

Abstract
Retinoic acid (RA), the active metabolite of vitamin A, is an important endogenous signaling molecule regulating cell cycle and maintenance of epithelia. RA isomers are also used as drugs to treat various cancers and dermatological diseases. However, the therapeutic uses of RA isomers are limited due to side effects such as teratogenicity, and resistance to treatment emerging mainly from autoinduction of RA metabolism. To improve the therapeutic usefulness of retinoids, RA metabolism blocking agents (RAMBAs) have been developed. These inhibitors generally target the cytochrome P450 (CYP) enzymes because RA clearance is predominantly mediated by P450s. Since the initial identification of inhibitors of RA metabolism, CYP26 enzymes have been characterized as the main enzymes responsible for RA clearance. This makes CYP26 enzymes an attractive target for the development of novel therapeutics for cancer and dermatological conditions. The basic principle of development of CYP26 inhibitors is that endogenous RA concentrations will be increased in the presence of a CYP26 inhibitor, thus, potentiating the activity of endogenous RA in a cell-type specific manner. This will reduce side effects compared to administration of RA and allow for more targeted therapy. In clinical trials, inhibitors of RA metabolism have been effective in treatment of psoriasis and other dermatological conditions as well as in some cancers. However, no CYP26 inhibitor has yet been approved for clinical use. This review summarizes the history of development of RAMBAs, the clinical and preclinical studies with the various structural series and the available knowledge of structure activity relationships of CYP26 inhibitors.
Keywords: Retinoic acid, Cytochrome P450, CYP26, azole, cancer, psoriasis, metabolism, inhibition, RAMBA
1. General biology of retinoic acid
Retinoic acid (RA) is the active metabolite of vitamin A (retinol) and a critical signaling molecule during embryonic development and post-natal life in all chordates. Chemically, RA exists as at least five different isomers: all-trans-RA (ATRA), 9-cisRA, 13-cisRA, 11-cisRA and 9,13-dicisRA (Figure 1) [1]. Of these, endogenous ATRA, 13-cisRA, and 9,13-dicisRA are commonly found in biological matrices whereas 9-cisRA is undetectable in many tissues [2] and very low in human serum [1]. 11-cisRA is not detected in human serum or reported in animal tissues [1, 2]. However, 11-cis-retinal, the precursor of 11-cisRA is a necessary component for vision [3].
Figure 1.
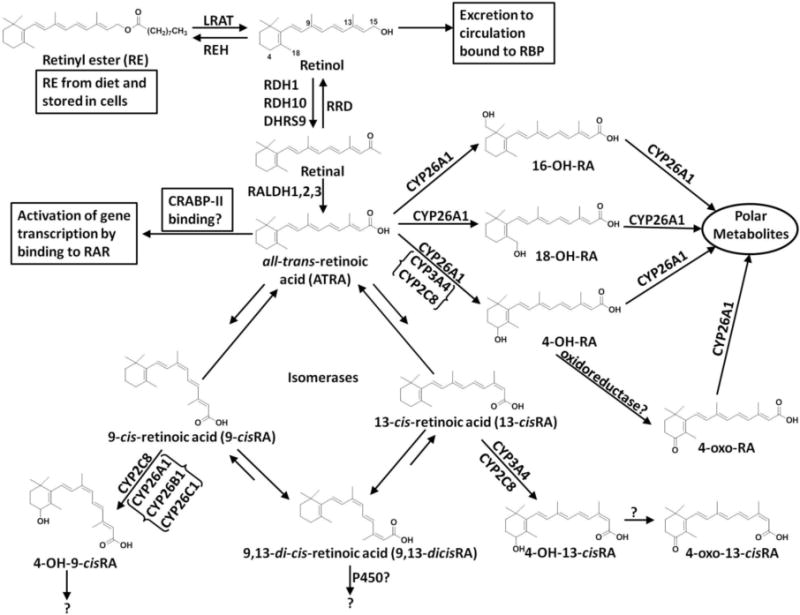
Metabolism of vitamin A. CYP26 isoforms appear to be the only cytochrome P450 dedicated to the metabolism of RA, as no other endogenous substrate has been identified. The metabolites 16-hydroxy-retinoic acid (16-OH-RA) and 18-hydroxy-retinoic acid (18-OH-RA) appear to be formed exclusively by CYP26. It has been proposed that CYP26 is the predominant P450 involved in ATRA metabolism, while CYP2C8 and CYP3A4 may play a bigger role in the metabolism of other RA isoforms.
Endogenous RA isomers are important in maintaining general health. They are involved in maintenance of healthy skin, epithelia and immune system [4, 5], in regulating insulin stimulated glucose secretion [6], in embryonic development [7], in stem cell differentiation [8], in neuronal differentiation [9] and in spermatogenesis [10]. RA isomers also regulate cell cycle and apoptosis [11]. In early studies, RA deficiency was shown to cause hyperkeratinization in rodents [12] and fetal malformations including blindness and lack of eyes in pigs [13]. Excessive intake of vitamin A on the other hand leads to a common syndrome known as hypervitaminosis A. Symptoms of hypervitaminosis A include erythema, weight loss, hair loss, changes to skin and mucus membranes, bone pain, and teratogenicity [14]. The exact role of each RA isomer in the above processes is not fully understood. Generally, ATRA is considered to be the biologically active isomer of RA, but 9-cisRA and 13-cisRA also appear to have biological activity and are marketed as drugs [15]. The biological importance of 9,13-dicisRA is unclear; it is inactive in cell culture [16, 17] and did not support growth in vitamin A deficient rats [18]. As a drug, ATRA is approved for use in acute promyelocytic leukemia (APL) [19] and is used off-label in other cancers. Isotrentinoin (13-cisRA) is approved for the treatment of acne, and high risk neuroblastoma in children [20]. Alitrentinoin (9-cisRA) is approved for use as a topical treatment for Kaposi’s sarcoma [15] and is used orally to treat chronic hand eczema unresponsive to other treatments [21]. The use and development of RA isomers and synthetic retinoids for treatment of cancer and metabolic disease has been recently reviewed by others [15]. While the exact biochemical mechanisms of how retinoids function in cancer and in dermatological diseases are not known, the therapeutic potential of retinoids is well established.
The biological activity of retinoids is largely mediated by their binding to the nuclear retinoic acid receptors (RARs) [11, 22, 23], but other mechanisms by which RA isomers cause changes in cell cycle and differentiation have also been shown [24, 25]. Binding of retinoids to RARs results in increased transcription of target genes [19] and, hence, the observed effects of RA on gene transcription are dependent on the cellular concentrations of ATRA as well as on the expression levels of RAR isoforms. There are three RAR isoforms, RARα, RARβ and RARγ, which each play different roles in the body. However, RA and its isomers bind to all three isoforms [26]. As such, specific agonists and antagonists of each RAR isoform have been developed and evaluated for various therapeutic indications [15]. Of the selective RAR agonists, many are in clinical development and Tazarotene and Adapalene have been approved for topical use. Interestingly, some RAR agonists also inhibit retinoic acid metabolism [27] and these compounds could be used to design more potent inhibitors of ATRA metabolism.
2. Regulation of retinoic acid homeostasis and importance of CYP26 enzymes
The concentrations of RA isomers in specific tissues and cells are collectively regulated by the dietary intake of RA precursors such as vitamin A, vitamin A palmitate and carotenoids, esterification of RA by LRAT (lecithin retinol acyltransferase) in the liver, synthesis of RA from retinol and retinal by retinol and retinal dehydrogenases (RDH and RALDH), and the metabolism of RA by P450 enzymes (Figure 1) [28–30]. Current literature shows that vitamin A intake and supplementation increase circulating RA concentrations [31–34]. Vitamin A supplements are generally in the form of retinyl esters, such as retinyl palmitate and retinyl acetate, which are subsequently converted to other retinoids (Figure 1). Daily supplements of 25–75,000 IU of retinyl palmitate increased the average plasma 9-cisRA (1.6-fold), 13-cisRA (2.5-fold) and ATRA (3-fold) in 41 volunteers [34]. In six healthy males, 0.46 mg/kg retinyl palmitate/day increased circulating ATRA and caused accumulation of 13-cisRA and 13cis-4oxoRA [31]. The dose of vitamin A correlates with the AUC of 13-cisRA, 13cis-4oxoRA, and retinyl palmitate [32], but the AUC of ATRA in plasma increases only up to the intake of 60,000 IU (18 mg) of vitamin A [32, 33], and then plateaus suggesting that either formation of ATRA from retinol and retinal is saturable at biological concentrations and/or the elimination of ATRA is rapidly induced. Whether inhibition of RA isomer clearance increases RA exposure after dietary vitamin A supplementation is not known and requires further study.
The clearance of ATRA appears to be mediated predominantly by cytochrome P450 family 26 enzymes (CYP26) in all chordates, and the role of CYP26 enzymes in RA clearance has been reviewed recently [35–37]. The quantitative importance of CYP26 enzymes in the clearance of 9-cisRA and 13-cisRA or the 4-oxo-RA metabolites is not known. All RA isomers are metabolized by CYP3A4 and CYP2C8, and these cytochromes P450 likely play a role in the clearance of exogenously administered ATRA and 13-cisRA [38–40]. While 9-cisRA has been shown to be a substrate of CYP26A1, 13-cisRA is a poor substrate of recombinant CYP26A1 and CYP26B1 [27], and the clearance pathways of ATRA and 13-cisRA appear to be different in humans. When ATRA is administered to humans, it induces its own metabolism likely via CYP26, which is believed to lead to resistance to ATRA treatment [41, 42]. RA resistance could be a result of either local induction of CYP26 in cancer cells or induction of the systemic clearance of ATRA. In rats, ATRA shows concentration-dependent nonlinear kinetics most likely due to saturation of CYP26A1 [43]. In contrast, 13-cisRA clearance is linear and not subject to autoinduction when it is administered to humans [36] despite the fact that 13-cisRA also induces the expression of CYP26A1 in HepG2 cells. The clearance of ATRA in vivo is 10–fold greater than that of 13-cisRA [36] despite their similar intrinsic clearances by CYP3A4 and CYP2C8. As such, CYP26 enzymes appear to be important in the clearance of ATRA but not 13-cisRA. Finally, based on in vitro data and in vitro-to-in vivo extrapolation, at biologically relevant concentrations, CYP26A1 is predicted to be responsible for the majority of ATRA clearance in the liver [44].
The CYP26 family has three isoforms: CYP26A1, CYP26B1 and CYP26C1 [45, 46], but the specific roles of each isoform during childhood and adult life are not well understood. There is only 40–50% sequence similarity between the three isoforms [47, 48], which suggests the isoforms have distinct roles. Both CYP26A1 and CYP26B1 are efficient ATRA hydroxylases [48, 49], whereas CYP26C1 appears to prefer 9-cisRA as a substrate [47]. During mouse embryo development, the expression of CYP26 isoforms is distinct in a spatio-temporal manner [50, 51]. Both Cyp26a1−/− and Cyp26b1−/− mice die during gestation or at birth and have severe, but specific, malformations [51–54]. Cyp26c1−/− mice, on the other hand, are viable and do not have malformations [53]. In adult rodents, CYP26 expression has been detected in lungs and liver and the expression levels of CYP26A1 and CYP26B1 were shown to correlate with dietary intake of vitamin A [35, 55, 56]. CYP26 enzymes also appear to be ubiquitously expressed in different rat tissues since ATRA metabolism was shown in microsomes from rat testes, kidney, and lung which do not have expression of other RA metabolizing cytochrome P450s [57]. However, the isoform specific tissue expression patterns have not been comprehensively characterized in rodents.
Based on single donor mRNA detection, CYP26B1 has fairly ubiquitous expression in adult human tissues [46]. It was found that CYP26A1 is present in the human liver, but CYP26B1 mRNA is low or undetected and CYP26B1 protein is undetectable [44, 58, 59]. In other adult human tissues, broad mRNA and protein expression of CYP26A1 and CYP26B1 was detected [48], but these studies were limited to single donors. This is a major limitation because the expression of CYP26A1 in the liver is subject to considerable inter-individual variability [58], and it is likely that CYP26A1 expression is variable in extrahepatic tissues as well. In a limited number of human fetal tissues of different gestational stages, CYP26A1 was expressed exclusively in the brain, whereas CYP26B1 was not present in the brain but found in all other tissues tested [59]. Based on studies in knock-out mice, CYP26 enzymes are believed to contribute to regulation of ATRA concentrations, homeostasis and signaling in specific cells and in circulation. However, the fact that the knock-out mice are not viable, together with the lack of potent selective inhibitors of CYP26A1 and CYP26B1 has limited the understanding of the importance of CYP26 enzymes in mediating RA isomer homeostasis.
Since CYP26A1 and CYP26B1 are expressed in a variety of extrahepatic tissues as well as in the liver, inhibition of these enzymes in a tissue specific fashion may have pharmacologically different consequences (Figure 2). Overall, it is not clear whether inhibition of CYP26A1 and/or CYP26B1 will increase circulating concentrations of ATRA and subsequently lead to higher tissue concentrations of ATRA. While inhibition of liver CYP26A1 as well as CYP3A4 and CYP2C8 is expected to decrease the systemic clearance of ATRA, the effect of hepatic inhibition of ATRA metabolism to tissue specific concentrations of ATRA has not been shown. Furthermore, the role of hepatic clearance to the regulation of ATRA concentrations in target tissues is unclear. A CYP26 inhibitor may, alternatively, only inhibit ATRA metabolism in target cells (Figure 2) affecting only local concentrations. Such tissue-specific inhibition of ATRA metabolism is predicted to result in lower side effects related to increased ATRA concentrations. However, taking advantage of tissue-specific inhibition of ATRA metabolism requires thorough characterization of the expression profiles of the individual CYP26 enzymes.
Figure 2.
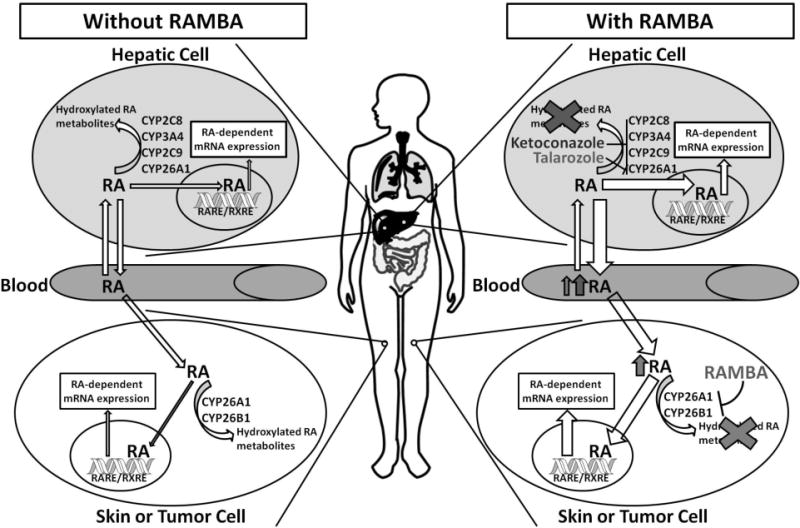
Effects of RAMBAs on in vivo concentrations on RA. Azole compounds such as ketoconazole inhibit a wide variety of cytochromes P450 in hepatic cells in addition to CYP26 in other cells. RAMBAs such as talarozole tend to be more CYP26-specific which results in less of an increase in circulating RA, but still leads to an increase in RA in CYP26-expressing cells. The increase in intracellular RA leads to expression of RA-regulated mRNA, including induction of CYP26A1.
The effect of decreased hepatic metabolism of RA isomers is expected to affect tissue RA isomer concentrations in a tissue and cell type specific manner. Based on uptake of administered radiolabeled RA, some tissues, such as the testes, pancreas and spleen, appear to possess a specific barrier for exposure to circulating ATRA, instead relying on synthesis of ATRA within the tissue [60–63]. Whether this functional barrier is due to RA metabolism or efflux transport is not known, but metabolism has been suggested to impair the access of RA to the testes [60]. Other tissues, such as the brain and the liver seem to be in equilibrium with blood, with 80–90% of the tissue RA pool being obtained from plasma [61]. While it is not known what role CYP26 isoforms play in regulating ATRA uptake to various tissues, it is likely that they contribute to regulating tissue- and cell-specific clearance of ATRA. Hence, inhibition of RA clearance via selective inhibition of CYP26 enzymes is expected to have tissue-specific effects both by changing the access of circulating ATRA to the target cells and by decreasing the clearance in the target organ.
The primary metabolite formed by CYP26A1 and CYP26B1 from ATRA is 4-OH-RA, but both enzymes also form other primary oxidation products including 18-OH-RA and 16-OH-RA and further metabolize these primary oxidation products to more polar secondary metabolites including diols and 4-oxo-alcohols (Figure 1) [27, 48]. While it is generally believed that the metabolites of ATRA do not play a role in regulating fetal development [64] and CYP26 enzymes function predominantly to eliminate ATRA, the possibility that ATRA metabolites play a role in retinoid signaling in some tissues cannot be completely dismissed. The oxidized metabolites of RA including 4-hydroxy-RA (4-OH-RA), 4oxo-RA, 18-OH-RA and RA-5,6-epoxide possess biological activity and inhibit cell proliferation in in vitro models [26, 65, 66]. As such, while inhibition of CYP26 enzymes is expected to result in increased concentrations of ATRA, the simultaneous decrease in ATRA metabolites could potentially decrease the net pharmacological effect of CYP26 inhibition.
RA clearance has also been suggested to be mediated by the expression of cellular retinoic acid binding proteins (CRABP-I and CRABP-II) that bind ATRA with high affinity (Kd = 0.06 nM for CRABP-I and Kd = 0.13 nM for CRABP-II [67]). CRABP-I also binds 13-cisRA but with lower affinity [68]. CRABP-I is expressed ubiquitously, whereas expression of CRABP-II is limited to skin, uterus, ovary, choroid plexus and embryonic tissues [69]. Based on current knowledge, CRABPs serve multiple functions in the cell in addition to stabilizing and solubilizing RA [69]. CRABP-II delivers ATRA to the nucleus and to RAR [67, 70], whereas CRABP-I appears to facilitate ATRA metabolism as demonstrated in rat testes microsomes [68] and in F9 teratocarcinoma cells [71]. It remains to be determined how expression of CRABPs affects overall RA homeostasis and the role of individual P450 enzymes in RA isomer clearance. However, it is possible that ATRA binding to CRABPs decreases the role of CYP3A4 and CYP2C8 in ATRA clearance.
3. Pharmacological effects of inhibitors of retinoic acid hydroxylation
As previously mentioned, retinoids are involved in a variety of cellular processes including cellular differentiation [22]. As such, pharmacological doses of RA isomers have been used to treat cancer [72, 73] and dermatological diseases such as acne and psoriasis [74, 75]. RA has been particularly successful in the treatment of acute promyelocytic leukemia (APL), but this success has been dampened by the development of retinoid resistance most likely due to an induction of RA metabolism [73, 76]. Furthermore, it has been shown that RA concentrations in prostate tumor tissues are lower than in healthy prostate tissue, possibly due to increased RA catabolism in these cells [77, 78]. Thus, an inhibitor of RA metabolism could be beneficial in the treatment of cancer either alone by increasing cellular ATRA concentrations or in combination with exogenous RA by combatting resistance related to induced ATRA metabolism.
Both oral and topical retinoids are highly effective in the treatment of acne, but are often used as a last resort due to safety concerns such as teratogenicity and depression with the oral formulate and inflammation and rashes with the topical formulate [74]. Teratogenicity is particularly a concern with some of the synthetic retinoids, which tend to have a long tissue half-life, e.g. 80–175 days for etretinate [79]. Thus a CYP26 inhibitor that has the potential to increase local concentrations of RA (Figure 2), rather than whole body concentrations, and has a shorter half-life than synthetic retinoids could result in effective treatment of dermatological diseases with an improved safety profile [76]. Based on this hypothesis, agents that inhibit RA clearance have been developed, collectively called RAMBAs (retinoic acid metabolism blocking agents). There have been various other reviews on RAMBAs that are excellent resources on the chemistry and synthesis of these inhibitors [80–82]. The remainder of this review will primarily focus on the pharmacological effects and clinical results of both older and more recently developed RAMBAs.
