

Abstract
Ewing sarcoma (ES) is rare in Japanese people, and only 30–40 patients develop the disease annually. To diagnose ES, molecular techniques that aim to detect characteristic fusion genes are commonly used in combination with conventional histological and immunohistochemical examinations. The treatment strategy for ES is characterized by multi-disciplinary collaboration between pediatric oncologists, medical oncologists, radiation oncologists, and orthopedic surgeons. In recent years, numerous large-scale national or international multi-institutional studies of ES have been performed. Pre- and postoperative intensive systemic chemotherapy with multiple anticancer drugs is the standard treatment method for ES. Depending on the obtained surgical margin, postoperative radiation might also be performed. If preoperative radiological examinations indicate that surgical excision would be difficult, preoperative radiation can be administered. As the treatment outcomes of ES have improved, late complications and secondary malignancies have become a problem. After treatment, patients with ES require very long-term follow-up in order to detect secondary malignancies and growth-related musculoskeletal complications.
Introduction
Ewing sarcoma (ES) is a highly malignant tumor composed of small round cells. The origin of this tumor was unclear until recently, when electron microscopic and immunohistochemical analyses suggested that it is of neurogenic origin [1–3]. ES tumors often express a balanced translocation involving the EWS gene on chromosome 22 and a member of the ETS family of transcription factors [4, 5]. With the development of diagnostic radiological techniques such as magnetic resonance imaging (MRI), extraskeletal masses can be depicted clearly and the tumor area can be accurately evaluated. Due to improvements in intensive chemotherapy, the prognosis of ES patients has improved markedly [6–16]. The current chemotherapy protocols used to treat ES include various combinations of the following six drugs: doxorubicin (DOX), cyclophosphamide (CPM), vincristine (VCR), actinomycin-D (ACT-D), ifosfamide (IFO), and etoposide (ETO). Local ES lesions are usually treated via surgical excision or radiotherapy, or a combination of both. In cases in which surgical excision is not possible due to the large size of the tumor, its anatomical location, or the fact that the acquired surgical margin is not sufficient to achieve local control, pre- or postoperative radiotherapy is usually selected [14]. Better understanding of how local control can be achieved has helped to improve the oncological outcomes of ES. Although the survival rate of ES patients has improved, their prognosis remains unsatisfactory, and the treatment of ES is still challenging to the medical teams involved, which include orthopedic surgeons, pediatric oncologists, and radiotherapists.
This review was undertaken to update our current knowledge regarding the diagnosis and treatment of ES. The author hopes that this article will help to summarize the latest findings about ES for treatment teams and lead to further improvements in the prognosis of ES.
History of ES
Lücke is considered to have been the first to describe this small round cell tumor [17, 18]. In 1918, Stout et al. [19] reported an undifferentiated round cell tumor that originated in the ulnar nerve and exhibited rosette formation, and he later proposed the concept of neuroepithelioma [20].
In 1921, Ewing reported a round cell sarcoma of the radius in a 14-year-old girl as a “diffuse endothelioma of bone” [21]. Radiotherapy produced a good response according to physical and plain X-ray examinations. In his report, Ewing reviewed six similar cases that he had experienced in the previous 4 months [21]. Ewing also documented the following specific radiographic findings: “A large portion or the whole of the shaft is involved, but the ends are generally spared. The shaft is slightly widened, but the main alteration is a gradual diffuse fading of the bone structure. Bone production is entirely absent”. Many of the features of ES described by Ewing in his original report are still very important, even after 90 years.
Angervall and Enzinger were the first to report the existence of ES of extraskeletal origin [22]. In 1976, Nesbitt and Vidone described a neuroepithelioma as a primitive neuroectodermal tumor (PNET) [23]. Since the latter report, the name PNET has been commonly used instead of neuroepithelioma. Askin et al. [24] described a small round cell tumor that originated in the thoracopulmonary region in 1979. In 1984, Jaffe et al. [1] reported a PNET of the bone composed of small round cells that exhibited rosette formation and were positive for the neural marker neuron-specific enolase.
