotherapy, a randomized trial. Clin Nutr. 2013;32:338–345. doi: 10.1016/j.clnu.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 99.Warstedt K, Furuhjelm C, Duchen K, Falth-Magnusson K, Fageras M. The effects of omega-3 fatty acid supplementation in pregnancy on maternal eicosanoid, cytokine, and chemokine secretion. Pediatr Res. 2009;66:212–217. doi: 10.1203/PDR.0b013e3181aabd1c. [DOI] [PubMed] [Google Scholar]
- 100.Trebble TM, Wootton SA, Miles EA, Mullee M, Arden NK, Ballinger AB, Stroud MA, Burdge GC, Calder PC. Prostaglandin E2 production and T cell function after fish-oil supplementation: response to antioxidant cosupplementation. Am J Clin Nutr. 2003;78:376–382. doi: 10.1093/ajcn/78.3.376. [DOI] [PubMed] [Google Scholar]
- 101.Dawczynski C, Massey KA, Ness C, Kiehntopf M, Stepanow S, Platzer M, Grun M, Nicolaou A, Jahreis G. Randomized placebo-controlled intervention with n-3 LC-PUFA-supplemented yoghurt: effects on circulating eicosanoids and cardiovascular risk factors. Clin Nutr. 2013;32:686–696. doi: 10.1016/j.clnu.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 102.Rees D, Miles EA, Banerjee T, Wells SJ, Roynette CE, Wahle KW, Calder PC. Dose-related effects of eicosapentaenoic acid on innate immune function in healthy humans: a comparison of young and older men. Am J Clin Nutr. 2006;83:331–342. doi: 10.1093/ajcn/83.2.331. [DOI] [PubMed] [Google Scholar]
- 103.La Guardia M, Giammanco S, Di Majo D, Tabacchi G, Tripoli E, Giammanco M. Omega 3 fatty acids: biological activity and effects on human health. Panminerva Med. 2005;47:245–257. [PubMed] [Google Scholar]
- 104.Kobayashi N, Barnard RJ, Henning SM, Elashoff D, Reddy ST, Cohen P, Leung P, Hong-Gonzalez J, Freedland SJ, Said J, Gui D, Seeram NP, Popoviciu LM, Bagga D, Heber D, Glaspy JA, Aronson WJ. Effect of altering dietary omega-6/omega-3 fatty acid ratios on prostate cancer membrane composition, cyclooxygenase-2, and prostaglandin E2. Clin Cancer Res. 2006;12:4662–4670. doi: 10.1158/1078-0432.CCR-06-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Petrik MB, McEntee MF, Chiu CH, Whelan J. Antagonism of arachidonic acid is linked to the antitumorigenic effect of dietary eicosapentaenoic acid in Apc (Min/+) mice. J Nutr. 2000;130:1153–1158. doi: 10.1093/jn/130.5.1153. [DOI] [PubMed] [Google Scholar]
- 106.Bose M, Hao X, Ju J, Husain A, Park S, Lambert JD, Yang CS. Inhibition of tumorigenesis in ApcMin/+ mice by a combination of (−)-epigallocatechin-3-gallate and fish oil. J Agric Food Chem. 2007;55:7695–7700. doi: 10.1021/jf071004r. [DOI] [PubMed] [Google Scholar]
- 107.Hillier K, Jewell R, Dorrell L, Smith CL. Incorporation of fatty acids from fish oil and olive oil into colonic mucosal lipids and effects upon eicosanoid synthesis in inflammatory bowel disease. Gut. 1991;32:1151–1155. doi: 10.1136/gut.32.10.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murphey LJ, Williams MK, Sanchez SC, Byrne LM, Csiki I, Oates JA, Johnson DH, Morrow JD. Quantification of the major urinary metabolite of PGE2 by a liquid chromatographic/mass spectrometric assay: determination of cyclooxygenase-specific PGE2 synthesis in healthy humans and those with lung cancer. Anal Biochem. 2004;334:266–275. doi: 10.1016/j.ab.