3.1 RAMBAs that have reached human studies
Ketoconazole and Liarozole
The imidazole derivative ketoconazole (Figure 3) is a broad-spectrum antifungal that was developed in the 1970s as an inhibitor of fungal cytochrome P450 enzymes [83]. After noting that some male patients developed gynecomastia while taking ketoconazole, the possibility that ketoconazole inhibits steroid synthesis in humans was investigated (Table 1). Following a high dose of ketoconazole (400 or 600 mg po), cortisol response to adrenocorticotropic hormone (ACTH) was found to be inhibited, leading to the hypothesis that ketoconazole may be useful as an inhibitor of steroid biosynthesis as a treatment for prostate cancer [84]. In subsequent clinical trials, ketoconazole (400 mg, tid) was found to inhibit both testicular and adrenal androgen biosynthesis, when used in the treatment of prostate cancer (Table 1) [85–87]. A compound similar to ketoconazole, liarozole (R75251, Liazal™), was developed by Janssen Research Foundation as an inhibitor of cytochrome P450 mediated androgen biosynthesis in testes and adrenals (Table 1) [88]. Liarozole (Figure 3) was shown to be twice as potent as ketoconazole at inhibiting testosterone biosynthesis in male rats and dogs [89] in addition to being well tolerated by humans in a small pilot study (Table 1) [89]. Subsequently, it was shown that liarozole suppressed tumor growth of androgen-dependent R3327 prostatic carcinoma in rats (Table 2) despite the fact that there was little change in testosterone levels [90]. This suggested that the inhibition of prostate tumor growth by liarozole was due to a mechanism independent of androgen biosynthesis. In a follow-up study in rats, liarozole was shown to inhibit tumor growth in androgen-dependent and -independent prostate carcinoma models (Table 2) [91], further supporting the hypothesis that the mechanism of action for liarozole, and possibly ketoconazole, was not solely due to inhibition of androgen biosynthesis.
Figure 3.
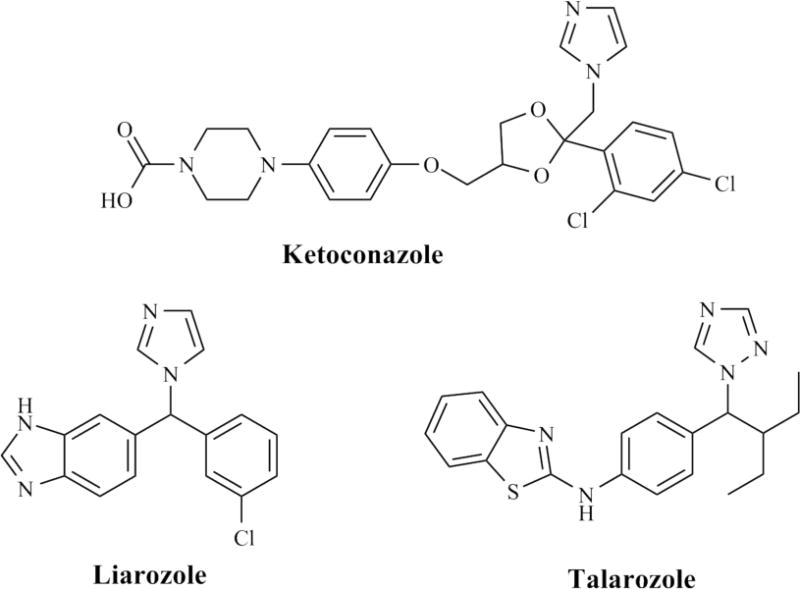
Structures of azole compounds that have been tested in humans for inhibition of ATRA metabolism.
Table 1.
Summary of clinical studies relating to inhibition of ATRA metabolism with ketoconazole, liarozole and talarozole. PSA, prostate specific antigen; PASI, Psoriasis Area and Severity index
| Ketoconazole (KTZ) | |||
|---|---|---|---|
| Dose | Population | Clinical Effects | Ref. |
| 400 mg or 600 mg po, single dose | Healthy men (n = 21) | Blunted cortisol response to ACTH infusion at 4 hrs up to 8 hrs; Returned to pre-treatment response after 16 hrs | [84] |
| 400 mg po, tid | Metastatic prostate cancer (n = 8) with previous orchiectomy (n = 10) | No complete remissions; Stabilization or partial response achieved in 60% of patients; Pain relief occurred in most patients | [86] |
| 400 mg po, tid | Previously untreated advanced prostate cancer (n = 12) | Significantly reduced testosterone levels which rebounded within 8 to 12 weeks; Most patients had decreased adrenal androgens and increased LH; 1 patient had partial response and 2 had improvement of symptoms | [87] |
| 200, 400, 800 or 1200 mg KTZ po 1 hr prior to ATRA on d. 2 and 29; 45 mg/m2 ATRA po, bid until disease progression | Early-stage or advanced non-small-cell lung cancer (n = 45) | Mean plasma AUC levels of RA were significantly reduced on d. 28 compared to d. 1; pretreatment with KTZ had no effect on plasma ATRA AUC on d. 2; 400 mg dose increased d. 29 AUC of RA by 115% | [121] |
| 400 mg KTZ po loading dose, then 200 mg po, tid with or without ATRA (45 mg/m2 po, bid) for 14 d. with crossover to ATRA alone | Advanced solid tumors (n = 13) | No objective tumor responses documented; 2 patients had stable disease and 8 had tumor progression; Cmax of KTZ >1000 ng/mL; d. 15 ATRA plasma AUC were significantly reduced compared to d. 1 with and without KTZ | [120] |
| 200 mg KTZ po on d. 1, 12 d. washout, 200 mg K + 30 mg alitretinoin po on d. 14, 30 mg A d. 15–26, KTZ + A on d. 27, A on d. 43–56 | Healthy men (n = 18) | KTZ significantly increased AUC (40%) and Cmax (60%) of alitretinoin; 4-oxometabolites not affected; Alitretinoin had no significant effect on AUC or Cmax of KTZ | [122] |
| Liarozole (LRZ) | |||
| Dose | Population | Clinical Effects | Ref. |
| 300 mg po, single dose | Healthy men (n = 12) | Significantly lowered plasma testosterone for 24 hr (<2 nmol/L in 3 volunteers); Significantly reduced estradiol and aldosterone; Significantly increased LH, FSH, progesterone, 17α-hydroxyprogesterone; Cmax = 4.92 μg/ml at 1 hr; t1/2 = 3 hr | [88] |
| 300 mg po, bid for 28 d. (0.75 mg dexamethasone po, bid for d. 14–28) | Metastatic prostate cancer (n = 6) | 2 patients had >50% decrease in PSA, 2 patients had no change in PSA; 1 patient had significant volume reduction of tumor and lymph nodes; Pain was reduced in 5 patients | [89] |
| 300 mg po, bid (n = 31); 150 mg po, bid (n = 11) | Metastatic prostate cancer in clinical relapse | Tumor volume reductions in 30% of patients; ≥50% reduction in PSA levels in 50% of patients; Pain relief in most patients; Elevated tissue RA levels | [95] |
| 150 mg po, bid for one month, then increased to 300 mg po, bid until disease progression or severe toxicity | Metastatic prostate cancer (n = 16) | 12 patients showed disease stabilization, median time to progression was 34 weeks; All patients with bone pain had pain relief in one month; 3 patients had >50% reduction in PSA and 4 patients had 25–50% reduction in PSA levels; 2 patients had reductions in size of metastatic lesions | [97] |
| 37.5 mg po, bid at the start with dose doubling until 300 mig po, bid was reached | Metastatic prostate cancer after androgen-ablative therapy (n = 38) | Dose-escalation study to produce grade 3 toxicity; no change in testosterone or cortisol levels; dose-related increase in 11-deoxycorticosterone; there was a negative correlation between dose of LRZ and PSA levels | [103] |
| 150 or 300 mg po, bid up to 10 months | Hormone-resistant prostate cancer (n = 55) | 7 out of 15 assessable patients had ≥50% decrease in prostate volume in 300 mg group; 41% had a response in PSA levels; >50% of patients had decreased pain | [102] |
| 300 mg LRZ po, bid compared to 100 mg cyproterone acetate (CPA) po, bid for 12–42 months | Metastatic prostate cancer in relapse (n = 321) | 26% lower risk of death in patients treated with LRZ compared to CPA; 20% of patients treated with LRZ had >50% reduction in PSA levels compared to 4% in CPA group; Pain improved more in LRZ group | [100] |
| Study 1 – 300 mg po, bid for 6 months; Study 2 – 150 mg po, bid and increased to 300 mg po, bid at week 4 | Prostate cancer; Study 1 – n = 31; Study 2 – n = 69 | 6 of 13 patients in Study 1 and 4 of 9 patients in Study 2 had reduced prostate size; 29% of patients had a partial or complete normalization of PSA levels; Near peak average plasma concentration was 4.67 μg/mL LRZ; Tmax for RA = 4 hrs; RA returned to pretreatment values by 8 hrs | [101] |
| 75, 150 or 300 mg LRZ po on d. 2 and 29 1 hr prior to ATRA; 45 mg/m2 ATRA po, bid, d. 3–27 and 45 mg/m2 ATRA po, qd on d. 1,2, 28, and 29; | Patients with solid tumors (n = 26) | LRZ had no effect on ATRA AUC on d. 2 but significantly increased AUC on d. 29; LRZ may be effective in attenuating retinoid resistance when using RA to treat cancer | [111] |
| 300 mg po, bid | Non-small cell lung cancer (n = 14) | Median survival for all patients was 3 months; the authors suggested LRZ as a single agent is ineffective for treating NSCLC | [105] |
| 150 mg po, bid for 2 weeks, then attempted dose escalation to 300 mg po, bid until disease progression | Advanced breast cancer (postmenopausal) (n = 29) | 4 patients responded to treatment and 7 patients had stabile disease; 11 patients had pain relief; estradiol and estrone levels decreased; trough drug levels ranged from 0.1 – 3.79 μg/mL at 1 month | [106] |
| 150 mg po, bid for 2 weeks, then attempted dose escalation to 300 mg po, bid until disease progression | ER negative (n = 16), ER positive, tamoxifen-refractory (n = 16), and chemotherapy-resistant (n = 27) breast cancer | 25% of patients in the first two groups were responders and 3 patients responded in the third group; Pain relief scores improved in 11 patients; Estradiol levels were suppresed; trough LRZ plasma levels ranged from 0 – 3 ng/mL at 1 month | [107] |
| 150 mg po, bid for 2 weeks, then attempted dose escalation to 300 mg po, bid until disease progression | Chemotherapy-resistant (n = 34) and potentially hormone-sensitive (n =37) metastatic breast cancer | 4 patients in chemotherapy-resistant group were partial responders; 2 complete responders and 6 partial responders in the potentially hormone sensitive group; Side effects were considered worse than hormonal agents | [108] |
| 75 or 150 mg po, bid for 12 weeks | Severe psoriasis (n = 31) | Decreased PASI score by 45% at week 4, 60% at week 8, and 77% at week 12; 9 patients dropped out because of insufficient response or adverse event | [125] |
| 75 mg LRZ po, bid or 25 mg acitretin po, qd for 24 weeks | Severe plaque psoriasis (n = 20) | Significant improvements in PASI score in both groups at 12 weeks; Reduction in inflammation and epidermal proliferation | [126] |
| 75 mg po, bid for at least 2 months | Extensive plaque psoriasis (n = 7) | 5 patients had improved PASI scores at 8 weeks; In responder patients, the number of actively cycling epidermal cells decreased to normal levels and the expression of differentiation markers decreased to levels of healthy volunteers; Markers of inflammation decreased with treatment | [127] |
| 50, 75, or 150 mg po for 12 weeks | Psoriasis (n = 139) | Only 150 mg group had response rate significantly different from placebo (PASI score changed from 15.8 to 8.8 at 12 weeks) | [123] |
| 75 mg LRZ po, bid (n = 7) or placebo (n = 8) for 12 weeks | Palmoplantar pustular psoriasis (n = 15) | 4 of 7 patients on LRZ were responders; Decreased number of pustules and significantly decreased fresh pustules; Decreased severity of disease | [124] |
| 150 mg po, bid for 12 weeks | Ichthyosis (n = 12) | Significantly reduced scaling and roughness, reduction in the extent and severity of skin lesions; Significant increase in keratin-4 after 12 weeks | [129] |
| 5% topical LRZ on one-half of body for 6 wks then both sides for 4 wks | Hereditary ichthyosis (n = 12) | Reduced scaling and roughness of LRZ-treated skin | [130] |
| 75–225 mg LRZ po, bid (n = 15) or 35–60 mg acitretin po (n = 17) for 12 weeks | Ichthyosis (n = 32) | 10 patients in LRZ group and 13 in acitretin group showed marked improvement; No differences in symptom scores for scaling, erythema or bullae between groups at end of treatment | [128] |
| 75 mg/d. or 150 mg/d. po LRZ or placebo | Lamellar ichthyosis vs healthy epidermis (n = 12) | Improvements in ichthyosis scores were seen at both doses; Reduction in thickness of stratum corneum and viable epidermis compared to baseline; CRABPII and KRT4 mRNA expression increased in 7 patients; KRT2 decreased in all patients; Increased CYP26A1 mRNA | [131] |
| 300 mg po, single dose | Healthy men (n = 12) | Significantly lowered plasma testosterone for 24 hr (<2 nmol/L in 3 volunteers); Significantly reduced estradiol and aldosterone; Significantly increased LH, FSH, progesterone, 17α-hydroxyprogesterone; Cmax = 4.92 μg/ml at 1 hr; t1/2 = 3 hr | [88] |
| 3.0% topical LRZ for 48 hrs | Healthy volunteers (n = 17) | 1.3 μg LRZ/g weight wet epidermis after 18 hr and 3 μg LRZ/g after 48 hr; 19 ng RA/g wet weight at 18 hr, and 6 ng RA/g at 48 hr (not detectable in control); 3% LRZ alone increased RA 4-hydroxylase activity with a synergistic increase when co-adminstered with 0.001% RA or 0.025% retinol | [181] |
| Talarozole (TLZ) | |||
| Dose | Population | Clinical Effects | Ref. |
| 0.5 to 4 mg po, bid for 8 d. | Healthy subjects | TLZ Tmax = 1 hr; t1/2 = 1.1 to 2.1 hrs; Css achieved with 24 hr of multiple dosing; Increased Cmax of RA on d. 1 and 8 at all TLZ doses compared to control | [141] |
| 0.07 or 0.35% topical for 9 d. | Healthy subjects (n = 15) | Elevated skin levels of CRABPII, cytokeratin K4, and CYP26A1 expression and reduced cytokeratin K2e and IL-1 expression; Epidermal thickness increased with 0.35% TLZ | [140] |
| 1 mg/d. po for 8 weeks (follow-up at 10 weeks) | Moderate to severe plaque psoriasis (n = 19) | TLZ Cp = 0.14 to 1.39 ng/ml at 2 to 4.5 hr post-dosing; >50% reduction in PASI score in 38% of patients at 8 weeks and 54% of patients at 2-week follow-up; ATRA Cp increased by 0.5 to 1.5 ng/ml from baseline 2 to 4.5 hr after dosing | [138] |
| 0.5, 1 or 2 mg/d. po for 12 weeks | Moderate to severe plaque psoriasis (n = 176) | 18.4%, 29.4%, and 37.1% of patients achieved 75% reduction in PASI score with 0.5, 1, and 2 mg doses, respectively | [141] |
| 1 mg/d. po for 12 weeks (follow-up at 16 weeks) | Moderate to severe facial acne (n = 17) | 31.5% reduction in total lesion count at 4 weeks, 76% reduction at 12 weeks, and 77.2% reduction at 16 weeks | [137] |
Table 2.