In the 1980s, chromosome analysis detected the t(11;22)(q24;q12) translocation in ES cell lines [25, 26]. The EWS-FLI1 chimeric gene, which is produced by a balanced translocation between FLI1 on chromosome 11q24 and EWS on chromosome 22q12, was identified in 1992 after the cloning of translocation breakpoints [5]. As the abovementioned small round cell tumors; i.e., ES, PNET, neuroepithelioma, and Askin’s tumor, exhibit similar types of chromosomal translocation, e.g., t(11;22)(q24;q12), they are classified as a single disease entity; i.e., the ES family of tumors. In this review paper, the name ES is used for all of these tumors.
Characteristics of ES
ES is the second most common bone tumor in childhood and adolescence. In Japan, approximately 30–40 patients with ES of the bone are registered in the bone tumor registry run by the Japanese Orthopaedic Association (JOA) Musculoskeletal Tumor Committee annually [27]. Males are affected more frequently than females (ratio 3:2). The peak age of ES patients ranges from 10 to 24 years (Fig. 1).
Fig. 1.
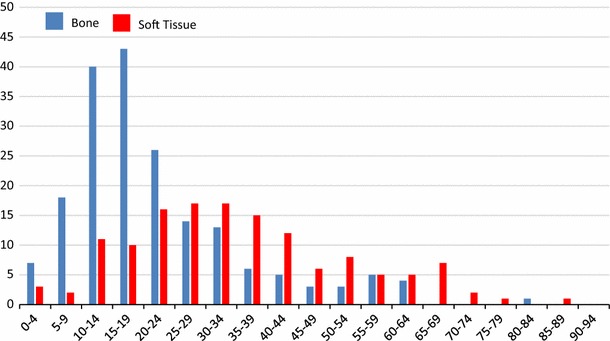
The age distribution of patients with ES of the bone (blue bar) and extraskeletal ES (red bar) [27, 28]
A total of 188 cases of ES of the bone were registered between 2006 and 2011 [27]. The sites that are most commonly affected by ES of the bone are the pelvis, ribs, and femur (Table 1). By comparison, 138 extraskeletal ES were registered in the same period, with 65 of these affecting males and 73 affecting females. The sites that were most frequently affected by extraskeletal ES were the thigh, gluteal region, back, lower leg, and the retroperitoneal region [28].
Table 1.
ES in Japan (2006–2011)
| Bone 188 | ||
| Cranium | 2 | Skull 1, mandible 1 |
| Trunk | 112 | Pelvis 43, ribs 29, spine 23, scapula 9, clavicle 6 and sternum 2 |
| Upper extremities | 13 | Humerus 10, ulna 2 and phalanges 1 |
| Lower extremities | 50 | Femur 25, tibia 10, fibula 10, tarsals 3 and metatarsals 2 |
| Multiple sites | 11 | |
| Soft tissue 138 | ||
| Head and neck | 12 | Head 5, neck 3, face 3 and maxilla 1 |
| Trunk | 68 | Gluteal region 11, back 10, retroperitoneal 9, lower back 8, inguinal region 8, thorax 7, pleural cavity 4, axilla 3, abdomen 3, shoulder 2, abdominal cavity 2 and perineal region 1 |
| Upper extremities | 12 | Upper arm 4, hand 4, elbow 2 and forearm 2 |
| Lower extremities | 41 | Thigh 30, lower leg 10 and foot 1 |
| Multiple sites | 5 | |
At the time of the diagnosis of ES of the bone, 34 % of patients had distant metastases and 5 % had regional lymph node metastases [27]. At the time of the diagnosis of extraskeletal ES, 29 % had distant metastases and 12 % had regional lymph node metastases [28].
The incidence of ES differs markedly among races [29]; i.e., ES is rare among black children [29–31] and the Chinese population [29]. Conversely, Caucasian children display a 6 times higher incidence of ES than black children [32]. The ratio of ES to osteosarcoma is around 0.5 among Caucasians in the United States and less than 0.15 among non-Caucasian children in Shanghai, Beijing, and the United States [29]. Among Japanese patients, the ratio of ES of the bone to osteosarcoma was 0.21 [27].
Symptoms and blood and serum findings
Patients with ES exhibit local symptoms such as tumor mass formation, induration, pain, swelling, venous dilation, and hyperemia. Pathological fractures sometimes occur due to bone metastasis, and spinal metastasis-associated back pain can progress to spinal paralysis. In cases in which an ES originates in the chest wall, pleural infiltration combined with carcinomatous pleurisy is often observed. The interval between the onset of the initial symptom and diagnosis has become shorter; i.e., it was 4.7 months in 2003, whereas it was 9.6 months in 1984 [33]. This change might have been due to improvements in diagnostic techniques.