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 109.Ward PS, Fuller RW, Ritter JM, Cashman SJ, Rees AJ, Dollery CT. Excretion of metabolites of prostacyclin and thromboxane by rats with nephrotoxic nephritis: effects of interleukin-1. Br J Pharmacol. 1991;103:1663–1668. doi: 10.1111/j.1476-5381.1991.tb09844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kivits GA, Nugteren DH. The urinary excretion of prostaglandins E and their corresponding tetranor metabolites by rats fed a diet rich in eicosapentaenoate. Biochim Biophys Acta. 1988;958:289–299. doi: 10.1016/0005-2760(88)90187-7. [DOI] [PubMed] [Google Scholar]
- 111.Ferretti A, Judd JT, Taylor PR, Schatzkin A, Brown C. Modulating influence of dietary lipid intake on the prostaglandin system in adult men. Lipids. 1989;24:419–422. doi: 10.1007/BF02535149. [DOI] [PubMed] [Google Scholar]
- 112.Ferretti A, Judd JT, Ballard-Barbash R, Nair PP, Taylor PR, Clevidence BA. Effect of fish oil supplementation on the excretion of the major metabolite of prostaglandin E in healthy male subjects. Lipids. 1991;26:500–503. doi: 10.1007/BF02536593. [DOI] [PubMed] [Google Scholar]
- 113.Murff HJ, Shrubsole MJ, Cai Q, Smalley WE, Dai Q, Milne GL, Ness RM, Zheng W. Dietary intake of PUFAs and colorectal polyp risk. Am J Clin Nutr. 2012;95:703–712. doi: 10.3945/ajcn.111.024000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Young LR, Kurzer MS, Thomas W, Redmon JB, Raatz SK. Effect of dietary fat and omega-3 fatty acids on urinary eicosanoids and sex hormone concentrations in postmenopausal women: a randomized controlled feeding trial. Nutr Cancer. 2011;63:930–939. doi: 10.1080/01635581.2011.589957. [DOI] [PubMed] [Google Scholar]
- 115.Fischer S, von Schacky C, Schweer H. Prostaglandins E3 and F3 alpha are excreted in human urine after ingestion of n-3 polyunsaturated fatty acids. Biochim Biophys Acta. 1988;963:501–508. doi: 10.1016/0005-2760(88)90318-9. [DOI] [PubMed] [Google Scholar]
- 116.Diczfalusy U, Hamberg M. Identification of the major urinary metabolite of prostaglandin E3 in the rat. Biochim Biophys Acta. 1986;878:387–393. doi: 10.1016/0005-2760(86)90247-x. [DOI] [PubMed] [Google Scholar]
- 117.Kuklev DV, Hankin JA, Uhlson CL, Hong YH, Murphy RC, Smith WL. Major urinary metabolites of 6-keto-prostaglandin F2alpha in Mice. J Lipid Res. 2013;54:1906–1914. doi: 10.1194/jlr.M037192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yin H, Liu W, Goleniewska K, Porter NA, Morrow JD, Peebles RS., Jr Dietary supplementation of omega-3 fatty acid-containing fish oil suppresses F2-isoprostanes but enhances inflammatory cytokine response in a mouse model of ovalbumin-induced allergic lung inflammation. Free Radic Biol Med. 2009;47:622–628. doi: 10.1016/j.freeradbiomed.2009.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Monneret G, Cossette C, Gravel S, Rokach J, Powell WS. 15R-methyl-prostaglandin D-2 is a potent and selective CRTH2/DP2 receptor agonist in human eosinophils. J Pharmacol Exp Ther. 2003;304:349–355. doi: 10.1124/jpet.102.042937. [DOI] [PubMed] [Google Scholar]
- 120.Kobzar G, Mardla V, Jarving I, Samel N. Comparison of anti-aggregatory effects of PGI2, PGI3 and iloprost on human and rabbit platelets. Cell Physiol Biochem. 2001;11:279–284. doi: 10.1159/000047814. [DOI] [PubMed] [Google Scholar]
- 121.Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–153. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]