Effects of select RAMBAS on ATRA metabolism in animal models and pharmacological outcomes of animal studies
| Ketoconazole (KTZ) | |||
|---|---|---|---|
| Dose | Animal model | Pharmacological Effects | Ref. |
| 2.5 – 40 mg/kg KTZ po 1 hr prior to iv injection of 200 ng [3H]RA | Male and female Wistar rats | 40 mg/kg KTZ inhibited the formation of polar RA metabolites compared to vehicle treated control at all time points between 10 and 120 min; 2.5 – 10 mg/kg KTZ partially suppressed the formation polar RA metabolites; 20 mg/kg and 40 mg/kg suppressed RA metabolism by 75%; 1–2 hr pre-treatment had the most inhibitory effect with decreasing effect with increasing pre-treatment times | [182] |
| 40 mg/kg KTZ po 1 hr prior to iv injection of 0.1 mg/kg RA | Male Wistar rats | KTZ increased the plasma AUC and t1/2 of RA and decreased the clearance; KTZ increased endogenous plasma RA from non-detectable (<0.5 ng/mL) to 1.3 ng/mL at 2 hrs | [94] |
| 320 mg/kg KTZ po and 0.48 mg/hr•kg RA | Male Wistar rats | Increased RA Cmax and Tmax more than 1.3-fold and increased AUC more than 2.6-fold | [183] |
| Liarozole (LRZ) | |||
| Dose | Animal model | Pharmacological Effects | Ref. |
| 40, 80, 120, and 160 mg LRZ per 100 g food for 72 d. | Intact and castrated male Copenhagen × Fisher F1 rats bearing R3327G sc implanted tumor | Doses of 80 mg and above were as effective as castration at reducing tumor weight; testes, prostate, and seminal vesicles were less effected by LRZ than castration; increased LH; LRZ was effective despite testosterone supplementation | [94] |
| 5 and 10 mg LRZ per 100 g food for 28 d. | Androgen-independent MatLu prostatic adenocarcinoma injected sc in intact and castrated male rats | LRZ reduced tumor volume by 79% and 72% for 5 and 10 mg doses; castration had no effect | [90] |
| 20, 40 or 80 mg/kg po | Skin carcinogenesis induced by DMBA in female CD-1 mice | Reduced tumor incidence and tumor burden | [91] |
| 0.3, 1.25, 5, and 20 mg/kg po | Female Wistar rats | Increased endogenous plasma RA from undetectable (<0.5 ng/mL) to 1.4 and 1.9 ng/mL for 5 and 20 mg/kg doses | [91] |
| 20 mg/kg po | Ovariectomized female Wistar rats challenged with estradiol undecylate | LRZ treatment completely inhibited vaginal keratinization similar to RA (20 mg/kg); reduced 57–60 kDa keratin and increased 45–47 kDa keratin protein expression similar to RA (20 mg/kg) | [93] |
| 40 mg/kg LRZ po 1 hr prior to iv injection of 0.1 mg/kg RA | Male Wistar rats | LRZ increased the plasma AUC and t1/2 of RA and decreased clearance more than 2.5-fold; LRZ increased endogenous plasma RA from non-detectable (<0.5 ng/mL) to 2.5 ng/mL at 2 hrs with RA detectable at all time points between 0.5 and 6 hrs | [93] |
| 40 mg/kg po | Mice injected sc with PC-3 tumor cells | Prevented tumor incidence in all but two of the mice inoculated 5 or 10 d. prior; Reduced collagenase IV levels in PC-3 ML-B tumors | [184] |
| 40 mg/kg LRZ dosed 15 min prior to 10 mg/kg ATRA or 9-cisRA | Male Balb/c nude mice | LRZ significantly increased ATRA mean peak plasma levels, Tmax, and plasma AUC after first dose and to a lesser extent 2 d. later; LRZ had less of an effect on 9-cisRA, only increased AUC after second dose | [185] |
| 20 to 120 mg LRZ/100 g food | Male Copenhagen × Fisher F1 rats bearing Dunning androgen-dependent and -independent prostate tumors | Inhibited the growth of androgen-dependent (H and G sublines) and androgen-independent (PIF-1 and AT-6 sublines) tumors but not the androgen-independent AT-6 sq tumor | [186] |
| 40 mg/kg LRZ 1 hr prior to 20 μg 4-keto-atRA | Male Wistar rats | Pretreatment with LRZ inhibited the metabolism of 4-keto-RA; Almost doubled t1/2 | [113] |
| Decreasing dose of 80, 60, 30, 15, 7.5, and 3.75 mg/kg po, bid | Male Copenhagen × Fisher F1 rats bearing Dunning AT-6sq androgen-independent prostate tumor | Reduced mean tumor weight at all doses; significantly increased plasma RA concentrations; Dose-related RA accumulation of RA above 7.5 mg/kg | [187] |
| 20 mg/kg or 80 mg/kg LRZ with and without 100 μg/kg tamoxifen | Female Sprague-Dawley rats bearing MNU-induced mammary tumors | LRZ stopped tumor growth; When combined with tamoxifen, LRZ led to tumor shrinkage while inhibiting the uterotrophic effect of tamoxifen | [188] |
| Talarozole (TLZ) | |||
| Dose | Animal model | Pharmacological Effects | Ref. |
| 2.5 mg/kg po | Male and female Wistar rats | Mean RA levels in plasma, skin, fat, kidney, and spleen were significantly elevated 2- to 4-fold from 2 to 8 hrs; Liver RA levels were significantly elevated 1.4-fold at 6 hrs; RA levels returned to baseline in all tissues by 18 hrs; elevated liver CYP26A1 mRNA expression after 4 d. and induction was reduced when RAR antagonist AGN193109 was co-administered; Cp of TLZ ranged from 14.8 to 67.7 ng/mL over 8 hrs | [133] |
| 0.04 – 10 mg/kg TLZ or 0.6 –20 mg/kg RA for 3 d. | Estrogen-exposed ovariectomized female Wistar rats | TLZ inhibited vaginal keratinization with ED50 of 1 mg/kg; RA had ED50 of 5.1 mg/kg; co-adminstration of the RAR antagonist AGN193109 reduced keratinization | [133] |
| 2.5 mg/kg TLZ or 2.5 mg/kg RA for 14 d. | Female skh: HR1 hairless mice | TLZ and RA increased thickness of pinnal epidermis ~2-fold; TLZ and RA increased the percentage of tail orthokeratosis from 30.3% to 90% | [133] |
| Other RAMBAs in development | |||
| Dose | Animal model | Pharmacological Effects | Ref. |
| 0.033 μmol/kg/d. compound 1a, 6 d./week for 6 weeks | Ovariectomized nude mice bearing MCF-7 tumor xenografts | 85.2% reduction in the mean final tumor volume compared to vehicle-treated control mice | [142] |
| 0.033 μmol/kg/d. of compounds 1, 3, and 4 for 6 weeks | Male SCID mice bearing LNCaP tumor xenografts | Tumor growth with compound 1a was similar to vehicle-treated control; Compounds 3 and 4 reduced tumor growth by 44% and 47%, respectively; No effect on body weight | [157] |
| 16.5, 33 or 66 μmol/kg/d. compound 1a and 66 μmol/kg/d. compound 3 for 6 weeks | Ovariectomized female athymic nude mice bearing MCF-7 tumor xenografts | Compound 1a had a dose-dependent inhibition of tumor growth (66.5%, 79.4%, and 92.6%); Compound 3 had an effect similar to 20 mg/kg ATRA on growth inhibition; Compound 1a inhibited growth 120% when combined with 33 μmol/kg/d. ATRA | [159] |
| 5, 10, or 20 mg/kg/d. compound 1a po, 5 d./week for 8 weeks | Female Sprague-Dawley rats bearing MNU-induced mammary tumors | Compound 1a reduced mean tumor volume and increased tumor apoptosis in a dose-dependent manner; Increased plasma RA concentration in dose-dependent manner (from 0.71 ng/ml in control to 5.16 ng/ml with highest dose of compound 1a) | [160] |
| 2.5 or 5 mg/kg compound 1b or 5 mg/kg ATRA alone or with 50 mg/kg chloroquine sc, biw | Female SCID mice bearing SKBR-3 tumor xenografts | 5 mg/kg compound 1b alone or with chloroquine was more effective than ATRA or 2.5 mg/kg compound 1b at inhibiting tumor growth; compound 1b was more effective in combination with chloroquine (92.6% growth inhibition vs 81.4% alone) | [163] |
| 2.5 or 5 mg/kg sc biw or 10 or 20 mg/kg sc qd of compound 1b or ATRA with or without 50 mg/kg sc biw chloroquine | Female SCID mice | Two highest doses of compound 1b were toxic (75% and 100% mortality rates); Severity of alopecia and skin scaling increased with dose; PK data based on 20 mg/kg sc; Cmax = 41.38 μg/ml, tmax = 2 h, t1/2 = 6 h, AUC 83.78 h•μg/ml; Three polar metabolites were observed, with one being identified as compound 1a; AUC of all metabolites 89.24 h•μg/ml | [189] |
| 0.08 – 1.25 mg/kg R116010 or 1.25–5 mg/kg RA po, bid for 21 d. | Mice inoculated with oestrogen-independent TA3-Ha carcinoma cells | 0.16 mg/kg R116010, bid was lowest effective dose to inhibit tumor growth, with toxicity at 5 mg/kg, bid; 2.5 mg/kg RA, bid was lowest effective dose with toxicity at 5 mg/kg, bid | [190] |
| 10 mg/kg compound 9 with 5 mg/kg ATRA | Female CD-1 mice | Inhibitor increased the plasma AUC of ATRA by 5.9-fold and increased ATRA half-life | [170] |
Several results led to the hypothesis that liarozole was exhibiting its effects through inhibition of RA metabolism. Liarozole treatment resulted in an accumulation of retinoic acid (RA) in epithelial tissues [92], and treatment with RA inhibited tumor growth of androgen-dependent prostatic carcinoma in nude mice similar to liarozole [91]. When tested in vitro, RA but not liarozole induced plasminogen activator, a marker of differentiation in F9 mouse teratocarcinoma cells [91]. However, when cells were treated with both liarozole and RA, liarozole potentiated the effects of RA, resulting in a 2.2-fold greater induction of plasminogen activator than RA alone [91]. Liarozole was shown to inhibit ATRA metabolism (Table 3) and enhance the antiproliferative effects of ATRA and its catabolites, 4-oxo-ATRA and 5,6-epoxy-ATRA (Table 4), but not the stereoisomers 9-cisRA or 13-cisRA in MCF-7 cells [66]. The lack of effect on 9-cisRA and 13-cisRA activity is most likely because there is little to no metabolism of these isomers in MCF-7 cells. Therefore, liarozole would not be expected to affect their intracellular clearance [66].
Table 3.
In vitro data for inhibition of ATRA metabolism by select lead compounds that have been evaluated or developed as CYP26 inhibitors. The structures of compounds 1–13 are shown in figures 3, 4 and 5.
| Compound | ATRA Concentration | Enzyme system | IC50 | Ref |
|---|---|---|---|---|
| Ketoconazole | 50 nM [3H]ATRA for 4 hrs | F9 embryonal carcinoma cells | 42% inhibition of RA metablism at 1 μM | [191] |
| 100 nM 9-cisRA | Recombinant CYP26A1 | 550 nM | [27] | |
| 3 μM RA with 10 nM [11,12-3H]RA for 30 min | Rat liver microsomes and rat skin homogenates | 22 μM (liver) 86 μM (skin) |
[176] | |
| 80–800 μM of ATRA with 25 nCi of [15-14C]RA | Hamster liver microsomes | 750 nM | [182] | |
| Liarozole | 100 nM ATRA | MCF7 cells pretreated with ATRA | 1 μM | [66] |
| [11,12-3H (N)]ATRA for 90 min | Rat Dunning R3327G prostate tumor homogenates | 260 nM | [112] | |
| 100 nM [3H]ATRA for 90 min | MCF-7 cells pretreated with ATRA | 0.44 μM | [115] | |
| 20 nM [3H]ATRA | Human endothelial and HepG2 cells | 10 μM increased t1/2 of RA 3- to 12-fold | [192] | |
| 10 μM ATRA | Caco-2 cells pretreated with ATRA | 90% inhibition of RA metabolism at 50 μM | [193] | |
| 0.25 μM retinol | RA accumulation in Sertoli, peritubular, and myoid cells isolated from rat testes | 10 μM increased RA conc. 2.5-fold in Sertoli cells; no effect in peritubular or myoid cells | [194] | |
| 7.72 pmol [11,12-3H]ATRA | Hamster liver microsomes | 6 μM | [157] | |
| 100 nM [11,12-3H]ATRA for 9 hrs | MCF-7 cells pretreated with ATRA | 7 μM | [116,171,172] | |
| 3 μM ATRA | Rat liver microsomes | 4.2 μM | [155] | |
| 100 nM 9-cisRA | Recombinant CYP26A1 microsomes | 2 μM | [27] | |
| [11,12-3H]RA (100 μCi/ml) for 30 min | Recombinant CYP26A1 microsomes | 3 μM | [133] | |
| Talarazole | [11,12-3H]RA (100 μCi/ml) | Recombinant CYP26 microsomes | 4 nM | [133] |
| 100 nM 9-cisRA | Recombinant CYP26A1 | 5.1 nM | [27] | |
| R116010 | 100 nM 9-cisRA | Recombinant CYP26A1 | 4.3 nM | [27] |
| 0.1 μM [11, 12-3H]RA | T47D cells pretreated with 1μM ATRA for 16 hrs | 8.7 nM | [142] | |
| 10 nM ATRA with 0.1 μCi [11,12-3H]ATRA | Microsomes from MCF-7 cells pretreated with ATRA | 10 nM | [167,168] | |
| 1a | 7.7 pmol [11,12-3H]ATRA | Hamster liver microsomes | 2.3 nM | |
| 1 nM [11,12-3H]ATRA for 5 hrs | MCF-7 and T47D cells pretreated with 1 μM ATRA 12–15 hrs | 10.9 nM (MCF-7) 6.3 nM (T47D) |
[157] | |
| 20 μCi/ml [11,12-3H]ATRA | T47D microsomes pretreated with | 2.4 nM | ||
| for 30 min | ATRA | |||
| 0.1 or 0.8 μM [11,12-3H]ATRA | LNCaP microsomes pretreated with 1 μM ATRA for 24 hrs | 6.5 nM | [159] | |
| 1b | 7.7 pmol [11,12-3H]ATRA | Hamster liver microsomes | 9 pM | [157] |
| 1 nM [11,12-3H]ATRA for 5 hrs | MCF-7 and T47D cells pretreated with 1 μM ATRA 12–15 hrs | 200 nM (MCF-7) 215 nM (T47D) |
||
| 20 μCi/ml [11,12-3H]ATRA for 30 min | T47D microsomes pretreated with ATRA | 40 nM | ||
| 2 | 7.7 pmol [11,12-3H]ATRA | Hamster liver microsomes | 0.05 nM | [157] |
| 1 nM [11,12-3H]ATRA for 5 hrs | MCF-7 and T47D cells pretreated with 1 μM ATRA 12–15 hrs | 24.7 nM (MCF-7) 10 nM (T47D) |
||
| 20 μCi/ml [11,12-3H]ATRA for 30 min | T47D microsomes pretreated with ATRA | 5.2 nM | ||
| 0.1 or 0.8 μM [11,12-3H]ATRA | LNCaP microsomes pretreated with 1μM ATRA for 24 hrs | 90 nM | [159] | |
| 3 | 7.7 pmol [11,12-3H]ATRA | Hamster liver microsomes | 43.7 nM | |
| 1 nM [11,12-3H]ATRA for 5hrs | MCF-7 and T47D cells pretreated with 1 μM ATRA 12–15 hrs | 56 nM (MCF-7) 24 nM (T47D) |
[157] | |
| 0.1 or 0.8 μM [11,12-3H]ATRA | LNCaP microsomes pretreated with 1μM ATRA for 24 hrs | 62.5 nM | [159] | |
| 4 | 7.7 pmol [11,12-3H]ATRA | Hamster liver microsomes | 76.7 nM | [157] |
| 0.1 or 0.8 μM [11,12−3H]ATRA | LNCaP microsomes pretreated with 1μM ATRA for 24 hrs | 90 nM | [159] | |
| 5 | 40 nM [3H]ATRA for 30 min | Microsomes from T47D cells pretreated with ATRA | 20 nM | [169] |
| 6 | 100 nM ATRA with 0.1 μCi [11,12-3H]ATRA for 9 hrs | MCF-7 cells pretreated with ATRA | 5 μM | [172] |
| 7 | 100 nM ATRA with 0.1 μCi [11,12-3H]ATRA for 24 hrs | MCF-7 cells pretreated with ATRA | 900 nM | [172] |
| 8 | 10 nM [11,12-3H]ATRA for 9 hrs | MCF-7 microsomes pretreated with ATRA | 500 nM | [173] |
| Imidazole of compound 9 | 10 nM [11,12-3H]ATRA for 9 hrs | MCF-7 microsomes pretreated with ATRA | 3 nM | [167] |
| 9 | 100 nM [11,12-3H]ATRA for 9 hrs | MCF-7 microsomes pretreated with ATRA | 0.35 nM | [168] |
| 10 | 0.05 μCi [3H]ATRA for 3 hrs | HeLa cells pretreated with ATRA | 14 nM | [175] |
| 11 | 3 μM RA with 10 nM [11,12-3H]RA for 30 min | Rat liver microsomes and rat skin homogenates | 99 μM (liver) 212 μM (skin) |
[176] |
| 12 | 10 nM [11,12-3H]RA for 30 min | Rat liver microsomes and rat skin homogenates; fibroblast and HaCat cells pretreated with ATRA | 12.8 – 19 μM | [177] |
| 13 | 3 μM ATRA and 10 nM [11,12-3H]ATRA for 30 min | Rat liver microsomes | 400 nM | [116] |
| 100 nM ATRA and 0.1 μCi [11,12-3H]ATRA for 9 hrs | MCF-7 cells pretreated with ATRA | 5 μM |
Table 4.
In vitro data for inhibitors of cell proliferation by selected lead compounds in the development of CYP26 Inhibitors. (GI50 is the concentration at which growth is inhibited by 50%). The structures of compounds 1–13 are shown in figures 3, 4 and 5.
| Compound | ATRA Conc. | Cell Line | Effects on Cell Growth | Ref |
|---|---|---|---|---|
| Liarazole | 1 – 10 nM | MCF-7 | LRZ concentration-dependent growth inhibition up to >90% inhibition (0.01 – 10 μM liarozole) | [109] |
| Not supplemented | MCF-7 DU145 LNCaP | No cytostatic properties at 10 μM LRZ | [91] | |
| 10 pM – 1 μM | MCF-7 | Enhances antiproliferative effects of ATRA at 1 μM LRZ | [66, 195] | |
| 1 nM – 1 μM | C3H/10T1/2 | 10 μM LRZ shifted the RA concentration required to inhibit MCA-induced transformation from 1 μM to 1 nM | [110] | |
| R116010 | 10 pM – 10 μM | T47D | 10 nM R116010 decreased ATRA GI50 1.25-fold to 1.6 nM | [142] |
| 100 nM R116010 decreased ATRA GI50 2.6-fold to 0.77 nM | ||||
| 1 μM R116010 decreased ATRA GI50 3-fold to 0.6 nM | ||||
| 1a | Not supplemented | MCF-7 | GI50 = 0.49 μM | [157] |
| T47D | GI50 = 0.003 μM | |||
| MDA-MB-231 | No inhibition of growth up to 10 μM I | |||
| LNCaP | GI50 = 10 μM | |||
| PC-3 | GI50= 7.7 μM | |||
| 1a | Not supplemented | LNCaP | GI50 = 10 μM | [159, 161] |
| 2 | GI50 = 6 μM | |||
| 3 | GI50 = 4.5 to 5.5 μM | |||
| 4 | GI50 = 5 μM | |||
| 1a | 0.01 – 1 μM | LNCaP | 1 μM compound 1a decreased ATRA GI50 to 92 nM | [159] |
| 2 | 1 μM compound 2 decreased ATRA GI50 to 71 nM | |||
| 3 | 1 μM compound 3 decreased ATRA GI50 to 61 nM | |||
| 4 | 1 μM compound 4 decreased ATRA GI50 to 66 nM | |||
| 1b | Not supplemented | MCF-7 | GI50 = 0.57 μM | [189] |
| SKBR-3 | GI50 = 5.38 μM | |||
| MDA-MB-231 | GI50 = 7.37 μM | |||
| 5 | 0.001 – 1 μM | T47D | GI50 = 300 nM | [169] |
| AT6.1 | GI50 = 125 nM |
In a series of studies in normal rats and mice, liarozole and ketoconazole increased the half-life and AUC of exogenously administered RA and increased the concentration of endogenous RA in plasma (Table 2). This suggests that liarozole and ketoconazole inhibit a P450-mediated pathway of RA elimination [93, 94]. Indeed, liarozole and ketoconazole have been shown to inhibit ATRA oxidation in vitro in hamster liver microsomes with IC50 values of 0.2–86 μM (Table 3) [93]. Taken together, these studies served as the basis to redefine liarozole and ketoconazaole as inhibitors of P450-mediated metabolism of RA.
To test whether inhibition of P450s would have pharmacological effects similar to those of RA in vivo, estrogen-stimulated ovariectomized rats were dosed with liarozole (20 mg/kg) or RA (20 mg/kg) (Table 2) [93]. Liarozole and ATRA both prevented vaginal keratinization and altered the keratin protein expression profile of vaginal epithelium, a marker of cellular differentiation. The retinoid mimetic effects of liarozole were believed to be caused by inhibition of ATRA metabolism in the epithelial tissue, thus causing a local increase in ATRA concentrations. However, liarozole also appears to have systemic effects, as co-administration of liarozole and ATRA increased the plasma AUC of ATRA and 9-cisRA (Table 2). In follow-up animal studies, liarozole was effective in decreasing tumor growth in androgen-dependent and – independent prostate tumors and in mammary tumors in rats (Table 2).
In small pilot studies, liarozole was shown to be moderately effective in treating prostate cancer (Table 1) and more tolerable than high-doses of ketoconazole [89, 95–97]. While both ketoconazole and liarozole had side effects attributed to hypervitaminosis A (dry skin, pruritis, rash, itchy skin, hypertriglyceridemia, etc.), ketoconazole caused more GI discomfort and an elevation in liver enzymes. Retinoids were known to have antitumoral properties, but tended to be rapidly metabolized in vivo [98, 99]. Once it was determined that liarozole was primarily mediating its antitumoral effects through increased ATRA concentrations and there was no correlation between testosterone level and anticancer effects [97], a series of clinical trials were done with liarozole in patients with prostate cancer (Table 1). A complete or partial response to liarozole treatment (150–300 mg po, bid) was seen in 20–30% of patients with prostate cancer; responses included tumor volume reduction, decreased levels of prostate specific antigen (PSA), and increased time to disease progression [89, 95, 97, 100–103]. Additionally, almost all of the patients studied had a reduction in pain to the point where they no longer needed analgesics [89, 95, 97, 100–103]. Despite promising results seen in clinical trials, the FDA did not approve liarozole for treatment of prostate cancer in the US due to methodological problems with the clinical trials [104]. Since then, researchers have explored the possibility that liarozole could be used in the treatment of other types of cancer. However, liarozole was ineffective in non-small cell lung cancer [105] and was beneficial only in a small fraction of breast cancer patients (10–35% of patients were responsive, [106–108]) when administered as single agent (Table 1). Whether efficacy of liarozole would be improved with co-administration with ATRA is not known. The antiproliferative activity of ATRA on MCF-7 breast cancer cells was enhanced by co-administration of liarozole (Table 4), while liarozole had no effect on cell growth in the absence of ATRA treatment [109]. Liarozole was found to potentiate the inhibitory activity of ATRA on carcinogen-induced neoplastic transformation of C3H mouse 10T1/2 embryonal fibroblast cells, a model system used to study chemotherapeutics (Table 4) [110]. Hence, the pharmacological effects of liarozole seem to be a result higher intracellular concentration of ATRA, most likely due to inhibition of ATRA metabolism.
In clinical trials (Table 1), liarozole was shown to be effective in attenuating retinoic acid resistance [111], increasing ATRA AUC. Unfortunately, liarozole has not been pursued further as cotherapy with ATRA. In a similar manner, ketoconazole increased the AUC of ATRA and 9-cisRA in cancer patients and healthy men (Table 1). Overall, liarozole appears to be moderately successful in preventing the progression of prostate cancer and highly effective in reducing pain associated with prostate cancer (Table 1), as well as decreasing plasma PSA levels more than cyproterone acetate, an FDA-approved treatment for prostate cancer [100]. However, the effectiveness of liarozole might partially be due to inhibition of androgen biosynthesis, and it remains to be seen if more CYP26-selective RAMBAs will be effective in prostate cancer treatment.
Liarozole has been studied in several in vitro model systems as a P450 inhibitor to further elucidate its mechanism of action (Table 3). Liarozole inhibited the metabolism of RA in rat liver homogenates (IC50 = 0.14 μM) and Dunning R3327G prostate tumor homogenates (IC50 = 0.26 μM) [112]. Liarozole also inhibited the metabolism of 4-oxo-ATRA in hamster liver microsomes (IC50 of 1.3 μM) and prolonged the half-life of the retinoid in rats (Table 2) [113]. Subsequently, treatment of T47D and MCF-7 breast cancer cells with ATRA was shown to result in an induction of RA metabolism [114]. This induced activity was inhibited by liarozole (IC50 of 0.44 to 7 μM [115, 116]), but not by inhibitors or substrates of CYP1A, CYP2A, CYP3A, CYP2B or CYP2C [114] At the time, the target P450 of liarozole was not known but eventually CYP26 enzymes were found to be the cytochromes P450 responsible for the induced RA metabolism, based on mRNA induction in MCF-7 cells and lack of expression of other cytochromes P450 in MCF-7 cells [37, 46, 116–118].