Anemia and leukocytosis are often observed in ES, as are increases in the white blood cell count, blood sedimentation rate; and the serum levels of lactate dehydrogenase (LDH), alkaline phosphatase, and C-reactive protein. An elevated LDH level is associated with a poor prognosis [33]. Physicians must be aware that ES patients often exhibit similar blood and serum biochemical findings to those displayed by patients with inflammatory conditions such as osteomyelitis.
Radiological findings
In ES of the bone, plain radiographs exhibit permeative and infiltrative destruction of the affected bone (often in the diaphysis of the long bone) (Fig. 2). In addition, an onion skin-like appearance and spiculae are indicative of periosteal reactions. The pelvis is the most commonly affected site; however, it is difficult to identify pelvic tumors on plain X-rays alone. Computed tomography (CT) is useful for depicting extraskeletal soft tissue masses, destruction of the bone cortex, and pulmonary metastasis (Fig. 3). On MRI, ES of the bone exhibits low signal intensity on T1-weighted images and high signal intensity on T2-weighted images, and appears as large extraskeletal soft tissue masses derived from bone. Skip lesions affecting the bone are often clearly depicted on MRI. On bone scans, ES of the bone demonstrates high 99mTc-MDP uptake. Similarly, it displays high 18F-fluorodeoxy glucose (18F-FDG) uptake on 18F-FDG-positron emission tomography (PET), [34] (Fig. 4).
Fig. 2.
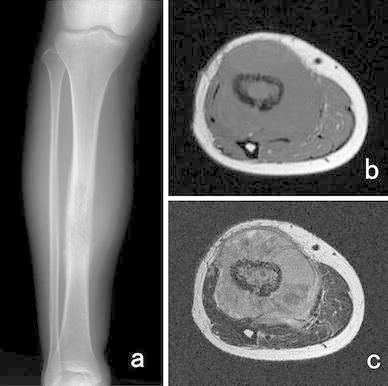
A 38-year-old woman with ES of the right tibia. a Plain X-ray showing permeative bone destruction. MRI detected a tumor that displayed b, low signal intensity and c, heterogeneous high signal intensity on T1- and T2-weighted imaging, respectively. The tumor was located both around and within the tibia. This case was presented by Dr Akira Kawai, Department of Musculoskeletal Oncology and Rehabilitation, National Cancer Center Hospital, Tokyo, Japan
Fig. 3.
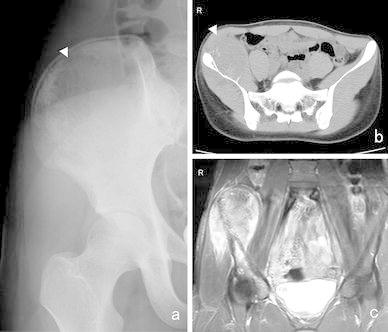
A 16-year-old patient with ES of the ilium. a A radiolucent lesion was visible in the right ilium on a plain X-ray (arrowhead). b CT detected a large mass that exhibited a periosteal reaction in the right ilium (arrowhead) and c, T2-weighted MRI depicted a mass that displayed heterogeneous high signal intensity in and around the right ilium and acetabulum
Fig. 4.
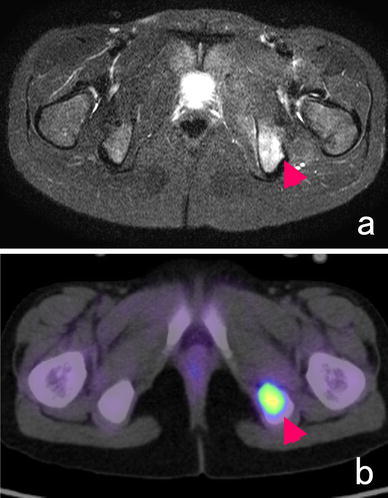
A 9-year-old girl with ES developed a metastasis in her left ischium. a T2-weighted MRI (T2-STIR) demonstrated an area of high signal intensity in the ischium, and 18F-FDG-PET detected high 18F-FDG uptake in the same region
From a radiological point of view, the differential diagnoses of ES include osteomyelitis, Langerhans cell histiocytosis, skeletal metastatic neuroblastoma, and malignant lymphoma of the bone.
Histology
Histologically, ES is composed of sheets of uniform small round tumor cells with round nuclei and little cytoplasm that do not exhibit matrix formation (Fig. 5a). ES tumor cells are characterized by uniform findings with few mitoses. ES also demonstrates morphological features that are indicative of neural differentiation, such as rosette formation. Since ES tumor cells contain abundant amounts of glycogen, periodic acid-Schiff (PAS)-positive granules are seen in their cytoplasm. Immunohistochemically, positivity for neural markers such as S-100 protein and PGP9.5 is sometimes detected, and vimentin positivity is also commonly observed. In over 90 % of ES cases, positivity for CD99, a product of the MIC2 gene, is detected on the cytoplasmic membrane (Fig. 6) [35]; however, CD99 expression is not specific to ES, as it is also detected in synovial sarcoma [36], lymphoblastic leukemia, lymphoma, and other tumors [4]. Atypical ES, a variant of ES composed of large cells, has also been reported [37, 38]. In addition, ES composed of spindle-shaped and adamantinoma-like cells, as well as other variants have also been reported [39].
Fig. 5.
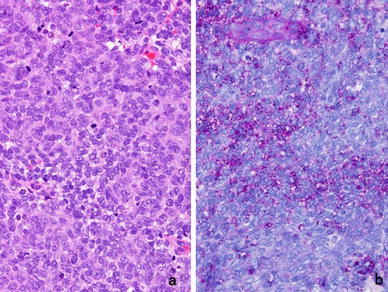
Histology of ES. a ES involves the sheet-like proliferation of small round cells with little cytoplasm (hematoxylin-eosin stain). b Periodic acid-Schiff staining detected small positive granules, which were considered to be glycogen molecules
Fig. 6.
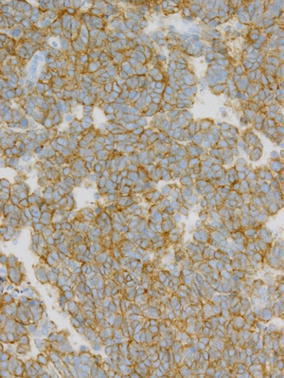
Membranous expression of CD99, a product of the MIC2 gene, is observed in ES cells
Fusion gene analysis
In recent years, molecular techniques have been commonly used to diagnose ES because it often exhibits specific chromosomal translocations. Reverse transcription polymerase chain reaction (RT-PCR) and fluorescent in situ hybridization (FISH) are very useful methods for detecting fusion genes (Fig. 7). Table 2 shows the most common chromosomal translocations found in ES. The EWS-FLI1 fusion gene, which is caused by the t(11;22)(q24;q12) translocation, is the most common type of fusion gene: 85 % of ES tumors that exhibit the EWS gene rearrangement were found to possess this type of translocation [4, 5, 26].
Fig. 7.
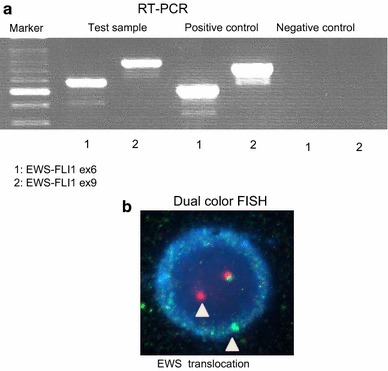
a RT-PCR. The left lane shows a marker, and the next three pairs of lanes (in a left-to-right direction) show the test samples, positive controls, and negative controls (1: EWS-FLI1 ex6, 2: EWS-FLI1 ex9), respectively. This figure shows that the test sample was positive for both EWS-FLI1 ex6 and EWS-FLI1 ex9, which means that the tumor possessed the EWS-FLI1 ex6 fusion gene. b FISH. An abnormal cell that has been hybridized with the Vysis LSI EWSR1 (22q12) dual color, break apart rearrangement probe. In a normal cell that lacks a t(22q12) translocation in the EWSR1 gene region, two fusion signals will be observed, reflecting the presence of two intact copies of EWSR1. The cell in this image shows one fusion, one orange, and one green signal, which is indicative of the rearrangement of one copy of the EWSR1 gene region
Table 2.