Based on in vitro studies, ketoconazole appears to be a more potent inhibitor of CYP26A1 than liarozole (Table 3). In a study with recombinant CYP26A1, liarozole inhibited 9-cisRA metabolism with an IC50 value of 2.1 μM while the IC50 value for ketoconazole was 550 nM [27]. This IC50 value for ketoconazole is nearly identical to the IC50 value of 588 nM for inhibition of CYP26A1 and 592 nM for CYP26B1 by ketoconazole determined using radiolabeled ATRA as a substrate, and five-fold lower than the IC50 for liarozole in the same system [119]. However, liarozole appears to be superior to ketoconazole in vivo as many of the human studies listed in Table 1 resulted in elevated liver enzymes with ketoconazole treatment but not with liarozole. Furthermore, while ketoconazole seems to be effective at increasing the AUC of exogenously administered RA [120–122], it seems to be less effective than liarozole in the treatment of prostate cancer (Table 1).
As mentioned previously, liarozole inhibited RA metabolism in vivo in animal models, resulting in inhibition of vaginal keratinzation and an increase in plasma RA levels to those seen with exogenous RA administration [93]. It has also long been known that retinoids are effective in treating a variety of dermatological diseases such as acne vulgaris and psoriasis [75], so it was logical to assess the ability of liarozole to treat skin conditions such as psoriasis and ichtyosis (Table 1). In several clinical trials of patients with psoriasis, liarozole (50 mg to 150 mg po, bid) improved the symptoms of psoriasis in more than 50% of treated patients as determined by their Psoriasis Area and Severity Index (PASI) score (Table 1) and side effects were generally mild and similar to those of hypervitaminosis A [123–127]. Liarozole was as effective as acitretin in treating ichthyosis [128] and reduced scaling and roughness in most patients [129–131]. Based on the results of the clinical trials with liarozole, RAMBAs are worth pursuing in the treatment of dermatological diseases [132]. Interestingly, in contrast to cancer treatment, in skin diseases RAMBAs such as liarozole appear to be effective as single agents without cotreatment with ATRA.
3.2 Talarozole
Both ketoconazole and liarozole are potent inhibitors of the 17α-hydroxylase/17,20-lyase (CYP17) as well as other hepatic cytochromes P450 (Figure 2). This is a potentially undesirable trait for a RAMBA resulting in adverse effects. To avoid off-target P450 inhibition, the more CYP26-specific inhibitor talarozole (R115866, Rambazole™) [132] was developed. The chemical structure for talarozole contains a triazole, a benzothiazole, and an aniline rather than an imidazole, a benzoimidazole, and a chlorobenzene like liarozole (Figure 3). Talarozole is a significantly more potent CYP26A1 inhibitor (IC50 of 4–5 nM) than liarozole (IC50 of 2–3 μM) when tested using recombinant human CYP26A1 (Table 3) [27, 133]. In addition, talarozole showed superior specificity for CYP26 as compared to liarozole with greater than 300-fold higher IC50 values for CYP19, CYP17, CYP2C11, CYP3A, and CYP2A1 than for CYP26A1 [133]. In MCF-7 breast cancer cells in which CYP26A1 mRNA was induced by ATRA, talarozole had an IC50 of 5 nM against ATRA metabolism, but in rat liver microsomes talarozole inhibited ATRA metabolism with an IC50 of 9 μM [116]. The more than 1000-fold difference in the IC50 values was attributed to the fact that inhibition of RA metabolism in rat liver microsomes requires inhibition of CYP1A1/2, CYP2A6, and CYP3A4, whereas inhibition in the MCF-7 cells would be more specific to CYP26 [116]. Therefore, the differences in IC50 values between the liver microsomes and MCF-7 cells would give an indication of the selectivity of talarozole for CYP26A1 over other CYP isoforms.
Following in vitro characterization of talarozole, animal studies were conducted to test the effectiveness of talarozole in vivo (Table 2). After a single oral dose of talarozole, endogenous RA levels in plasma, skin, fat, kidney, spleen and liver were significantly increased (1.5- to 4-fold) in rats, with levels returning to baseline after 18 hrs [133]. Similar to treatment with RA alone, talarozole was able to inhibit vaginal keratinization, induce hyperplasia in ear epidermis, increase orthokeratosis in the tail, and induce hepatic CYP26 mRNA expression in rats [133]. Furthermore, co-administration of the RAR antagonist, AGN193109, significantly reduced the ability of talarozole to inhibit vaginal keratinization [133]. The data suggest that the pharmacological effects of talarozole are due to increased RA concentrations through the inhibition of CYP26A1.
CYP26A1 protein was shown to be constitutively expressed in vivo in basal epidermal keratinocytes in human skin, thus verifying the ability of skin to metabolize RA [134]. Based on this information, the effects of talarozole were studied in cultured human epidermal keratinocytes when co-administered with low concentrations of ATRA [135]. The aim was to determine whether talarozole could be useful in the treatment of dermatological conditions such as psoriasis, ichthyosis and acne. Talarozole alone had no effect on autocrine cultures of keratinocytes in the absence of ATRA. When administered with low doses of ATRA (10 nM), talarozole potentiated the effects of ATRA on epidermal growth and differentiation as evidenced by increased HB-EGF and involucrin mRNA expression and a reduction in keratin 10 mRNA and protein expression [135]. This most likely occurred through the sustained concentrations of ATRA due to talarozole preventing ATRA catabolism. In an organotypic epidermis cell culture model, ATRA (1 nM) altered the mRNA expression of biomarkers of keratinization (KRT2, KRT4) and CRABPII, but not CYP26A1 [136]. When talarozole (1 μM) was combined with low level RA (1 nM), KRT2, KRT4 and CRABPII were not further affected, but CYP26A1 expression was significantly induced [136]. The mRNA expression levels of CYP26B1, LRAT, RALDH2 and RDH16 were not significantly altered by 1 nM RA alone or in combination with talarozole [136]. The proposed reason for these differences is that the nuclear receptors for KRT2, KRT4 and CRABPII expression in previously vitamin A deficient cells are extremely sensitive to the addition of RA, whereas a more sustained 1 nM RA concentration is required to induce CYP26A1 mRNA expression and an even higher concentration of RA is required to alter the expression profile of other biomarkers. When cells were pretreated with radiolabeled retinol, followed by talarozole (1 μM), there was a six-fold increase in cell-associated [3H]-RA compared to treatment with retinol alone [136], further suggesting that talarozole has the ability to alter intracellular concentrations of endogenous RA through inhibition of RA metabolism. In the same study [136], treatment with liarozole (1 μM) resulted in only a two- to three-fold increase in [3H]-RA, which was suggested to be due to the weaker inhibition of CYP26A1 by liarozole.
Up to this point, the inhibitory activity of talarozole has been attributed to its potency toward CYP26A1, however, according to comparative mRNA expression data, CYP26B1 expression in the skin may be much higher than that of CYP26A1 [48]. While talarozole is a known potent inhibitor of recombinantly expressed CYP26A1 [133], this data suggests it may also be a potent inhibitor of CYP26B1. However, there is currently no published data on CYP26B1 inhibition by xenobiotics.
Following these in vitro and animal model assessments, several clinical trials were conducted for 8- to 12-weeks with 1 mg oral daily doses of talarozole for the treatment of psoriasis and acne vulgaris, respectively [137–139]. As summarized in Table 1, talarozole was efficacious in all studies with mainly mild to moderate adverse events. The plasma RA levels were only measured in the 8-week study. They were increased but within normal or slightly above normal biological range and returned to baseline levels once talarozole was cleared from the body [138]. A phase I clinical trial was conducted to examine the use of talarozole as a topical treatment [140]. Topical treatment with talarozole did not result in skin inflammation or epidermal thickness as is often seen with topical retinoid treatment. When mRNA expression levels were examined in treated skin, talarozole increased the expression of CRABP2, KRT4, CYP26A1 and CYP26B1 dose dependently [140]. When protein expression levels were examined using immunofluorescence analysis, KRT4 protein levels were increased consistent with the mRNA expression data [140]. However, using both immunofluorescence analysis and Western blot analysis, there was no significant change in CYP26A1 or CYP26B1 protein expression following topical treatment with talarozole [140]. Despite the potential for talarozole to be a new oral or topical treatment for psoriasis or acne, Barrier Therapeutics suspended their development of talarozole in 2008 as a cost-cutting measure [141].
Interestingly, while there were numerous studies evaluating the antitumor efficacy of ketoconazole and liarozole, the potential for talarozole as an anticancer agent is yet to be explored. The lack of data on the effect of talarozole on cell proliferation may be because of concurrent development of a similar compound R116010, which has been shown to have antitumor activity [142], and is discussed in detail in section 3.2.
Other azole antifungals
There are a variety of other azoles, such as fluconazole, itraconazole and voriconazole, on the market for the treatment of fungal infections. These compounds were designed to inhibit ergosterol biosynthesis through the inhibition of lanosterol 14α-demethylase, but have also been shown to inhibit RA metabolism as evidenced by case reports of hypervitaminosis A symptoms upon co-treatment with RA and these antimycotics [143–148]. Whether this is due to inhibition of CYP26A1 is not known. Fluconazole was shown to increase plasma AUC of exogenous ATRA administered to patients with APL [149]. In human liver microsomes, fluconazole was approximately half as potent as ketoconazole in inhibiting radiolabeled ATRA metabolism [149]. Both ketoconazole and fluconazole are inhibitors of CYP3A4 in vivo [150–153], but only ketoconazole inhibits CYP26A1 in vitro [27]. Since the contribution of CYP3A4 is predicted to be relevant in ATRA metabolism following exogenous administration of ATRA [44], fluconazole likely exerts its effect on exogenously administered ATRA metabolism mostly through inhibition of CYP3A4. Interestingly, a high dose of fluconazole (700 mg/kg) was shown to induce CYP26A1 and CYP26B1 mRNA expression in mouse embryos, probably through increased endogenous RA levels sufficient to activate RARs in the embryos [154]. While there have not been clinical trials with itraconazole or voriconazole as inhibitors of RA metabolism, there are several case reports of these compounds resulting in hypervitaminosis A symptoms, particularly in patients receiving exogenous RA [144–148]. Furthermore, there is in vitro evidence of these compounds inhibiting RA metabolism, although they are not particularly potent [27, 155]. Thus, while these compounds are capable of inhibiting RA metabolism, they have not been pursued for this purpose, most likely because of their lack of specificity and their weak inhibition of CYP26A1. However, the azole scaffold appears to be a good starting point for finding a more specific inhibitor of RA metabolism and has been further evaluated by a number of different groups.
3.2. Preclinical CYP26 inhibitors
There are number of RAMBAs developed by several academic groups as well as pharmaceutical companies that have been tested for their ability to inhibit ATRA metabolism in vitro (Table 3). Since recombinant CYP26 enzymes have not been widely available, rat and hamster liver microsomes and cell models have been commonly used to evaluate the potency of RAMBAs. In cell models, CYP26A1 mRNA has been shown to be inducible by ATRA treatment and this induction correlates with increased ATRA metabolism in the cells [116]. As such, inhibition of ATRA metabolism is usually tested following CYP26A1 induction by ATRA. Overall, ATRA-induced cells and microsomes from ATRA-induced cells have gained wide acceptance as testing systems for novel RAMBAs. Since the expression of other ATRA metabolizing P450 enzymes such as CYP3A4 and CYP2C8 is negligible or nonexistent in MCF-7 cells, it is likely that the induced ATRA metabolism in the cells is due to induction of CYP26 enzyme expression. However, it is possible that both CYP26A1 and CYP26B1 are significantly induced in these cells in response to ATRA treatment. Whether isoform selectivity of the inhibitors can be assessed in the induced cell systems is not well established. In contrast to the ATRA-induced cells, the enzymes in liver microsomes contributing to ATRA metabolism will be substrate concentration-dependent. At low (nM) concentration of RA, CYP26 enzymes are likely to predominate ATRA metabolism [44], but as these enzymes are saturated at micromolar substrate concentrations, the CYP3A and CYP2C enzymes are likely to be the major ATRA hydroxylases. Hence, the different IC50 values observed in the various systems for the same inhibitor-substrate pair can most likely be explained by the relative contributions of different enzymes to ATRA clearance.
For pharmacological endpoints, the new compounds developed as RAMBAs have most commonly been tested for inhibition of cell proliferation in various cancer cell lines with cotreatment with ATRA. Overall, most of the CYP26 inhibitors appear to be devoid of antiproliferative activity in the absence of ATRA. When ATRA is added to the cell culture, the predominant effect of RAMBAs is to cause a shift in the IC50 of ATRA thus potentiating its effects. Hence, it is unlikely that these RAMBAs will have considerable effect in cells that have little or low ATRA metabolizing activity since they will have negligible effects on intracellular ATRA concentration.
4-Imidazoyl-retinoic acid and its derivatives
ATRA is known to be a high affinity substrate of CYP26 enzymes, and it is metabolized by CYP26 enzymes and other P450s to 4-OH-RA (Figure 1). The hypothesized orientation of ATRA in the P450 active site with the C-4 close to the heme iron was derived from the oxidation of ATRA at the C-4 of the β-ionone ring. Based on this, a hydrogen atom at the C-4 position was replaced with a triazole or imidazole, and a series of triazole- and imidazole-containing inhibitors were developed (Figure 4 and Table 3) [81, 156, 157]. Two key modifications made in these compounds included: 1) addition of an imidazole or triazole moiety to the β-ionone ring at the 4-position and 2) the carboxylic acid was replaced or derivatized to an azole, methyl ester, or hydroxyphenyl amide (Figure 4). In hamster liver microsomes the methyl ester of 4-imidazoyl-retinoic acid (compound 1b in Figure 4, VN/12-1) was the most potent inhibitor of ATRA metabolism with an IC50 value of 9 pM (Table 3) while the 4-imidazoyl-retinoic acid (compound 1a in Figure 4, VN/14-1) was 250-fold less potent with an IC50 value of 2.3 nM [157]. This greater potency of the ester was surprising as the acid moiety of RA is generally believed to play an important role in binding to CYP26 and interact with positively charged arginines in the CYP26 active site [158]. The greater potency of the ester could potentially be due to inhibition of multiple RA metabolizing P450s in hamster liver microsomes and nonselective inhibition of other P450s instead of CYP26. To increase the assay specificity for CYP26 inhibition, MCF-7 and T47D cells were pretreated with ATRA to induce CYP26 expression and inhibition of ATRA metabolism was measured [157]. In contrast to the hamster liver microsomes, the 4-imidazoyl-retinoic acid (compound 1a) demonstrated the most potent inhibition of ATRA metabolism with IC50 values between 2.4 nM and 10.9 nM in ATRA-i6nduced MCF-7, T47D, and LNCaP cells (Table 3). Compound 1b showed greatly decreased potency (IC50 = 200 nM) when compared to hamster liver microsomes in the induced cells. This observation supports the hypothesis that the initial potency of the ester was due to inhibition of other P450 enzymes involved in ATRA metabolism and that the acid moiety improves CYP26 binding affinity [157]. 4-Imidazoyl-retinoic acid, was also effective in inhibiting the growth of MCF-7, T47D, LNCaP, and PC-3 cells but not that of MDA-MB-231 cells (Table 4) and the inhibitory potency varied greatly between the different cell lines [157].
Figure 4.
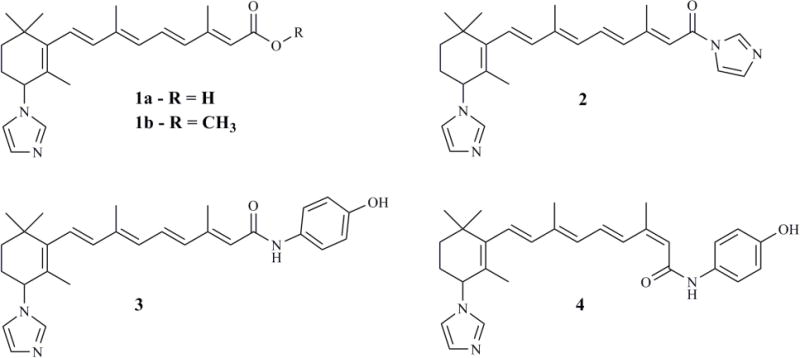
Structure of 4-imidazoyl retinoic acid (compound 1a) and its lead derivatives developed and tested as RAMBAs applicable for cancer treatment.
To follow up on compound 1a as a lead, the series of azoyl-retinoids was extended by synthesis of 4-imidazoyl-retinyl imidazole (compound 2, Figure 4, VN/50-1), 4′-hydroxyphenyl-4-imidazoyl-retinamide (compound 3, Figure 4, VN/66-1), and 4′-hydroxyphenyl-4-imidazoyl-13-cis-retinamide (compound 4, Figure 4, VN/69-1) [159, 160]. These compounds inhibited ATRA metabolism in microsomes prepared from CYP26-expressing LNCaP cells with IC50 values of 63–90 nM, demonstrating slightly lower potency than the lead 4-imidazoyl-retinoic acid as CYP26 inhibitors (Table 3). However, in the LNCaP cells, in the absence of ATRA, compounds 2, 3, and 4 inhibited cell proliferation slightly more potently (IC50 values of 4.5–6 μM) when compared to compound 1a (IC50 = 10 μM) (Table 4) [159, 160]. The potencies of these RAMBAs were similar to that of ATRA (IC50 = 4.3 μM) in inhibiting cell growth both in MCF-7 and LNCaP cells. In the MCF-7 cells, compound 2 was the most potent compound in inhibiting cell growth (IC50 = 126 nM); while in the T47D cells, compounds 1 and 2 were equipotent (3–9 nM IC50) [159, 160]. When the RAMBAs were combined with ATRA, a 47- to 70-fold enhancement in the potency of RA was observed (Table 4). The IC50 values of ATRA for inhibition of cell growth were between 61 and 92 nM in LNCaP cells [165], demonstrating the beneficial effect of inhibiting ATRA metabolism.
The anticancer activities of these RAMBAs cannot be explained completely by inhibition of ATRA metabolism and the subsequent increase in ATRA concentrations [161, 162]. These compounds are considered atypical RAMBAs because they inhibit growth in the absence of ATRA in T47D, MCF-7, and LNCaP cells (Table 4) and have effects similar to ATRA on cell cycle arrest. In T47D and LNCaP cells, these RAMBAs as well as ATRA caused a significant increase in the percentage of cells in the Sub-G1 phase and a decrease in the percentage of cells in the S phase, but did not affect the percentage of cells in the G0/G1 phase [159, 160]. However, the effect on the percentage of cells in the S-phase was greater in the T47D cells than in the LNCaP cells and the increase of cells in the G2/M phase was greater in LNCaP cells than in T47D cells [159, 160]. It has been shown that the 4-imidazoyl retinoic acid (compound 1a) binds RARs with IC50 values of 16–200 nM while compounds 2, 3 and 4 did not bind RARs [159, 160]. It is also possible that these RAMBAs have retinoid like metabolites that are responsible for some of their actions, but it is not known whether these RAMBAs are substrates of CYP26 or metabolically stable in the cell models.
As a result of the promising activity of these RAMBAs in the cell proliferation assays, they were also tested in various mouse xenograft models. Distinct model and compound specific effects were observed. In rats and mice with MNU-induced and MCF-7 xenograft mammary tumors, compound 1a decreased tumor size when administered on its own or in combination with ATRA (Table 2) [160, 163]. However, the effect of compound 1a co-administered with ATRA was greater than that observed with either ATRA or compound 1a alone (Table 2). Conversely, in mice with LNCaP prostate cancer xenograft tumors, ATRA efficiently decreased tumor size, while compound 1a was ineffective in decreasing tumor size [159]. Surprisingly, in the same model and study, compounds 3 and 4 decreased tumor size, demonstrating the fact that although these RAMBAs are part of the same series they have differences in their activity between models (Table 2). It is possible that the differences in the activity of compound 1a among tumor types is dependent on both tumor responsiveness to androgens, as seen in the androgen-sensitive breast and prostate tumors, as well as the histone acetylation state regulating transcription within the tumor [159, 164]. Compound 1a also inhibits CYP19 (aromatase) and CYP17 suggesting this compound can also affect steroid metabolism [82, 165, 166] and potentially explains its activity in the absence of RA. Because the MNU-induced mammary carcinomas are sensitive to androgens, it is possible that the antiproliferative effects are due to, at least in part, changes in androgen levels from the inhibition of CYP17 in addition to the changes in RA concentration that result from CYP26 inhibition.
In subsequent studies, these RAMBAs have been combined with histone deacetylase (HDAC) inhibitors and they have been shown to have a synergistic effect with HDAC inhibitors [161, 162]. In LNCaP prostate cancer cells, combination of SAHA (HDAC inhibitor) and compound 3 resulted in significant increase in the growth inhibitory action when compared to either compound alone [161, 162]. In the hormone insensitive PC-3 prostate cancer cell line, compound 3 had synergistic effects with another HDAC inhibitor, MS-275, both in in vitro cell systems as well as in the mouse xenograft model [161, 162]. The synergistic effects were observed through the induction of RARβ as well as in measurement of molecular markers of apoptosis. Interestingly, a serial treatment with the two agents was antagonistic [161, 162]. These results suggest that combination of RAMBAs with HDAC inhibitors in the clinic may be beneficial in cancer treatment.
Liarozole and talarozole derivatives
The chemical structure of R116010 (Figure 5) is highly similar to that of talarozole (Figure 3). The triazole moiety of talarozole was replaced with an imidazole and the pentyl was replaced with a dimethylethanamine to make R116010. These minor changes did not significantly affect CYP26A1 inhibitory potency (Table 3) [27]. Because of the extensive number of studies done on liarozole, the efficacy of R116010 is often compared to that of liarozole in in vitro studies [27, 142, 167, 168]. R116010 was approximately 100-fold more potent than liarozole in inhibiting cell growth in human T47D breast cancer cells pretreated with ATRA (Table 4). R116010 inhibited ATRA metabolism with an IC50 value of 8.4 nM in comparison to the IC50 of 1.4 μM for liarozole (Table 3) [142]. Co-incubation of R116010 (1 μM) with ATRA (0.1 μM) has also been shown to enhance the potency of ATRA in MCF-7 cells, as evidenced by a 4.7-fold greater induction of CYP26A1 mRNA expression in the presence of R116010 than with RA alone [167, 168]. In T47D microsomes with induced CYP26A1 expression, R116010 had an IC50 of 4 nM against ATRA metabolism (Table 3), whereas it showed little inhibition of CYP2C11, CYP2A1, CYP3A or CYP2B1/2 catalyzed reactions in rat liver microsomes or CYP19 in human placental microsomes [142]. While R116010 did slightly inhibit CYP17 in rat testicular S10 fraction, the IC50 (250 nM) was more than 60-fold greater than the IC50 for CYP26A1 [142], suggesting R116010 is a highly selective inhibitor of CYP26 catalyzed metabolism of ATRA. R116010 had no antiproliferative activity in the absence of ATRA, but did enhance the antiproliferative activity of ATRA in a dose-dependent manner in T47D cells (Table 4) [142]. The IC50 values of ATRA against cell proliferation decreased from 2 nM to 0.62 nM in the presence of 1 μM R116010 [142]. Lower concentrations of R116010 also decreased the IC50 of ATRA but to a lesser extent. In mice inoculated with TA3-Ha cells, R116010 significantly reduced tumor weight at a dose as low as 0.16 mg/kg po, bid. Toxicity in the form of hypervitaminosis A was not observed until a dose of 5 mg/kg po, bid (Table 2) [142]. In comparison, in mice treated with ATRA, the lowest active dose was 2.5 mg/kg po, bid and toxicity was seen at 5 mg/kg po, bid [142]. While there is little difference in the therapeutic and toxic doses of ATRA, the dose of R116010 causing hypervitaminosis A was about 20-fold higher than lowest therapeutic dose, demonstrating that R116010 had a much broader therapeutic window than ATRA.
Figure 5.
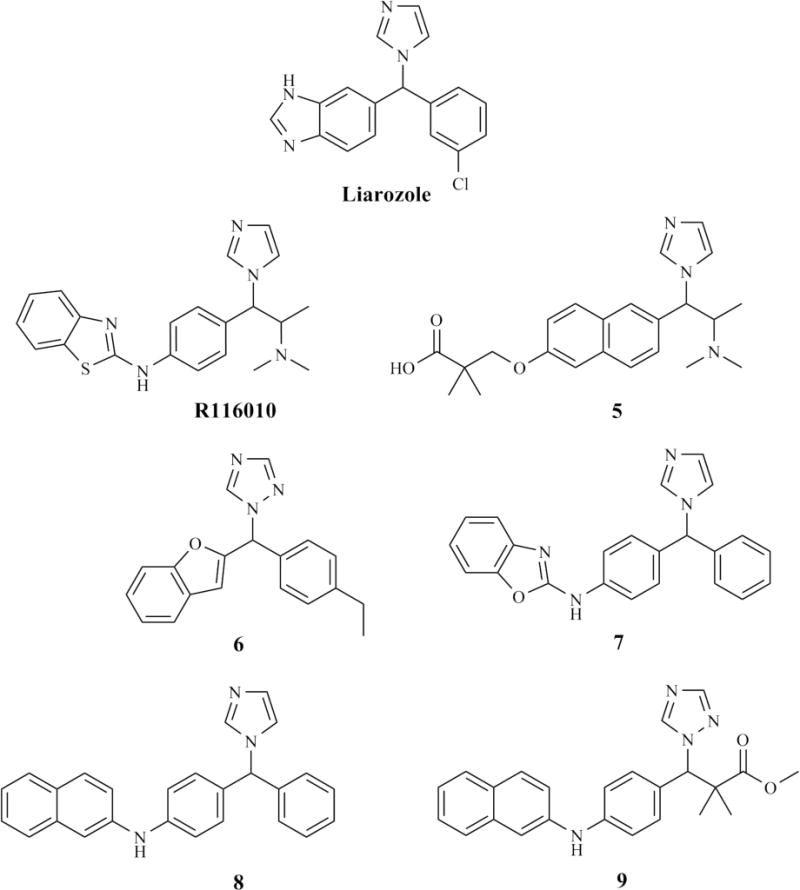
Structures of the lead azole inhibitors developed and evaluated as inhibitors of ATRA metabolism.
Using liarozole and R116010 as lead compounds, novel imidazole-containing naphthyl derivatives were synthesized by OSI Pharmaceuticals and found to be selective and potent inhibitors of ATRA metabolism [169]. In the first series, [2-imidazol-1-yl-2-(6-alkoxy-naphtalen-2-yl)-1-methyl-ethyl]-dimethyl-amines were synthesized with the lead inhibitor 3-[6-[2-dimethylamino-1-imidazol-1-yl-butyl)-naphthalen-2-yloxy]-2,2-methyl, 2-ethyl-propionic acid (compound 5, Figure 5) having a 20 nM IC50 for CYP26A1 (Table 3). This compound was relatively selective for CYP26A1 as its IC50 for CYP3A4 was 300-fold higher (6.3 μM), and the IC50 values for CYP1A2, CYP2D6 and CYP2C19 were >5 μM. Interestingly, the anti stereoisomer of compound 5 was an approximately 2-fold weaker inhibitor of CYP26 than the syn isomer but a more potent inhibitor of CYP3A4 than the anti isomer. When T47D breast cancer cells and AT6.1 adenocarcinoma cells were cotreated with ATRA and compound 5, compound 5 inhibited cell proliferation with IC50 values of 125 nM and 300 nM, respectively (Table 4) [169]. This in vitro data is promising because, following a 5 mg/kg oral dose to mice, the Cmax of compound 5 was 6 μM and the half-life was 2.5 hours, demonstrating that potentially efficacious concentrations could be achieved in vivo.
To follow-up these studies, further modifications of compound 5 were made and structure activity relationships were studied to develop a more potent and selective RAMBA [169, 170]. Four CYP26 inhibitors were identified in this series that had suitable potency, selectivity, and in vivo pharmacokinetic characteristics in mice. Interestingly, when the methyl group adjacent to the imidazole in compound 5 was changed to an ethyl, the inhibitory potency towards CYP26A1 in microsomes from ATRA-treated T47D breast cancer cells increased, yielding an IC50 value of 1.3 nM. The ethyl derivative was more than 1000-fold less potent as an inhibitor of CYP3A4 (IC50 of 3.3 μM) than CYP26A1 demonstrating high selectivity for CYP26 [169, 170]. Similar to compound 5, the ethyl derivative of compound 5 also significantly increased the plasma AUC of exogenously administered ATRA in mice, demonstrating the ability of RAMBAs to suppress CYP26-mediated metabolism of ATRA.
In an ongoing research effort to develop CYP26A1 inhibitors, the Simons laboratory at Cardiff University has evaluated a number of compounds with diverse structures as inhibitors of ATRA metabolism. They have also used homology models of CYP26A1, built using the homologous RA metabolizing P450s, CYP2C8, CYP2C9, and CYP3A4 [158], with molecular docking to improve the understanding of CYP26A1 active site features [116, 167, 168, 171]. Interestingly, only the homology model built using CYP3A4 as a template could accommodate talarozole binding to the active site [158]. Hence, this model was used for further evaluation of CYP26A1 structural features.
In a series of synthetic efforts, liarozole and talarozole structures were used as leads and new triazole containing benzofuranyl derivatives were synthesized [171]. The first set of compounds with the lead compound 6 (Figure 5) in this series had micromolar inhibitory potency towards CYP26A1 when tested in ATRA induced MCF-7 cells (Table 3) [171]. Upon further optimization, potency was improved by changing the benzofuran moiety to a naphthalene, benzoimidazole or benzooxazole and by adding a benzyl linker between the imidazole moiety and the benzofuran group (compounds 7 and 8, Figure 5) [172, 173]. N-{4-[1H-Imidazol-1-yl(phenyl)methyl]phenyl}-naphthalen-2-amine (compound 8) was identified as a lead compound (IC50 = 0.5 μM, in MCF-7 whole cells pretreated with ATRA) together with the benzoimidazole and benzooxazol (compound 7) analogs of this compounds (IC50 =1.5 and 0.9 μM in MCF-7 cells, respectively) (Figure 5 and Table 3) [173]. The binding interactions of these compounds with CYP26A1 were evaluated via docking to the CYP26A1 homology model, allowing identification of key hydrogen bonding interactions of the inhibitors with purported CYP26A1 active site residues. Interestingly, these three lead compounds were also tested in a neuroblastoma cell line for their ability to potentiate the ATRA-induced expression of CYP26B1 mRNA [173]. All three compounds potentiated the ability of ATRA to induce CYP26B1 mRNA suggesting that they may also inhibit CYP26B1.
To further improve these imidazole containing inhibitors, further modifications were made using compound 8 as a lead compound by varying the composition of the linker between the naphthyl and benzyl groups, and by substituting the phenyl ring with a flexible C3 chain such as propionic acid methyl ester (the imidazole analogue of compound 9) [167, 168]. While altering the composition of the linker group decreased the binding affinity to CYP26A1 (IC50 >1 μM), replacing the naphthyl with a phenyl generally preserved the nanomolar binding affinity of the inhibitors [167, 168]. Replacement of the benzyl moiety with a propanoate led to about 10-fold increased potency towards CYP26A1 (Table 3). The most promising inhibitor in this series was 3-imidazol-1-yl-2-methyl-3-[4-(naphthalene-2-ylamino)-phenyl]-propionic acid methyl ester (imidazole analog of compound 9). This compound was a micromolar inhibitor of CYP1A2, CYP2C9, and CYP2C19, but had an IC50 of 600 nM towards CYP2D6 and <100nM towards CYP3A4 demonstrating a lack of selectivity towards CYP26A1 [167, 168].
Further optimization of these compounds for increased CYP26A1 selectivity led to the replacement of the imidazole with a triazole (compound 9, Figure 5). Surprisingly, this resulted in a 10-fold increase in potency in inhibiting ATRA metabolism in MCF-7 cells (IC50 = 3 nM for the imidazole analog and 0.35 nM for the triazole compound 9) [168]. In addition, while the triazole containing inhibitor was more potent as a CYP26A1 inhibitor than the imidazole containing lead compound, it was significantly weaker inhibitor of CYP3A4 (IC50 of 330 nM), yielding a greatly increased selectivity. In addition, it was devoid of the nanomolar inhibition potency of CYP2D6 that was observed with the imidazole containing inhibitor (IC50 of 1.8 μM for compound 9) and was a micromolar inhibitor of CYP1A2, CYP2C9 and CYP2C19 [168]. As such, the selectivity of the triazole, compound 9, was similar to that observed with R116010. In the absence of ATRA these compounds had no effect on CYP26A1 expression in the SH-SY5Y cells, while in the presence of ATRA they significantly increased the magnitude of CYP26A1 mRNA induction caused by ATRA treatment [167, 168]. Therefore, all of these compounds are expected to exhibit RA-dependent actions in cell models. However, no data on their effect on cell proliferation or on keratinization in animals is currently available.
Nonazole inhibitors
Using a structural design similar to the known RAR agonist tazarotenic acid [174], novel inhibitors of ATRA metabolism were discovered in an effort by Allergan to pursue the strategy of retinoid receptor and function selectivity for various dermatological applications. Using HeLa cells stably transfected to express CYP26, a series of phenyl acetic acid derivatives were tested for inhibition of ATRA metabolism [175]. Interestingly, despite the structural similarity of these compounds to the RAR agonist tazarotenic acid, these compounds have been shown not to be RAR or RXR agonists or antagonists [175]. Overall, however, the size and composition of the molecules seems to mimic the size and shape of ATRA. The most potent inhibitor in the series for CYP26A1 in the transfected HeLa cells was compound 10 (Figure 6), 2-[4-(2-{8-cyclopropyl-4,4-dimethyl-3,4-dihydrospiro[1-benzopyran-2,1′-cyclopropane]-6-yl}ethynyl)-2-fluorophenyl]acetic acid, (IC50 = 14 nM, Table 3) which is comparable in potency to the azole-based compounds talarazole and R116010 [175]. Other compounds in the series had IC50 values between 22 and 1700 nM [82].
Figure 6.
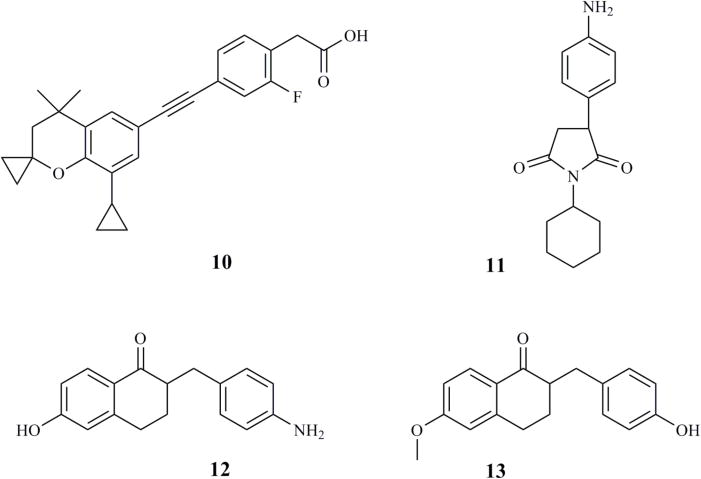
Strcutures of non-azole compounds developed as RAMBAs and tested as inhibitors of ATRA metabolism.
Other non-azole containing inhibitors have met with limited success. Compound 11 ((4-aminophenyl) pyrrolidine-2,5-dione, Figure 6) was shown to be a weaker inhibitor of RA metabolism than ketoconazole in rat liver microsomes and rat skin homogenates (Table 3) [176]. In an effort to increase potency, some 1,2-diphenylethane derivatives and a series of larger, tetralone containing compounds were synthesized (compounds 12 and 13, Figure 6) [176]. Compounds 12 and 13 were shown to inhibit the metabolism of ATRA in rat liver microsomes (Table 3) with a potency comparable to that of liarozole and ketoconazole [116, 177, 178]. However, only a subset of the synthesized compounds were able to effectively inhibit ATRA metabolism in fibroblasts, where CYP26 enzymes are presumably expressed [178]. In contrast, the 2-benzylidenetetralones were poor inhibitors of ATRA metabolism in rat liver microsomes, but inhibited ATRA metabolism in MCF-7 cells expressing ATRA-induced CYP26 with IC50 values between 7 and 100 μM [116]. This inhibition was comparable to ketoconazole and liarozole but much weaker than what was observed with talarozole (IC50 = 5 nM). The tetralones appeared to have a broader spectrum of P450 inhibition as they inhibited ATRA metabolism in rat liver microsomes with IC50 values of 0.5–18 μM while the IC50 values in MCF-7 cells were higher, 5–100 μM [116]. Despite the promising data, when the lead tetralones were tested with recombinant CYP26A1, they were weak inhibitors resulting in incomplete inhibition [27].
4. Concluding remarks and future directions
Since the discovery of the antiproliferative effects of ketoconazole and liarozole in the 1980s and the hypothesis that this was due to inhibition of ATRA metabolism, a plethora of different inhibitors of ATRA metabolism have been synthesized and tested preclinically. Some applications of these RAMBAs include their ability to increase the antiproliferative effects of ATRA, their ability to increase endogenous or therapeutically administered ATRA concentrations, and their potential benefits in dermatological diseases. It is noteworthy that the CYP26 family of enzymes was only identified and cloned in the late 1990s and, as such, the number of inhibitors developed against CYP26A1 is considerable. Despite the identification of many promising compounds from these studies, no RAMBA by design has been approved for use in humans and only talarozole has been evaluated in human clinical studies. Based on the available data on talarozole, ketoconazole, and liarozole, it appears that potent RAMBAs that inhibit CYP26 enzymes will increase both endogenous and exogenous ATRA concentrations, but the importance of selective CYP26 isoform inhibition is not known. As such, the exact molecular target of RAMBAs is still poorly characterized. Extensive clinical data is available on the effects of liarozole and ketoconazole on cancer progression, but it is not clear what therapeutic effects of liarozole can be attributed to CYP26A1 inhibition in comparison to CYP17 and CYP19 inhibition. Whether ketoconazole and liarozole inhibit CYP26B1 and CYP26C1 as well is not known and further research in this area is needed.
A considerable limitation for the development of novel RAMBAs that are selective inhibitors of CYP26A1 is the lack of structural information on the CYP26A1 active site. No high-resolution crystal structures or low resolution structural data is currently available for any of the CYP26 enzymes. In the absence of a crystal (or NMR) structure, three different homology models of CYP26A1 have been generated using various P450 enzymes as templates [158, 179, 180]. While talarozole has been docked into the active site of all of these models and ATRA orientation in the active site appears acceptable, the validity of the models is difficult to evaluate. All of the template P450 enzymes have low homology with CYP26A1 and as such do not provide an ideal template for generation of homology models. However, an evaluation of the various azole inhibitors of CYP26A1 (Figures 3 and 5) that have been developed by different groups shows a convergence to very similar compounds identified as lead inhibitors. This observation is in agreement with the notion that CYP26A1 has a very small active site that can accommodate ATRA and its primary metabolites but very few other compounds in the active site. As such, the chemical space for developing new RAMBAs may be limited.
In addition to the lack of structural data, a challenge to the development of CYP26 inhibitors is the lack of understanding of the importance of the individual CYP26 isoforms in various biological processes and the role of tissue specific metabolism and barriers in ATRA pharmacology. Hence, it is not clear whether broad-spectrum inhibition of all the ATRA metabolizing enzymes would be more beneficial in development of new cancer agents than a CYP26A1-selective inhibitor. In contrast, for some applications a selective CYP26B1 inhibitor may be beneficial. The lack of knowledge of the expression patterns and biological importance of CYP26A1 and CYP26B1 during adult life highlights the difficulty of interpreting the preclinical and clinical data of RAMBAs. Similarly, the expression of CYP26B1 in the MCF-7 and T47D cells among others is not known. As such, the interpretation of the inhibitory potency of the existing RAMBAs against CYP26A1 specifically needs to be done with caution.
The potential of RAMBAs in cancer treatment and in dermatological diseases is well acknowledged. However, as we begin to understand more about the importance of ATRA signaling in mediating glucose homeostasis and neurogeneration, new therapeutic areas in which RAMBAs may be useful are likely to emerge. The existing arsenal of potent and selective inhibitors of ATRA metabolism will provide a great starting point for determining whether inhibition of ATRA metabolism and/or CYP26 enzymes is an appropriate target in these diseases.
Acknowledgments
This study was supported in part by a grant from the US National Institutes of Health R01-GM081569. The authors wish to thank Dr Wendel Nelson for helpful conversations during the preparation of this review.