Most common types of translocations found in Ewing sarcoma
| Translocation | Fusion gene | % of tumors exhibiting EWS gene rearrangement |
|---|---|---|
| t(11;22)(q24;q12) | EWSR1-FLI1 | 85 |
| t(21;22)(q22;q12) | EWSR1-ERG | 10 |
| t(7;22)(q22;q12) | EWSR1-ETV1 | rare |
| t(17;22)(q21;q12) | EWSR1-ETV4 | rare |
| t(2;22)(q35;q12) | EWSR1-FEV | rare |
EWS-ERG, which is caused by the t(21;22)(q22;q12) translocation, is observed in 10 % of cases [40, 41]. Other rare fusion genes include: EWS-ETV1, which is caused by the t(7;22)(q22;q12) translocation [42]; EWS-ETV4, which is caused by the t(17;22)(q21;q12) translocation [43, 44]; and EWS-FEV, which is caused by the t(2;22)(q35;q12) translocation [45]. These fusion products encode chimeric proteins that function as aberrant oncogenic transcription factors. Other cytogenetic abnormalities have also been reported [33, 46]. Subjecting bone marrow aspiration samples to RT-PCR might be useful for detecting fusion genes in the bone marrow of ES patients. Kunisada et al. [47] reported that 2 of 5 patients without radiologically detectable metastasis (M0) possessed fusion genes in their bone marrow.
The most common fusion gene involves the in frame fusion of exon 7 of the EWS gene with exon 6 of the FLI1 gene (the type 1 fusion gene). When the prognosis of 147 ES patients was examined according to fusion gene type in the Cooperative ES Study (CESS), patients with the type 1 fusion gene were reported to have a better prognosis than patients with other types of fusion gene [48]. In addition, in a study by the Memorial Sloan Kettering Cancer Center the 64 patients with the type 1 fusion gene exhibited a better prognosis than the 35 patients with other types of fusion gene [49]. However, in 1999 Ginsberg examined 136 cases of ES and found that patients with the EWS-FLI1 and EWS-ERG fusion genes had similar clinical courses [50]. In a recent prospective study (the European Ewing Tumor Working Initiative National Groups 99 study; the Euro-EWING 99 study), which examined 565 patients with primary ES tumors (1999–2007), including 296 patients with type 1 fusion genes and 269 patients with other types of fusion genes, a prospective evaluation did not find that the type 1 fusion gene confers a prognostic benefit [51]. In ES, the accumulation of genetic aberrations involving several genes on different chromosomes seems to have a greater influence than fusion gene type on the biological and clinical behavior of ES.
Treatment
Radiotherapy had been the standard treatment for ES, and until the 1960s radiotherapy or surgery were the only treatments for ES [15]. ES is currently treated in a multidisciplinary manner involving chemotherapy, surgery, and radiotherapy. If a tumor is considered to be difficult to excise with an adequate margin, preoperative radiotherapy involving 36–55 Gy is administered. Similarly, postoperative radiotherapy is performed if the margin achieved by surgery is inadequate for local control.
According to the bone tumor registry developed by the JOA [27], 173 of 188 patients with ES of the bone received treatments including chemotherapy, including 65 patients who underwent chemotherapy and surgery; 50 patients who received chemotherapy and radiotherapy; and 32 patients who were treated with chemotherapy, surgery, and radiotherapy (Fig. 8).
Fig. 8.
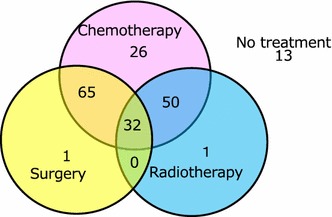
Treatments employed in 188 cases of ES (2006–2011). The data shown are derived from the Bone Tumor Registry in Japan, 2010, run by the JOA Musculoskeletal Tumor Committee
In the 1960s, a maximum of 10 % of ES patients survived [15], and VCR (a vinca alkaloid), ACT-D (an antitumor antibiotic), CPM (an alkylating agent), and DOX (an antitumor antibiotic) started to be included in treatment protocols for ES. After the 1980s, IFO (an alkylating agent) and ETO (a vinca alkaloid) were added to such protocols [15]. Currently, the 5-year overall survival rate of ES patients is approximately 60 % [10, 11, 14, 50, 52].