Nonstandard Abbreviations
- CYP
Cytochrome P450
- RA
retinoic acid
- ATRA
all trans retinoic acid
- APL
acute promyelocytic leukemia
- RAR
retinoic acid receptor
- RXR
retinoc × receptor
- LRAT
lecithin retinoic acyltransferase
- RDH
retinol dehydrogenase
- RALDH
retinal dehydrogenase
- IU
international units
- AUC
Area Under the Curve
- CRABP
cellular retinoic acid binding protein
- RAMBA
retinoc acid metabolism blocking agents
- IC50
half maximal inhibitory concentration
- PSA
prostate specific antigen
- PASI
Psoriasis Area and Severity Index
- HB-EGF
Heparin-binding EGF-like growth factor
- KRT
keratin
- LH
Luteinizing hormone
- FSH
follicle-stimulating hormone
- KTZ
ketoconazole
- Cmax
maximum plasma concentration
- LRZ
liarazole
- CPA
cyproterone acetate
- NSCLC
non-small cell lung cancer
- ER
estrogen receptor
- t1/2
half-life
- TLZ
talarazole
- Css
steady state concentration
- tmax
time till maximum plasma concentration
- DMBA
7,12-dimethylbenz(a)anthracene
- MNU
N-methyl-N-nitrosourea
4. BIBLIOGRAPHY
- 1.Arnold SL, Amory JK, Walsh TJ, Isoherranen N. A sensitive and specific method for measurement of multiple retinoids in human serum with UHPLC-MS/MS. J Lipid Res. 2011;53(3):587–598. doi: 10.1194/jlr.D019745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kane MA, Folias AE, Wang C, Napoli JL. Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal Chem. 2008;80(5):1702–1708. doi: 10.1021/ac702030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asselineau D, Bernard BA, Bailly C, Darmon M. Retinoic acid improves epidermal morphogenesis. Dev Biol. 1989;133(2):322–335. doi: 10.1016/0012-1606(89)90037-7. [DOI] [PubMed] [Google Scholar]
- 5.Ross AC. Vitamin A and retinoic acid in T cell-related immunity. Am J Clin Nutr. 2012;96(5):1166S–1172S. doi: 10.3945/ajcn.112.034637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kane MA, Folias AE, Pingitore A, Perri M, Obrochta KM, Krois CR, Cione E, Ryu JY, Napoli JL. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc Natl Acad Sci USA. 2010;107(50):21884–21889. doi: 10.1073/pnas.1008859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134(6):921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226(2):322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci. 2007;8(10):755–765. doi: 10.1038/nrn2212. [DOI] [PubMed] [Google Scholar]
- 10.Chung SS, Wolgemuth DJ. Role of retinoid signaling in the regulation of spermatogenesis. Cytogenet Genome Res. 2004;105(2–4):189–202. doi: 10.1159/000078189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noy N. Between death and survival: retinoic acid in regulation of apoptosis. Annu Rev Nutr. 2010;30:201–217. doi: 10.1146/annurev.nutr.28.061807.155509. [DOI] [PubMed] [Google Scholar]
- 12.Wolbach SB, Howe PR. Tissue changes following deprivation of fat-soluble a vitamin. J Exp Med. 1925;42(6):753–777. doi: 10.1084/jem.42.6.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hale F. The relation of maternal vitamin A deficiency to microphthalmia in pigs. Texas S J Med. 1937;33:228. [Google Scholar]
- 14.Kistler A. Hypervitaminosis A: side-effects of retinoids. Biochem Soc Trans. 1986;14(5):936–939. doi: 10.1042/bst0140936. [DOI] [PubMed] [Google Scholar]
- 15.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6(10):793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 16.Chen WC, Sass JO, Seltmann H, Nau H, Orfanos CE, Zouboulis CC. Biological effects and metabolism of 9-cis-retinoic acid and its metabolite 9,13-di-cis-retinoic acid in HaCaT keratinocytes in vitro: comparison with all-trans-retinoic acid. Arch Dermatol Res. 2000;292(12):612–620. doi: 10.1007/s004030000189. [DOI] [PubMed] [Google Scholar]
- 17.Nonnecke BJ, Horst RL, Dubeski PL, Reinhardt TA. Reactivity and phenotype of mononuclear leukocytes from nongravid heifers after in vitro exposure to 9,13-di-cis-retinoic acid. J Dairy Sci. 1997;80(11):2833–2841. doi: 10.3168/jds.S0022-0302(97)76248-9. [DOI] [PubMed] [Google Scholar]
- 18.Zile MH, Emerick RJ, DeLuca HF. Identification of 13-cis retinoic acid in tissue extracts and its biological activity in rats. Biochim Biophys Acta. 1967;141(3):639–641. doi: 10.1016/0304-4165(67)90194-8. [DOI] [PubMed] [Google Scholar]
- 19.Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Ann Rev Pathol. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 20.Veal GJ, Cole M, Errington J, Pearson AD, Foot AB, Whyman G, Boddy AV. Pharmacokinetics and metabolism of 13-cis-retinoic acid (isotretinoin) in children with high-risk neuroblastoma – a study of the United Kingdom Children’s Cancer Study Group. Br J Cancer. 2007;96(3):424–431. doi: 10.1038/sj.bjc.6603554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruzicka T, Larsen FG, Galewicz D, Horvath A, Coenraads PJ, Thestrup-Pedersen K, Ortonne JP, Zouboulis CC, Harsch M, Brown TC, Zultak M. Oral alitretinoin (9-cis-retinoic acid) therapy for chronic hand dermatitis in patients refractory to standard therapy: Results of a randomized, double-blind, placebo-controlled, multicenter trial. Arch Dermatol. 2004;140(12):1453–1459. doi: 10.1001/archderm.140.12.1453. [DOI] [PubMed] [Google Scholar]
- 22.Amann PM, Eichmuller SB, Schmidt J, Bazhin AV. Regulation of gene expression by retinoids. Curr Med Chem. 2011;18(9):1405–1412. doi: 10.2174/092986711795029618. [DOI] [PubMed] [Google Scholar]
- 23.Petkovich M, Brand NJ, Krust A, Chambon P. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature. 1987;330(6147):444–450. doi: 10.1038/330444a0. [DOI] [PubMed] [Google Scholar]
- 24.Aggarwal S, Kim SW, Cheon K, Tabassam FH, Yoon JH, Koo JS. Nonclassical action of retinoic acid on the activation of the cAMP response element-binding protein in normal human bronchial epithelial cells. Mol Biol Cell. 2006;17(2):566–575. doi: 10.1091/mbc.E05-06-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radominska-Pandya A, Chen G, Czernik PJ, Little JM, Samokyszyn VM, Carter CA, Nowak G. Direct interaction of all-trans-retinoic acid with protein kinase C (PKC). Implications for PKC signaling and cancer therapy. J Biol Chem. 2000;275(29):22324–22330. doi: 10.1074/jbc.M907722199. [DOI] [PubMed] [Google Scholar]
- 26.Idres N, Marill J, Flexor MA, Chabot GG. Activation of retinoic acid receptor-dependent transcription by all-trans-retinoic acid metabolites and isomers. J Biol Chem. 2002;277(35):31491–31498. doi: 10.1074/jbc.M205016200. [DOI] [PubMed] [Google Scholar]
- 27.Thatcher JE, Buttrick B, Shaffer SA, Shimshoni JA, Goodlett DR, Nelson WL, Isoherranen N. Substrate specificity and ligand interactions of CYP26A1, the human liver retinoic acid hydroxylase. Mol Pharmacol. 2011 doi: 10.1124/mol.111.072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol. 2006;66(7):606–630. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 29.Napoli JL. Biochemical pathways of retinoid transport, metabolism, and signal transduction. Clin Immunol Immunop. 1996;80(3 Pt 2):S52–62. doi: 10.1006/clin.1996.0142. [DOI] [PubMed] [Google Scholar]
- 30.Ross AC. Cellular metabolism and activation of retinoids: roles of cellular retinoid-binding proteins. FASEB J. 1993;7(2):317–327. doi: 10.1096/fasebj.7.2.8440409. [DOI] [PubMed] [Google Scholar]
- 31.Eckhoff C, Collins MD, Nau H. Human plasma all-trans-, 13-cis- and 13-cis-4-oxoretinoic acid profiles during subchronic vitamin A supplementation: comparison to retinol and retinyl ester plasma levels. J Nutr. 1991;121(7):1016–1025. doi: 10.1093/jn/121.7.1016. [DOI] [PubMed] [Google Scholar]
- 32.Hartmann S, Brors O, Bock J, Blomhoff R, Bausch J, Wiegand UW, Hartmann D, Hornig DH. Exposure to retinoic acids in non-pregnant women following high vitamin A intake with a liver meal. Int J Vitam Nutr Res. 2005;75(3):187–194. doi: 10.1024/0300-9831.75.3.187. [DOI] [PubMed] [Google Scholar]
- 33.Hartmann S, Brors O, Bock J, Blomhoff R, Bausch J, Wiegand UW, Hartmann D, Hornig DH. Exposure to retinyl esters, retinol, and retinoic acids in non-pregnant women following increasing single and repeated oral doses of vitamin A. Ann Nutr Metab. 2005;49(3):155–164. doi: 10.1159/000086879. [DOI] [PubMed] [Google Scholar]
- 34.Sedjo RL, Ranger-Moore J, Foote J, Craft NE, Alberts DS, Xu MJ, Giuliano AR. Circulating endogenous retinoic acid concentrations among participants enrolled in a randomized placebo-controlled clinical trial of retinyl palmitate. Cancer Epidem Biomar. 2004;13(11 Pt 1):1687–1692. [PubMed] [Google Scholar]
- 35.Ross AC, Zolfaghari R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu Rev Nutr. 2011;31:65–87. doi: 10.1146/annurev-nutr-072610-145127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thatcher JE, Isoherranen N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin Drug Metab Toxicol. 2009;5(8):875–886. doi: 10.1517/17425250903032681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White JA, Guo YD, Baetz K, Beckett-Jones B, Bonasoro J, Hsu KE, Dilworth FJ, Jones G, Petkovich M. Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J Biol Chem. 1996;271(47):29922–29927. doi: 10.1074/jbc.271.47.29922. [DOI] [PubMed] [Google Scholar]
- 38.Marill J, Cresteil T, Lanotte M, Chabot GG. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol. 2000;58(6):1341–1348. doi: 10.1124/mol.58.6.1341. [DOI] [PubMed] [Google Scholar]
- 39.McSorley LC, Daly AK. Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem Pharmacol. 2000;60(4):517–526. doi: 10.1016/s0006-2952(00)00356-7. [DOI] [PubMed] [Google Scholar]
- 40.Nadin L, Murray M. Participation of CYP2C8 in retinoic acid 4-hydroxylation in human hepatic microsomes. Biochem Pharmacol. 1999;58(7):1201–1208. doi: 10.1016/s0006-2952(99)00192-6. [DOI] [PubMed] [Google Scholar]
- 41.van der Leede BM, van den Brink CE, Pijnappel WW, Sonneveld E, van der Saag PT, van der Burg B. Autoinduction of retinoic acid metabolism to polar derivatives with decreased biological activity in retinoic acid-sensitive, but not in retinoic acid-resistant human breast cancer cells. J Biol Chem. 1997;272(29):17921–17928. doi: 10.1074/jbc.272.29.17921. [DOI] [PubMed] [Google Scholar]
- 42.Muindi J, Frankel SR, Miller WH, Jr, Jakubowski A, Scheinberg DA, Young CW, Dmitrovsky E, Warrell RP., Jr Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood. 1992;79(2):299–303. [PubMed] [Google Scholar]
- 43.el Mansouri S, Tod M, Leclerq M, Petitjean O, Perret G, Porthault M. Time- and dose-dependent kinetics of all-trans-retinoic acid in rats after oral or intravenous administration(s) Drug Metab Dispos. 1995;23(2):227–231. [PubMed] [Google Scholar]
- 44.Thatcher JE, Zelter A, Isoherranen N. The relative importance of CYP26A1 in hepatic clearance of all-trans retinoic acid. Biochem Pharmacol. 2010;80(6):903–912. doi: 10.1016/j.bcp.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White JA, Beckett-Jones B, Guo YD, Dilworth FJ, Bonasoro J, Jones G, Petkovich M. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J Biol Chem. 1997;272(30):18538–18541. doi: 10.1074/jbc.272.30.18538. [DOI] [PubMed] [Google Scholar]
- 46.White JA, Ramshaw H, Taimi M, Stangle W, Zhang A, Everingham S, Creighton S, Tam SP, Jones G, Petkovich M. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc Natl Acad Sci USA. 2000;97(12):6403–6408. doi: 10.1073/pnas.120161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taimi M, Helvig C, Wisniewski J, Ramshaw H, White J, Amad M, Korczak B, Petkovich M. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J Biol Chem. 2004;279(1):77–85. doi: 10.1074/jbc.M308337200. [DOI] [PubMed] [Google Scholar]
- 48.Topletz AR, Thatcher JE, Zelter A, Lutz JD, Tay S, Nelson WL, Isoherranen N. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem Pharmacol. 2012;83(1):149–163. doi: 10.1016/j.bcp.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lutz JD, Dixit V, Yeung CK, Dickmann LJ, Zelter A, Thatcher JE, Nelson WL, Isoherranen N. Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem Pharmacol. 2009;77(2):258–268. doi: 10.1016/j.bcp.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abu-Abed S, MacLean G, Fraulob V, Chambon P, Petkovich M, Dolle P. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech Dev. 2002;110(1–2):173–177. doi: 10.1016/s0925-4773(01)00572-x. [DOI] [PubMed] [Google Scholar]
- 51.MacLean G, Abu-Abed S, Dolle P, Tahayato A, Chambon P, Petkovich M. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development. Mech Dev. 2001;107(1–2):195–201. doi: 10.1016/s0925-4773(01)00463-4. [DOI] [PubMed] [Google Scholar]
- 52.Abu-Abed S, Dolle P, Metzger D, Beckett B, Chambon P, Petkovich M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001;15(2):226–240. doi: 10.1101/gad.855001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uehara M, Yashiro K, Mamiya S, Nishino J, Chambon P, Dolle P, Sakai Y. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev Biol. 2007;302(2):399–411. doi: 10.1016/j.ydbio.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 54.Yashiro K, Zhao X, Uehara M, Yamashita K, Nishijima M, Nishino J, Saijoh Y, Sakai Y, Hamada H. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev Cell. 2004;6(3):411–422. doi: 10.1016/s1534-5807(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 55.Ross AC, Cifelli CJ, Zolfaghari R, Li N-q. Multiple cytochrome P-450 genes are concomitantly regulated by vitamin A under steady-state conditions and by retinoic acid during hepatic first-pass metabolism. Physiol Genomics. 2011;43(1):57–67. doi: 10.1152/physiolgenomics.00182.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto Y, Zolfaghari R, Ross AC. Regulation of CYP26 (cytochrome P450RAI) mRNA expression and retinoic acid metabolism by retinoids and dietary vitamin A in liver of mice and rats. FASEB J. 2000;14(13):2119–2127. doi: 10.1096/fj.00-0061com. [DOI] [PubMed] [Google Scholar]
- 57.Fiorella PD, Napoli JL. Microsomal retinoic acid metabolism. Effects of cellular retinoic acid-binding protein (type I) and C18-hydroxylation as an initial step. J Biol Chem. 1994;269(14):10538–10544. [PubMed] [Google Scholar]
- 58.Tay S, Dickmann L, Dixit V, Isoherranen N. A comparison of the roles of peroxisome proliferator-activated receptor and retinoic acid receptor on CYP26 regulation. Mol Pharmacol. 2010;77(2):218–227. doi: 10.1124/mol.109.059071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xi J, Yang Z. Expression of RALDHs (ALDH1As) and CYP26s in human tissues and during the neural differentiation of P19 embryonal carcinoma stem cell. Gene Expr Patterns. 2008;8(6):438–442. doi: 10.1016/j.gep.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 60.Ahluwalia B, Gambhir K, Sekhon H. Distribution of labeled retinyl acetate and retinoic acid in rat and human testes. A possible site of retinyl acetate incorporation in rat testes. J Nutr. 1975;105(4):467–474. doi: 10.1093/jn/105.4.467. [DOI] [PubMed] [Google Scholar]
- 61.Kurlandsky SB, Gamble MV, Ramakrishnan R, Blaner WS. Plasma delivery of retinoic acid to tissues in the rat. J Biol Chem. 1995;270(30):17850–17857. doi: 10.1074/jbc.270.30.17850. [DOI] [PubMed] [Google Scholar]
- 62.McCormick AM, Kroll KDO, Napoli JL. 13-cis-Retinoic acid metabolism in vivo. The major tissue metabolites in the rat have the all-trans configuration. Biochemistry. 1983;22(16):3933–3940. doi: 10.1021/bi00285a032. [DOI] [PubMed] [Google Scholar]
- 63.Smith JE, Milch PO, Muto Y, Goodman DS. The plasma transport and metabolism of retinoic acid in the rat. Biochem J. 1973;132(4):821–827. doi: 10.1042/bj1320821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niederreither K, Abu-Abed S, Schuhbaur B, Petkovich M, Chambon P, Dolle P. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat Genet. 2002;31(1):84–88. doi: 10.1038/ng876. [DOI] [PubMed] [Google Scholar]
- 65.Van heusden J, Wouters W, Ramaekers FC, Krekels MD, Dillen L, Borgers M, Smets G. All-trans-retinoic acid metabolites significantly inhibit the proliferation of MCF-7 human breast cancer cells in vitro. Br J Cancer. 1998;77(1):26–32. doi: 10.1038/bjc.1998.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van heusden J, Wouters W, Ramaekers FC, Krekels MD, Dillen L, Borgers M, Smets G. The antiproliferative activity of all-trans-retinoic acid catabolites and isomers is differentially modulated by liarozole-fumarate in MCF-7 human breast cancer cells. Br J Cancer. 1998;77(8):1229–1235. doi: 10.1038/bjc.1998.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong D, Ruuska SE, Levinthal DJ, Noy N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem. 1999;274(34):23695–23698. doi: 10.1074/jbc.274.34.23695. [DOI] [PubMed] [Google Scholar]
- 68.Fiorella PD, Napoli JL. Expression of cellular retinoic acid binding protein (CRABP) in Escherichia coli. Characterization and evidence that holo-CRABP is a substrate in retinoic acid metabolism. J Biol Chem. 1991;266(25):16572–16579. [PubMed] [Google Scholar]
- 69.Noy N. Retinoid-binding proteins: mediators of retinoid action. Biochem J. 2000;348(Pt 3):481–495. [PMC free article] [PubMed] [Google Scholar]
- 70.Budhu AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol Cell Biol. 2002;22(8):2632–2641. doi: 10.1128/MCB.22.8.2632-2641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boylan JF, Gudas LJ. The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells. J Biol Chem. 1992;267(30):21486–21491. [PubMed] [Google Scholar]
- 72.Garattini E, Gianni M, Terao M. Retinoids as differentiating agents in oncology: a network of interactions with intracellular pathways as the basis for rational therapeutic combinations. Curr Pharm Des. 2007;13(13):1375–1400. doi: 10.2174/138161207780618786. [DOI] [PubMed] [Google Scholar]
- 73.Lazzarino M, Regazzi MB, Corso A. Clinical relevance of all-trans retinoic acid pharmacokinetics and its modulation in acute promyelocytic leukemia. Leuk Lymphoma. 1996;23(5–6):539–543. doi: 10.3109/10428199609054862. [DOI] [PubMed] [Google Scholar]
- 74.Rigopoulos D, Larios G, Katsambas AD. The role of isotretinoin in acne therapy: why not as first-line therapy? facts and controversies. Clin Dermatol. 2010;28(1):24–30. doi: 10.1016/j.clindermatol.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 75.Pilkington T, Brogden RN. Acitretin: A review of its pharmacology and therapeutic use. Drugs. 1992;43(4):597–627. doi: 10.2165/00003495-199243040-00010. [DOI] [PubMed] [Google Scholar]
- 76.Miller WH. The emerging role of retinoids and retinoic acid metabolism blocking agents in the treatment of cancer. Cancer. 1998;83(8):1471–1482. doi: 10.1002/(sici)1097-0142(19981015)83:8<1471::aid-cncr1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 77.Pasquali D, Thaller C, Eichele G. Abnormal level of retinoic acid in prostate cancer tissues. J Clin Endocrinol Metab. 1996;81(6):2186–2191. doi: 10.1210/jcem.81.6.8964849. [DOI] [PubMed] [Google Scholar]
- 78.Lotan R. Effects of vitamin A and its analogs (retinoids) on normal and neoplastic cells. Biochim Biophys Acta. 1980;605(1):33–91. doi: 10.1016/0304-419x(80)90021-9. [DOI] [PubMed] [Google Scholar]
- 79.Orfanos CE, Zouboulis CC, Almond-Roesler B, Geilen CC. Current use and future potential role of retinoids in dermatology. Drugs. 1997;53(3):358–388. doi: 10.2165/00003495-199753030-00003. [DOI] [PubMed] [Google Scholar]
- 80.Bruno RD, Njar VC. Targeting cytochrome P450 enzymes: a new approach in anticancer drug development. Bioorg Med Chem. 2007;15(15):5047–5060. doi: 10.1016/j.bmc.2007.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Njar VC. Cytochrome P450 retinoic acid 4-hydroxylase inhibitors: potential agents for cancer therapy. Mini-Rev Med Chem. 2002;2(3):261–269. doi: 10.2174/1389557023406223. [DOI] [PubMed] [Google Scholar]
- 82.Njar VC, Gediya L, Purushottamachar P, Chopra P, Vasaitis TS, Khandelwal A, Mehta J, Huynh C, Belosay A, Patel J. Retinoic acid metabolism blocking agents (RAMBAs) for treatment of cancer and dermatological diseases. Bioorg Med Chem. 2006;14(13):4323–4340. doi: 10.1016/j.bmc.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 83.Symoens J, Moens M, Dom J, Scheijgrond H, Dony J, Schuermans V, Legendre R, Finestine N. An evaluation of two years of clinical experience with ketoconazole [with discussion and concluding remarks] Rev Infect Dis. 1980;2(4):674–691. doi: 10.1093/clinids/2.4.674. [DOI] [PubMed] [Google Scholar]
- 84.Pont A, Williams PL, Loose DS, Feldman D, Reitz RE, Bochra C, Stevens DA. Ketoconazole blocks adrenal steroid synthesis. Ann Intern Med. 1982;97(3):370–372. doi: 10.7326/0003-4819-97-3-370. [DOI] [PubMed] [Google Scholar]
- 85.Williams G. Ketoconazole for prostate cancer. Lancet. 1984;2(8404):696. doi: 10.1016/s0140-6736(84)91253-4. [DOI] [PubMed] [Google Scholar]
- 86.Tapazoglou E, Subramanian MG, Al-Sarraf M, Kresge C, Decker DA. High-dose ketoconazole therapy in patients with metastatic prostate cancer. Am J Clin Oncol. 1986;9(5):369–375. doi: 10.1097/00000421-198610000-00001. [DOI] [PubMed] [Google Scholar]
- 87.Moffat LE, Kirk D, Tolley DA, Smith MF, Beastall G. Ketoconazole as primary treatment of prostatic cancer. Br J Urol. 1988;61(5):439–440. doi: 10.1111/j.1464-410x.1988.tb06593.x. [DOI] [PubMed] [Google Scholar]
- 88.Bruynseels J, De Coster R, Van Rooy P, Wouters W, Coene MC, Snoeck E, Raeymaekers A, Freyne E, Sanz G, Vanden Bussche G, et al. R75251, a new inhibitor of steroid biosynthesis. Prostate. 1990;16(4):345–357. doi: 10.1002/pros.2990160409. [DOI] [PubMed] [Google Scholar]
- 89.Mahler C, Denis L, De Coster R. The effects of a new imidazole derivative in advanced prostatic cancer. A preliminary report. Prog Clin Biol Res. 1989;303:205–209. [PubMed] [Google Scholar]
- 90.Van Ginckel R, De Coster R, Wouters W, Vanherck W, van der Veer R, Goeminne N, Jagers E, Van Cauteren H, Wouters L, Distelmans W, et al. Antitumoral effects of R 75251 on the growth of transplantable R3327 prostatic adenocarcinoma in rats. Prostate. 1990;16(4):313–323. doi: 10.1002/pros.2990160406. [DOI] [PubMed] [Google Scholar]
- 91.De Coster R, Wouters W, Van Ginckel R, End D, Krekels M, Coene MC, Bowden C. Experimental studies with liarozole (R75251): An antitumoral agent which inhibits retinoic acid breakdown. J Steroid Biochem Mol Biol. 1992;43(1–3):197–201. doi: 10.1016/0960-0760(92)90208-z. [DOI] [PubMed] [Google Scholar]
- 92.Denis LJ. Controversies in the management of localized and metastatic prostatic cancer. Eur J Cancer. 1991;27(3):333–341. doi: 10.1016/0277-5379(91)90542-l. [DOI] [PubMed] [Google Scholar]
- 93.Van Wauwe J, Van Nyen G, Coene MC, Stoppie P, Cools W, Goossens J, Borghgraef P, Janssen PA. Liarozole, an inhibitor of retinoic acid metabolism, exerts retinoid-mimetic effects in vivo. J Pharmacol Exp Ther. 1992;261(2):773–779. [PubMed] [Google Scholar]
- 94.Van Wauwe JP, Coene MC, Goossens J, Cools W, Monbaliu J. Effects of cytochrome P-450 inhibitors on the in vivo metabolism of all-trans-retinoic acid in rats. J Pharmacol Exp Ther. 1990;252(1):365–369. [PubMed] [Google Scholar]
- 95.Mahler C, Verhelst J, Denis L. Ketoconazole and liarozole in the treatment of advanced prostatic cancer. Cancer. 1993;71(3 Suppl):1068–1073. doi: 10.1002/1097-0142(19930201)71:3+<1068::aid-cncr2820711427>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 96.Dawson NA. Treatment of progressive metastatic prostate cancer. Oncology. 1993;7(5):17–24, 27. discussion 27–19. [PubMed] [Google Scholar]
- 97.Boccardo F, Cannata D, Guarneri D, Oneto F, Cortesi E, Bono A. R75251 in prostate cancer patients in progression after first-line hormonal treatment. Tumori. 1994;80(4):276–279. doi: 10.1177/030089169408000406. [DOI] [PubMed] [Google Scholar]
- 98.Muindi JR, Frankel SR, Huselton C, DeGrazia F, Garland WA, Young CW, Warrell RP., Jr Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res. 1992;52(8):2138–2142. [PubMed] [Google Scholar]
- 99.Lotan R, Lotan D, Sacks PG. Inhibition of tumor cell growth by retinoids. Methods Enzymol. 1990;190:100–110. doi: 10.1016/0076-6879(90)90014-r. [DOI] [PubMed] [Google Scholar]
- 100.Debruyne FJ, Murray R, Fradet Y, Johansson JE, Tyrrell C, Boccardo F, Denis L, Marberger JM, Brune D, Rassweiler J, Vangeneugden T, Bruynseels J, Janssens M, De Porre P. Liarozole-a novel treatment approach for advanced prostate cancer: results of a large randomized trial versus cyproterone acetate. Liarozole Study Group. Urology. 1998;52(1):72–81. doi: 10.1016/s0090-4295(98)00129-0. [DOI] [PubMed] [Google Scholar]
- 101.Denis L, Debruyne F, De Porre P, Bruynseels J. Early clinical experience with liarozole (Liazal) in patients with progressive prostate cancer. Eur J Cancer. 1998;34(4):469–475. doi: 10.1016/s0959-8049(97)10120-4. [DOI] [PubMed] [Google Scholar]
- 102.Dijkman GA, Fernandez del Moral P, Bruynseels J, De Porre P, Denis L, Debruyne FMJ. Liarozole (R75251) in hormone-resistant prostate cancer patients. Prostate. 1997;33(1):26–31. doi: 10.1002/(sici)1097-0045(19970915)33:1<26::aid-pros5>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 103.Seidmon EJ, Trump DL, Kreis W, Hall SW, Kurman MR, Ouyang SP, Wu J, Kremer AB. Phase I/II dose-escalation study of liarozole in patients with stage D, hormone-refractory carcinoma of the prostate. Ann Surg Oncol. 1995;2(6):550–556. doi: 10.1007/BF02307090. [DOI] [PubMed] [Google Scholar]
- 104.Liarozole. Liarozole fumarate, Liazal, R 75251, R 85246. Drugs R D. 1999;2(6):427–430. doi: 10.2165/00126839-199902060-00017. [DOI] [PubMed] [Google Scholar]
- 105.O’Byrne KJ, Han C, Mitchell K, Lane D, Carmichael J, Harris AL, Talbot DC. Phase II study of liarozole in advanced non-small cell lung cancer. Eur J Cancer. 1998;34(9):1463–1466. doi: 10.1016/s0959-8049(98)00144-0. [DOI] [PubMed] [Google Scholar]
- 106.Goss PE, Oza A, Goel R, Nabholtz JM, Coster RD, Bruynseels J, Reid C, Wadden N, Crump M, Tye LM. Liarozole fumarate (R85246): a novel imidazole in the treatment of receptor positive postmenopausal metastatic breast cancer. Breast Cancer Res Treat. 2000;59(1):55–68. doi: 10.1023/a:1006320122711. [DOI] [PubMed] [Google Scholar]
- 107.Goss PE, Strasser K, Marques R, Clemons M, Oza A, Goel R, Blackstein M, Kaizer L, Sterns EE, Nabholtz JM, De Coster R, Crump M, Abdolell M, Qi S. Liarozole fumarate (R85246): in the treatment of ER negative, tamoxifen refractory or chemotherapy resistant postmenopausal metastatic breast cancer. Breast Cancer Res Treat. 2000;64(2):177–188. doi: 10.1023/a:1006480504790. [DOI] [PubMed] [Google Scholar]
- 108.Hamilton A, Roy JA, Beex L, Piccart M, Mauriac L, Coleman R, Paridaens R, Boes GH, van Vreckem A, Palmer P, Klijn J. EORTC 10941: A phase II study of liarozole in postmenopausal patients with ‘Chemotherapy-Resistant’ or ‘Potentially Hormone Sensitive’ metastatic breast cancer. Breast Cancer Res Treat. 2000;60(2):181–188. doi: 10.1023/a:1006398518037. [DOI] [PubMed] [Google Scholar]
- 109.Wouters W, van Dun J, Dillen A, Coene MC, Cools W, De Coster R. Effects of liarozole, a new antitumoral compound, on retinoic acid-induced inhibition of cell growth and on retinoic acid metabolism in MCF-7 human breast cancer cells. Cancer Res. 1992;52(10):2841–2846. [PubMed] [Google Scholar]
- 110.Acevedo P, Bertram JS. Liarozole potentiates the cancer chemopreventive activity of and the up-regulation of gap junctional communication and connexin43 expression by retinoic acid and beta-carotene in 10T1/2 cells. Carcinogenesis. 1995;16(9):2215–2222. doi: 10.1093/carcin/16.9.2215. [DOI] [PubMed] [Google Scholar]
- 111.Miller VA, Rigas JR, Muindi JR, Tong WP, Venkatraman E, Kris MG, Warrell RP., Jr Modulation of all-trans retinoic acid pharmacokinetics by liarozole. Cancer Chemother Pharmacol. 1994;34(6):522–526. doi: 10.1007/BF00685665. [DOI] [PubMed] [Google Scholar]
- 112.Krekels MDWG, Zimmerman J, Janssens B, Van Ginckel R, Cools W, Van Hove C, Coene MC, Wouters W. Analysis of the oxidative catabolism of retinoic acid in rat Dunning R3327G prostate tumors. Prostate. 1996;29(1):36–41. doi: 10.1002/(SICI)1097-0045(199607)29:1<36::AID-PROS5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 113.Van Wauwe J, Coene MC, Cools W, Goossens J, Lauwers W, Le Jeune L, Van Hove C, Van Nyen G. Liarozole fumarate inhibits the metabolism of 4-keto-all-trans-retinoic acid. Biochem Pharmacol. 1994;47(4):737–741. doi: 10.1016/0006-2952(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 114.Han IS, Choi JH. Highly specific cytochrome P450-like enzymes for all-trans-retinoic acid in T47D human breast cancer cells. J Clin Endocrinol Metab. 1996;81(6):2069–2075. doi: 10.1210/jcem.81.6.8964830. [DOI] [PubMed] [Google Scholar]
- 115.Krekels MD, Verhoeven A, van Dun J, Cools W, Van Hove C, Dillen L, Coene MC, Wouters W. Induction of the oxidative catabolism of retinoid acid in MCF-7 cells. Br J Cancer. 1997;75(8):1098–1104. doi: 10.1038/bjc.1997.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yee SW, Jarno L, Gomaa MS, Elford C, Ooi LL, Coogan MP, McClelland R, Nicholson RI, Evans BA, Brancale A, Simons C. Novel tetralone-derived retinoic acid metabolism blocking agents: synthesis and in vitro evaluation with liver microsomal and MCF-7 CYP26A1 cell assays. J Med Chem. 2005;48(23):7123–7131. doi: 10.1021/jm0501681. [DOI] [PubMed] [Google Scholar]
- 117.Fujii H, Sato T, Kaneko S, Gotoh O, Fujii-Kuriyama Y, Osawa K, Kato S, Hamada H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997;16(14):4163–4173. doi: 10.1093/emboj/16.14.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ray WJ, Bain G, Yao M, Gottlieb DI. CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J Biol Chem. 1997;272(30):18702–18708. doi: 10.1074/jbc.272.30.18702. [DOI] [PubMed] [Google Scholar]
- 119.Helvig C, Taimi M, Cameron D, Jones G, Petkovich M. Functional properties and substrate characterization of human CYP26A1, CYP26B1, and CYP26C1 expressed by recombinant baculovirus in insect cells. J Pharmacol Toxicol Meth. 2011;64(3):258–263. doi: 10.1016/j.vascn.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 120.Lee JS, Newman RA, Lippman SM, Fossella FV, Calayag M, Raber MN, Krakoff IH, Hong WK. Phase I evaluation of all-trans retinoic acid with and without ketoconazole in adults with solid tumors. J Clin Oncol. 1995;13(6):1501–1508. doi: 10.1200/JCO.1995.13.6.1501. [DOI] [PubMed] [Google Scholar]
- 121.Rigas JR, Francis PA, Muindi JR, Kris MG, Huselton C, DeGrazia F, Orazem JP, Young CW, Warrell RP., Jr Constitutive variability in the pharmacokinetics of the natural retinoid, all-trans-retinoic acid, and its modulation by ketoconazole. J Natl Cancer Inst. 1993;85(23):1921–1926. doi: 10.1093/jnci/85.23.1921. [DOI] [PubMed] [Google Scholar]
- 122.Schmitt-Hoffmann AH, Roos B, Sauer J, Spickermann J, Maares J, Schoetzau A, Meyer I. Pharmacokinetic interactions between alitretinoin and ketoconazole or simvastatin or ciclosporin A. Clin Exp Dermatol. 2011;36:24–28. doi: 10.1111/j.1365-2230.2011.04034.x. [DOI] [PubMed] [Google Scholar]
- 123.Berth-Jones J, Todd G, Hutchinson PE, Thestrup-Pedersen K, Vanhoutte FP. Treatment of psoriasis with oral liarozole: a dose-ranging study. Brit J Dermatol. 2000;143(6):1170–1176. doi: 10.1046/j.1365-2133.2000.03884.x. [DOI] [PubMed] [Google Scholar]
- 124.Bhushan M, Burden AD, McElhone K, James R, Vanhoutte FP, Griffiths CEM. Oral liarozole in the treatment of palmoplantar pustular psoriasis: a randomized, doubleblind, placebo-controlled study. Brit J Dermatol. 2001;145(4):546–553. doi: 10.1046/j.1365-2133.2001.04411.x. [DOI] [PubMed] [Google Scholar]
- 125.Dockx P, Decree J, Degreef H. Inhibition of the metabolism of endogenous retinoic acid as treatment for severe psoriasis: an open study with oral liarozole. Br J Dermatol. 1995;133(3):426–432. doi: 10.1111/j.1365-2133.1995.tb02672.x. [DOI] [PubMed] [Google Scholar]
- 126.Kuijpers, Van P, Bergers, Boegheim, Den B, Siegenthaler, Van DEK, Schalkwijk The effects of oral liarozole on epidermal proliferation and differentiation in severe plaque psoriasis are comparable with those of acitretin. Brit J Dermatol. 1998;139(3):380–389. doi: 10.1046/j.1365-2133.1998.02399.x. [DOI] [PubMed] [Google Scholar]
- 127.van Pelt JP, de Jong EM, de Bakker ES, van de Kerkhof PC. Effects of systemic treatment with liarozole on cutaneous inflammation, epidermal proliferation and differentiation in extensive plaque psoriasis. Skin Pharmacol Appl Skin Physiol. 1998;11(2):70–79. doi: 10.1159/000029811. [DOI] [PubMed] [Google Scholar]
- 128.Verfaille CJ, Vanhoutte FP, Blanchet-Bardon C, Van Steensel MA, Steijlen PM. Oral liarozole vs. acitretin in the treatment of ichthyosis: a phase II/III multicentre, doubleblind, randomized, active-controlled study. Brit J Dermatol. 2007;156(5):965–973. doi: 10.1111/j.1365-2133.2006.07745.x. [DOI] [PubMed] [Google Scholar]
- 129.Lucker GP, Heremans AM, Boegheim PJ, van de Kerkhof PC, Steijlen PM. Oral treatment of ichthyosis by the cytochrome P-450 inhibitor liarozole. Brit J Dermatol. 1997;136(1):71–75. [PubMed] [Google Scholar]
- 130.Lucker GPH, Verfaille CJ, Heremans AMC, Vanhoutte FP, Boegheim JPJ, Steijlen PPM. Topical liarozole in ichthyosis: a double-blind, left–right comparative study followed by a long-term open maintenance study. Brit J Dermatol. 2005;152(3):566–569. doi: 10.1111/j.1365-2133.2005.06399.x. [DOI] [PubMed] [Google Scholar]
- 131.Pavez Lorie E, Ganemo A, Borgers M, Wouters L, Blockhuys S, van de Plassche L, Torma H, Vahlquist A. Expression of retinoid-regulated genes in lamellar ichthyosis vs. healthy control epidermis: changes after oral treatment with liarozole. Acta Derm Venereol. 2009;89(1):12–20. doi: 10.2340/00015555-0573. [DOI] [PubMed] [Google Scholar]
- 132.Verfaille CJ, Borgers M, van Steensel MA. Retinoic acid metabolism blocking agents (RAMBAs): a new paradigm in the treatment of hyperkeratotic disorders. J Dtsch Dermatol Ges. 2008;6(5):355–364. doi: 10.1111/j.1610-0387.2007.06541.x. [DOI] [PubMed] [Google Scholar]
- 133.Stoppie P, Borgers M, Borghgraef P, Dillen L, Goossens J, Sanz G, Szel H, Van Hove C, Van Nyen G, Nobels G, Vanden Bossche H, Venet M, Willemsens G, Van Wauwe J. R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodents. J Pharmacol Exp Ther. 2000;293(1):304–312. [PubMed] [Google Scholar]
- 134.Heise R, Mey J, Neis MM, Marquardt Y, Joussen S, Ott H, Wiederholt T, Kurschat P, Megahed M, Bickers DR, Merk HF, Baron JM. Skin retinoid concentrations are modulated by CYP26AI expression restricted to basal keratinocytes in normal human skin and differentiated 3D skin models. J Invest Dermatol. 2006;126(11):2473–2480. doi: 10.1038/sj.jid.5700432. [DOI] [PubMed] [Google Scholar]
- 135.Giltaire S, Herphelin F, Frankart A, Hérin M, Stoppie P, Poumay Y. The CYP26 inhibitor R115866 potentiates the effects of all-trans retinoic acid on cultured human epidermal keratinocytes. Brit J Dermatol. 2009;160(3):505–513. doi: 10.1111/j.1365-2133.2008.08960.x. [DOI] [PubMed] [Google Scholar]