Chemotherapy
The purpose of preoperative chemotherapy is (1) to eradicate any micrometastases that exist at the time of diagnosis, (2) to reduce the volume of the tumor in order to facilitate its excision, and (3) to provide information that will aid the selection of anti-tumor drugs for postoperative chemotherapy. In the USA, the Memorial Sloan-Kettering Cancer Center has developed a few treatment protocols for ES [7]. In addition, Rosen et al. [7] described an effective chemotherapy regimen called T-11.
IESS-1 (the Intergroup ES Study), which included 331 patients, compared the prognosis of patients that were treated with the VAC regimen (VCR, ACT-D, and CPM) and that of patients that were administered the VAC + DOX (VACD) regimen [8] (Table 3). As a result, it was found that the addition of DOX had a beneficial effect on patient survival. Patients treated with the VAC regimen alone, the VAC regimen and whole lung irradiation, or the VACD regimen alone exhibited 5-year disease-free survival rates of 24, 44, and 60 %, respectively. IESS-II, which included 293 patients, reported that patients that were treated with an intensified VACD regimen displayed a 5-year disease-free survival rate of 68 %, whereas that of the patients treated with a moderate dose VACD regimen was 48 % [9]. NCI INT-0091 (1988–1992) was a prospective randomized trial of a single VDC-containing regimen (VCR, DOX, and CPM) versus the administration of VDC and the IE regimen (IFO + ETO) [10]. The 5-year disease-free survival rate was 54 % in the VDC group and 69 % in the VDC + IE group; thus, VDC + IE had a significantly positive effective on patient survival in localized cases compared with VDC. The main concept behind this regimen was adopted by the Japanese Ewing Sarcoma Study (JESS) 04 protocol.
Table 3.
Studies of Ewing sarcoma conducted in the USA
| Study | Author | No. of patients | Chemotherapy | 5-year relapse-free survival (%) |
|---|---|---|---|---|
| IESS-I | Nesbit et al. [8] | 331 | VAC | 24 |
| VAC + whole lung RT | 44 | |||
| VACD | 60 | |||
| IESS-II | Burgert et al. [9] | 293 | VACD (intensified) | 68 |
| VACD (moderate) | 48 | |||
| NCI INT-0091 | Grier et al. [10] | 200 | VDC | 54 |
| 198 | VDC + IE | 69 | ||
| COG AEWS0031 | Womer et al. [11] | 587 | VDC + IE 3-week interval | 65 (5 year-EFS) |
| VDC + IE 2-week interval | 73 (5 year-EFS) |
VAC: VCR + ACT-D + CPM
VACD: VAC + DOX
VDC: VCR + DOX + CPM
IE: IFO + ETO
In recent years, the results of the AEWS0031 study performed by the Children’s Oncology Group, which included 587 patients, were reported [11]. The latter study prospectively compared the effects of VDC + IE between regimens involving administration intervals of 3 and 2 weeks. The 5-year disease-free survival rate was 65 % in the 3-week interval group and 73 % in the 2-week interval group (p = 0.048), and there was no difference in the incidence of toxicities between the 2 groups. At present, the administration of VDC + IE using a short administration interval is regarded as the standard chemotherapy regimen for treating ES in the USA.
In the CESS, a large-scale European study, 81 of 93 patients were treated with the VACD regimen, and the 5-year survival rate was 55 % (Table 4). The 5-year survival rate was 80 % in cases of tumor volume of <100 ml and was 31 % in cases of tumor volume ≥100 ml [12]. In the CESS-86 study, which included 301 patients, the VACD regimen was used to treat patients with tumor volumes of <100 ml, and the VAID regimen, in which the CPM included in the VACD regimen was replaced with IFO, was administered to patients with tumor volumes of ≥100 ml [13]. The 10-year disease-free survival rate was 52 % in the standard risk group treated with VACD and 51 % in the high risk group administered VAID. Thus, the VAID regimen was effective at increasing the survival of high risk patients. In the EICESS-92 study, which included 647 patients, patients with tumor volumes of <100 ml were randomly assigned to receive either the VAID or VACD regimen, and patients with tumor volumes of ≥100 ml with/without metastasis (including skip metastasis) were randomly assigned to receive either the VAID or EVAID regimen [14]. In the standard risk group, the 5-year disease-free survival rate was 68 % in the VAID group and 67 % in the VACD group. Therefore, in the standard risk group CPM had a similar effect on event-free survival to IFO; however, it was also associated with increased toxicity. In the high risk group, the 5-year disease-free survival rate was 44 % in the VAID group, and adding ETO to the VAID regimen resulted in an increase in the survival rate to 52 % in the EVAID group. Therefore, it was confirmed that ETO had a beneficial effect among high risk patients. In recent years, the Euro-EWING 99 study examined the utility of induction treatment with the VIDE regimen and one postoperative cycle of the VAI regimen [16]. Patients with localized disease or lung metastasis were classified into the R1 or R2 arm. The patients in the R1 group were randomly assigned to receive the VAC or VAI regimen, and the patients in the R2 group were randomly assigned to receive the VAI regimen or busulfan and melphalan after one postoperative cycle of VAI. In the R1 group, which contained 856 patients, it was found that there was no difference in survival between the VAC and VAI groups.