- 136.Pavez Loriè E, Chamcheu J, Vahlquist A, Törmä H. Both all-trans retinoic acid and cytochrome P450 (CYP26) inhibitors affect the expression of vitamin A metabolizing enzymes and retinoid biomarkers in organotypic epidermis. Arch Dermatol Res. 2009;301(7):475–485. doi: 10.1007/s00403-009-0937-7. [DOI] [PubMed] [Google Scholar]
- 137.Verfaille CJ, Coel M, Boersma IH, Mertens J, Borgers M, Roseeuw D. Oral R115866 in the treatment of moderate to severe facial acne vulgaris: an exploratory study. Brit J Dermatol. 2007;157(1):122–126. doi: 10.1111/j.1365-2133.2007.07896.x. [DOI] [PubMed] [Google Scholar]
- 138.Verfaille CJ, Thissen CA, Bovenschen HJ, Mertens J, Steijlen PM, van de Kerkhof PC. Oral R115866 in the treatment of moderate to severe plaque-type psoriasis. J Eur Acad Dermatol. 2007;21(8):1038–1046. doi: 10.1111/j.1468-3083.2007.02158.x. [DOI] [PubMed] [Google Scholar]
- 139.Bovenschen HJ, Otero ME, Langewouters AMG, Van Vlijmen-Willems IMJJ, Van Rens DWA, Seyger MMB, Van De Kerkhof PCM. Oral retinoic acid metabolism blocking agent Rambazole for plaque psoriasis: an immunohistochemical study. Brit J Dermatol. 2007;156(2):263–270. doi: 10.1111/j.1365-2133.2006.07660.x. [DOI] [PubMed] [Google Scholar]
- 140.Pavez Lorie E, Cools M, Borgers M, Wouters L, Shroot B, Hagforsen E, Torma H, Vahlquist A. Topical treatment with CYP26 inhibitor talarozole (R115866) dose dependently alters the expression of retinoid-regulated genes in normal human epidermis. Br J Dermatol. 2009;160(1):26–36. doi: 10.1111/j.1365-2133.2008.08895.x. [DOI] [PubMed] [Google Scholar]
- 141.Geria AN, Scheinfeld NS. Talarozole, a selective inhibitor of P450-mediated alltrans retinoic acid for the treatment of psoriasis and acne. Curr Opin Investig Drugs. 2008;9(11):1228–1237. [PubMed] [Google Scholar]
- 142.Van heusden J, Van Ginckel R, Bruwiere H, Moelans P, Janssen B, Floren W, van der Leede BJ, van Dun J, Sanz G, Venet M, Dillen L, Van Hove C, Willemsens G, Janicot M, Wouters W. Inhibition of all-trans-retinoic acid metabolism by R116010 induces antitumour activity. Br J Cancer. 2002;86(4):605–611. doi: 10.1038/sj.bjc.6600056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vanier KL, Mattiussi AJ, Johnston DL. Interaction of all-trans-retinoic acid with fluconazole in acute promyelocytic leukemia. J Pediatr Hematol Oncol. 2003;25(5):403–404. doi: 10.1097/00043426-200305000-00010. [DOI] [PubMed] [Google Scholar]
- 144.Cordoba R, Ramirez E, Lei S, Lopez de la Guia A, Sanjurjo M, Carcas A, Hernandez-Navarro F. Hypercalcemia due to an interaction of all-trans retinoic acid (ATRA) and itraconazole therapy for acute promyelocytic leukemia successfully treated with zoledronic acid. Eur J Clin Pharmacol. 2008;64(10):1031–1032. doi: 10.1007/s00228-008-0517-3. [DOI] [PubMed] [Google Scholar]
- 145.Ueno H, Kizaki M, Takayama N, Matsushita H, Muto A, Okamoto S, Ikeda Y. Itraconazole and retinoid resistance. Am J Hematol. 1995;50(4):319–320. doi: 10.1002/ajh.2830500426. [DOI] [PubMed] [Google Scholar]
- 146.Bennett MT, Sirrs S, Yeung JK, Smith CA. Hypercalcemia due to all-trans retinoic acid in the treatment of acute promyelocytic leukemia potentiated by voriconazole. Leuk Lymphoma. 2005;46(12):1829–1831. doi: 10.1080/10428190500235298. [DOI] [PubMed] [Google Scholar]
- 147.Cheng MP, Paquette K, Lands LC, Ovetchkine P, Théoret Y, Quach C. Voriconazole inhibition of vitamin A metabolism: Are adverse events increased in cystic fibrosis patients? Pediatr Pulm. 2010;45(7):661–666. doi: 10.1002/ppul.21234. [DOI] [PubMed] [Google Scholar]
- 148.Dixon KS, Hassoun A. Pseudotumor cerebri due to the potentiation of all-trans retinoic acid by voriconazole. J Am Pharm Assoc. 2010;50(6):742–744. doi: 10.1331/JAPhA.2010.09133. [DOI] [PubMed] [Google Scholar]
- 149.Schwartz EL, Hallam S, Gallagher RE, Wiernik PH. Inhibition of all-trans-retinoic acid metabolism by fluconazole in vitro and in patients with acute promyelocytic leukemia. Biochem Pharmacol. 1995;50(7):923–928. doi: 10.1016/0006-2952(95)00213-j. [DOI] [PubMed] [Google Scholar]
- 150.Brown HS, Galetin A, Hallifax D, Houston JB. Prediction of in vivo drug-drug interactions from in vitro data : factors affecting prototypic drug-drug interactions involving CYP2C9, CYP2D6 and CYP3A4. Clin Pharmacokinet. 2006;45(10):1035–1050. doi: 10.2165/00003088-200645100-00006. [DOI] [PubMed] [Google Scholar]
- 151.Varhe A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;56(6 Pt 1):601–607. doi: 10.1038/clpt.1994.184. [DOI] [PubMed] [Google Scholar]
- 152.Varhe A, Olkkola KT, Neuvonen PJ. Fluconazole, but not terbinafine, enhances the effects of triazolam by inhibiting its metabolism. Br J Clin Pharmacol. 1996;41(4):319–323. doi: 10.1046/j.1365-2125.1996.03189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang S, Pillai VC, Mada SR, Strom S, Venkataramanan R. Effect of voriconazole and other azole antifungal agents on CYP3A activity and metabolism of tacrolimus in human liver microsomes. Xenobiotica. 2011;0(0):1–8. doi: 10.3109/00498254.2011.631224. [DOI] [PubMed] [Google Scholar]
- 154.Tiboni GM, Marotta F, Carletti E. Fluconazole alters CYP26 gene expression in mouse embryos. Reprod Toxicol. 2009;27(2):199–202. doi: 10.1016/j.reprotox.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 155.Ahmad M. Study on cytochrome P-450 dependent retinoic acid metabolism and its inhibitors as potential agents for cancer therapy. Sci Pharm. 2011;79(4):921–935. doi: 10.3797/scipharm.1106-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Njar VC, Nnane IP, Brodie AM. Potent inhibition of retinoic acid metabolism enzyme(s) by novel azolyl retinoids. Bioorg Med Chem Lett. 2000;10(17):1905–1908. doi: 10.1016/s0960-894x(00)00391-7. [DOI] [PubMed] [Google Scholar]
- 157.Patel JB, Huynh CK, Handratta VD, Gediya LK, Brodie AM, Goloubeva OG, Clement OO, Nanne IP, Soprano DR, Njar VC. Novel retinoic acid metabolism blocking agents endowed with multiple biological activities are efficient growth inhibitors of human breast and prostate cancer cells in vitro and a human breast tumor xenograft in nude mice. J Med Chem. 2004;47(27):6716–6729. doi: 10.1021/jm0401457. [DOI] [PubMed] [Google Scholar]
- 158.Gomaa MS, Yee SW, Milbourne CE, Barbera MC, Simons C, Brancale A. Homology model of human retinoic acid metabolising enzyme cytochrome P450 26A1 (CYP26A1): active site architecture and ligand binding. J Enzym Inhib Med Ch. 2006;21(4):361–369. doi: 10.1080/14756360600742014. [DOI] [PubMed] [Google Scholar]
- 159.Huynh CK, Brodie AMH, Njar VCO. Inhibitory effects of retinoic acid metabolism blocking agents (RAMBAs) on the growth of human prostate cancer cells and LNCaP prostate tumour xenografts in SCID mice. Br J Cancer. 2006;94(4):513–523. doi: 10.1038/sj.bjc.6602971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Patel JB, Mehta J, Belosay A, Sabnis G, Khandelwal A, Brodie AM, Soprano DR, Njar VC. Novel retinoic acid metabolism blocking agents have potent inhibitory activities on human breast cancer cells and tumour growth. Br J Cancer. 2007;96(8):1204–1215. doi: 10.1038/sj.bjc.6603705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gediya LK, Chopra P, Purushottamachar P, Maheshwari N, Njar VC. A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells. J Med Chem. 2005;48(15):5047–5051. doi: 10.1021/jm058214k. [DOI] [PubMed] [Google Scholar]
- 162.Khandelwal A, Gediya L, Njar V. MS-275 synergistically enhances the growth inhibitory effects of RAMBA VN/66-1 in hormone-insensitive PC-3 prostate cancer cells and tumours. Br J Cancer. 2008;98(7):1234–1243. doi: 10.1038/sj.bjc.6604295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Goss PE, Qi S, Hu H, Gediya LK, Purushottamachar P, Godbole AM, Njar VC. Anti-tumor effects of a novel retinoic acid metabolism blocking agent VN/14-1 in the N-methyl-N-nitrosourea-induced rat mammary carcinoma model and its effects on the uterus. Breast Cancer Res Treat. 2012;133(1):137–144. doi: 10.1007/s10549-011-1724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gediya LK, Belosay A, Khandelwal A, Purushottamachar P, Njar VC. Improved synthesis of histone deacetylase inhibitors (HDIs) (MS-275 and CI-994) and inhibitory effects of HDIs alone or in combination with RAMBAs or retinoids on growth of human LNCaP prostate cancer cells and tumor xenografts. Bioorg Med Chem. 2008;16(6):3352–3360. doi: 10.1016/j.bmc.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Belosay A, Brodie AM, Njar VC. Effects of novel retinoic acid metabolism blocking agent (VN/14-1) on letrozole-insensitive breast cancer cells. Cancer Res. 2006;66(23):11485–11493. doi: 10.1158/0008-5472.CAN-06-2168. [DOI] [PubMed] [Google Scholar]
- 166.Vasaitis TS, Bruno RD, Njar VC. CYP17 inhibitors for prostate cancer therapy. J Steroid Biochem Mol Biol. 2011;125(1–2):23–31. doi: 10.1016/j.jsbmb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gomaa MS, Bridgens CE, Aboraia AS, Veal GJ, Redfern CP, Brancale A, Armstrong JL, Simons C. Small molecule inhibitors of retinoic acid 4-Hydroxylase (CYP26): Synthesis and biological evaluation of imidazole methyl 3-(4-(aryl-2-ylamino)phenyl)propanoates. J Med Chem. 2011;54(8):2778–2791. doi: 10.1021/jm101583w. [DOI] [PubMed] [Google Scholar]
- 168.Gomaa MS, Bridgens CE, Veal GJ, Redfern CPF, Brancale A, Armstrong JL, Simons C. Synthesis and biological evaluation of 3-(1H-imidazol- and triazol-1-yl)-2,2-dimethyl-3-[4-(naphthalen-2-ylamino)phenyl]propyl derivatives as small molecule inhibitors of retinoic acid 4-hydroxylase (CYP26) J Med Chem. 2011;54(19):6803–6811. doi: 10.1021/jm200695m. [DOI] [PubMed] [Google Scholar]
- 169.Mulvihill MJ, Kan JL, Beck P, Bittner M, Cesario C, Cooke A, Keane DM, Nigro AI, Nillson C, Smith V, Srebernak M, Sun FL, Vrkljan M, Winski SL, Castelhano AL, Emerson D, Gibson N. Potent and selective [2-imidazol-1-yl-2-(6-alkoxy-naphthalen-2-yl)-1-methyl-ethyl]-dimethyl-amines as retinoic acid metabolic blocking agents (RAMBAs) Bioorg Med Chem Lett. 2005;15(6):1669–1673. doi: 10.1016/j.bmcl.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 170.Mulvihill MJ, Kan JL, Cooke A, Bhagwat S, Beck P, Bittner M, Cesario C, Keane D, Lazarescu V, Nigro A, Nillson C, Panicker B, Smith V, Srebernak M, Sun FL, O’Connor M, Russo S, Fischetti G, Vrkljan M, Winski S, Castelhano AL, Emerson D, Gibson NW. 3-[6-(2-Dimethylamino-1-imidazol-1-yl-butyl)-naphthalen-2-yloxy]-2,2-dimethyl-propionic acid as a highly potent and selective retinoic acid metabolic blocking agent. Bioorg Med Chem Lett. 2006;16(10):2729–2733. doi: 10.1016/j.bmcl.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 171.Pautus S, Yee SW, Jayne M, Coogan MP, Simons C. Synthesis and CYP26A1 inhibitory activity of 1-[benzofuran-2-yl-(4-alkyl/aryl-phenyl)-methyl]-1H-triazoles. Bioorg Med Chem. 2006;14(11):3643–3653. doi: 10.1016/j.bmc.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 172.Pautus S, Aboraia AS, Bassett CE, Brancale A, Coogan MP, Simons C. Design and synthesis of substituted imidazole and triazole N-phenylbenzo[d]oxazolamine inhibitors of retinoic acid metabolizing enzyme CYP26. J Enzym Inhib Med Ch. 2009;24(2):487–498. doi: 10.1080/14756360802218334. [DOI] [PubMed] [Google Scholar]
- 173.Gomaa MS, Armstrong JL, Bobillon B, Veal GJ, Brancale A, Redfern CPF, Simons C. Novel azolyl-(phenylmethyl)]aryl/heteroarylamines: Potent CYP26 inhibitors and enhancers of all-trans retinoic acid activity in neuroblastoma cells. Bioorg Med Chem. 2008;16(17):8301–8313. doi: 10.1016/j.bmc.2007.06.048. [DOI] [PubMed] [Google Scholar]
- 174.Tang-Liu DD, Matsumoto RM, Usansky JI. Clinical pharmacokinetics and drug metabolism of tazarotene: a novel topical treatment for acne and psoriasis. Clin Pharmacokinet. 1999;37(4):273–287. doi: 10.2165/00003088-199937040-00001. [DOI] [PubMed] [Google Scholar]
- 175.Vasudevan J, Johnson AT, Huang D, Chandraratna RA. Compounds having activity as inhibitors of cytochrome P450RAI. US 6,291, 677, B1. 2001 Sep 18; [Google Scholar]
- 176.Kirby AJ, Lelain R, Mason P, Maharlouie F, Nicholls PJ, Smith HJ, Simons C. Some 3-(4-aminophenyl)pyrrolidine-2,5-diones as all-trans-retinoic acid metabolising enzyme inhibitors (RAMBAs) J Enzym Inhib Med Ch. 2002;17(5):321–327. doi: 10.1080/1475636021000059100. [DOI] [PubMed] [Google Scholar]
- 177.Kirby AJ, Lain RL, Maharlouie F, Mason P, Nicholls PJ, John Smith H, Simons C. Inhibition of retinoic acid metabolising enzymes by 2-(4-aminophenylmethyl)-6-hydroxy-3,4-dihydronaphthalen-1(2H)-one and related compounds. J Enzym Inhib Med Ch. 2003;18(1):27–33. doi: 10.1080/1475636021000049221. [DOI] [PubMed] [Google Scholar]
- 178.Greer VP, Mason P, Kirby AJ, Smith HJ, Nicholls PJ, Simons C. Some 1,2-diphenylethane derivatives as inhibitors of retinoic acid-metabolising enzymes. J Enzym Inhib Med Ch. 2003;18(5):431–443. doi: 10.1080/1475636031000155427. [DOI] [PubMed] [Google Scholar]
- 179.Karlsson M, Strid A, Sirsjo A, Eriksson LA. Homology models and molecular modeling of human retinoic acid metabolizing enzymes cytochrome P450 26A1 (CYP26A1) and P450 26B1 (CYP26B1) J Chem Theory Comput. 2008;4(6):1021–1027. doi: 10.1021/ct800033x. [DOI] [PubMed] [Google Scholar]
- 180.Ren JH, Xiong XQ, Sha Y, Yan MC, Lin B, Wang J, Jing YK, Zhao DM, Cheng MS. Structure prediction and R115866 binding study of human CYP26A1: homology modelling, fold recognition, molecular docking and MD simulations. Mol Simulat. 2008;34:337–346. [Google Scholar]
- 181.Kang S, Duell EA, Kim KJ, Voorhees JJ. Liarozole inhibits human epidermal retinoic acid 4-hydroxylase activity and differentially augments human skin responses to retinoic acid and retinol in vivo. J Invest Dermatol. 1996;107(2):183–187. doi: 10.1111/1523-1747.ep12329579. [DOI] [PubMed] [Google Scholar]
- 182.Van Wauwe JP, Coene MC, Goossens J, Van Nijen G, Cools W, Lauwers W. Ketoconazole inhibits the in vitro and in vivo metabolism of all-trans-retinoic acid. J Pharmacol Exp Ther. 1988;245(2):718–722. [PubMed] [Google Scholar]
- 183.Saadeddin A, Torres-Molina F, Carcel-Trullols J, Araico A, Peris JE. Pharmacokinetics of the time-dependent elimination of all-trans-retinoic acid in rats. AAPS J. 2004;6(1):1–9. doi: 10.1208/ps060101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stearns ME, Wang M, Fudge K. Liarozole and 13-cis-retinoic acid anti-prostatic tumor activity. Cancer Res. 1993;53(13):3073–3077. [PubMed] [Google Scholar]
- 185.Achkar CC, Bentel JM, Boylan JF, Scher HI, Gudas LJ, Miller WH. Differences in the pharmacokinetic properties of orally administered all-trans-retinoic acid and 9-cis-retinoic acid in the plasma of nude mice. Drug Metab Dispos. 1994;22(3):451–458. [PubMed] [Google Scholar]
- 186.Dijkman GA, Van Moorselaar RJ, Van Ginckel R, Van Stratum P, Wouters L, Debruyne FM, Schalken JA, de Coster R. Antitumoral effects of liarozole in androgen-dependent and independent R3327-Dunning prostate adenocarcinomas. J Urol. 1994;151(1):217–222. doi: 10.1016/s0022-5347(17)34920-0. [DOI] [PubMed] [Google Scholar]
- 187.Smets G, Van Ginckel R, Daneels G, Moeremans M, Van Wauwe J, Coene MC, Ramaekers FC, Schalken JA, Borgers M, De Coster R. Liarozole, an antitumor drug, modulates cytokeratin expression in the Dunning AT-6sq prostatic carcinoma through in situ accumulation of all-trans-retinoic acid. Prostate. 1995;27(3):129–140. doi: 10.1002/pros.2990270303. [DOI] [PubMed] [Google Scholar]
- 188.Goss PE, Strasser-Weippl K, Qi S, Hu H. Effects of liarozole fumarate (R85246) in combination with tamoxifen on N-methyl-N-nitrosourea (MNU)-induced mammary carcinoma and uterus in the rat model. BMC Cancer. 2007;7:26. doi: 10.1186/1471-2407-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Godbole AM, Purushottamachar P, Martin MS, Daskalakis C, Njar VC. Autophagy inhibition synergistically enhances anticancer efficacy of RAMBA, VN/12-1 in SKBR-3 cells, and tumor xenografts. Mol Cancer Ther. 2012;11(4):898–908. doi: 10.1158/1535-7163.MCT-11-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Godbole AM, Purushottamachar P, Martin MS, Njar VC. Murine toxicology and pharmacokinetics evaluation of retinoic acid metabolism blocking agent (RAMBA), VN/12-1. Cancer Chemother Pharmacol. 2012 doi: 10.1007/s00280-012-1877-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Williams JB, Napoli JL. Inhibition of retinoic acid metabolism by imidazole antimycotics in F9 embryonal carcinoma cells. Biochem Pharmacol. 1987;36(8):1386–1388. doi: 10.1016/0006-2952(87)90102-x. [DOI] [PubMed] [Google Scholar]
- 192.Lansink M, Bennekum AMV, Blaner WS, Kooistra T. Differences in metabolism and isomerization of all-trans-retinoic acid and 9-cis-retinoic acid between human endothelial cells and hepatocytes. Eur J Biochem. 1997;247(2):596–604. doi: 10.1111/j.1432-1033.1997.00596.x. [DOI] [PubMed] [Google Scholar]
- 193.Lampen A, Meyer S, Arnhold T, Nau H. Metabolism of vitamin A and its active metabolite all-trans-retinoic acid in small intestinal enterocytes. J Pharmacol Exp Ther. 2000;295(3):979–985. [PubMed] [Google Scholar]
- 194.Cavazzini D, Catizone A, Galdieri M, Ottonello S. Vitamin A metabolism in cultured somatic cells from rat testis. Mol Cell Biochem. 2003;252(1):165–171. doi: 10.1023/a:1025575521060. [DOI] [PubMed] [Google Scholar]
- 195.Van heusden J, Borgers M, Ramaekers F, Xhonneux B, Wouters W, De Coster R, Smets G. Liarozole potentiates the all-trans-retinoic acid-induced structural remodelling in human breast carcinoma MCF-7 cells in vitro. Eur J Cell Biol. 1996;71(1):89–98. [PubMed] [Google Scholar]