Table 4.
Studies of Ewing sarcoma conducted in Europe
| Study | Author | No. of patients | Protocol | Disease-free survival (%) |
|---|---|---|---|---|
| CESS81 | Jürgens et al. [12] | 93 | VACD | 80 (volume <100 ml) (5-year) |
| 31 (volume ≥100 ml) (5-year) | ||||
| CESS86 | Paulussen et al. [13] | 301 | SR: VACD | 52 (10-year) |
| HR: VAID | 51 (10-year) | |||
| EICESS92 | Paulussen et al. [14] | 647 | SR: VAID/VACD | 68/67 (5-year) |
| HR: VAID/EVAID | 44/52 (5-year) | |||
| EURO-EWING99 | Le Deley et al. [16] | 856 | Randomization in SR (R1 arm) | |
| 6 VIDE + 1 VAI | ||||
| + 7VAC(n = 431) or | 75 (3-year) | |||
| +7VAI (n = 425) | 78 (3-year) |
SR Standard risk, HR high risk (definitions are provided in the literature)
VACD: VAC (VCR + ACT-D + CPM) + DOX
VAID: VAI (VCR + ACT-D + IFO) + DOX
EVAID: VAID + ETO
VIDE: VCR + IFO + DOX + ETO
On the other hand, Obata et al. [52] conducted a Japanese study involving 243 patients from 29 institutions who exhibited a mean follow-up period of 66 months. Of the 243 patients, 183 underwent definitive surgery as the primary local treatment. The treatment protocols included many different regimens such as the T-11 and modified T-11 regimens in 47 patients, V(A)CD + IE-based regimens in 41 patients [10, 53, 54], the VAID/EVAID regimen in 22/3 patients [13, 14], and VACD-based regimens in 22 patients [9, 12], etc. In addition, high-dose chemotherapy with hematopoietic stem cell rescue was performed in 51 patients. In patients with localized disease (M0), the 5-year overall survival rate was 55 %, and the event-free survival rate was 47 %. In patients with metastatic disease (M1), the 5-year overall survival rate was 13 %, and the event-free survival rate was 7 %. The following characteristics were found to be associated with a poor prognosis: age ≥16 years, M1 disease, and a tumor located in the trunk [52]. The Japanese ES Study (JESS) group has not published the final results of the JESS04 study; however, satisfactory results have been obtained in similarly large studies conducted in the USA and Europe.
Intensive treatment
Meyers et al. [55] reported the results of intensive treatment in 32 patients with bone or bone marrow metastasis from ES . If a good response was obtained after 5 cycles of induction chemotherapy composed of a 3-drug regimen (CPM, DOX, and VCR) alone or the same 3-drug regimen combined with an additional 2-drug regimen (IFO and ETO), consolidation therapy including high-dose melphalan, ETO, and whole body irradiation was performed followed by stem-cell support. The event-free 2-year survival rate of all 32 cases was 20 %, and that of the 23 cases that received stem-cell support was 24 %; i.e., the latter regimen failed to improve the survival of high risk patients.
The treatment results for primary disseminated multifocal ES, excluding single pulmonary metastases that were present at diagnosis, were also reported in the Euro-EWING 99 trial [56]. In this group, 6 cycles of the VIDE regimen were administered preoperatively and then local therapy involving surgery and/or radiotherapy was performed. After one postoperative cycle of the VAI regimen had been administered, high-dose busulfan-melphalan was administered followed by an autologous stem cell transplantation (HDT/SCT). The median follow-up period was 3.8 years, and the 3-year disease-free survival rate was 27 %.
Patients with pulmonary metastasis are administered chemotherapy based on the first-line regimens for patients without metastasis. Single pulmonary metastases can be controlled with intensive chemotherapy; however, the prognosis of patients with bone or bone marrow metastasis remains poor.
Local treatment: radiotherapy and tumor excision
As ES is sensitive to radiotherapy, radiotherapy was performed as a local treatment before the introduction of chemotherapy. It was reported that ES is usually treated with radiation doses ranging from 36 to 60 Gy [12, 14, 57]. Two irradiation techniques are commonly used to treat ES. Definitive radiotherapy aims to destroy all viable tumor cells and is used to treat centrally located lesions, such as those located in the spinal column (Fig. 9). On the other hand, pre- or postoperative adjuvant radiotherapy is administered in cases in which the risk of local relapse is considered to be high, e.g., because an insufficient margin was acquired or it is considered that a safe margin would be difficult to obtain. In the CESS86 study [57], 46 Gy were delivered to patients with positive surgical margins, and 60 Gy were delivered to the non-surgical group. There was no significant difference in the treatment results of the two groups; thus, 60 Gy successfully obtained local control of ES.
Fig. 9.
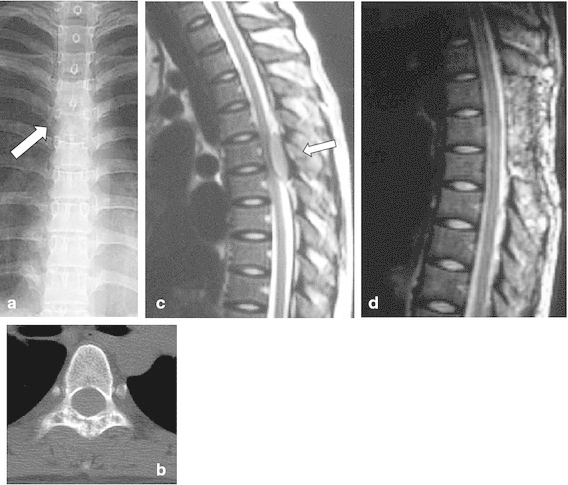
An 18-year-old girl had a 1-month history of back pain without any obvious cause and experienced muscle weakness in both legs The patient suffered numbness and urinary disturbances. a Plain X-ray showing the loss of spinous processes at the T5 level (arrow). b CT detected a spotted osteolytic lesion in the lamina of the T5 vertebra. c T2-weighted MRI showed a mass that exhibited iso-signal intensity and was compressing the spinal cord from the posterior direction (arrow). d Emergent decompression of the T4-6 vertebrae was performed to rescue the spinal cord from compression. Preoperative chemotherapy composed of VDC + IE was performed, 45 Gy radiation was administered as a local treatment, and VDC-IE was administered as a post-local therapy. The patient became completely free from spinal palsy and was able to walk without any support. She has been disease-free for 6 years after the local treatment
Several studies have detected higher local relapse rates after radiotherapy alone than after combined therapy involving surgery and radiotherapy. In the CESS studies, the local control rate obtained in the patients who received definitive radiotherapy was significantly worse than that achieved in the patients who underwent surgery with/without additional radiotherapy [58]. Bacci et al. [59] reported that the local control rate of ES was 100 % in patients who underwent surgery and radiotherapy and 67 % in patients that were treated with radiotherapy alone. Wilkins et al. [60] found that the 5-year survival rate of patients that received radiotherapy alone as a local treatment was 27 %, whereas that of the patients treated with radiation and surgery was 72 %. In patients with ES, surgery improves the safety of local control. As for safe surgical margins, it has been suggested that tumors must be excised together with 2 cm of normal tissue [61]. In other sarcomas, membranous structures around the tumor can function as a barrier against tumor extension; however, the barrier function of such structures seems to be insufficient to prevent growth in ES.
In 2003, based on the results of the CESS and EICESS trials, Schuck et al. [62] reported that postoperative radiotherapy improves local control in poor responders to chemotherapy, even after wide resection . Currently, the standard treatment strategy for surgically accessible primary lesions is to surgically resect the tumor as much as possible together with a safe margin, although radiotherapy might be selected depending on the surgical margin and the histological effects of chemotherapy. If a tumor is inoperable, radiotherapy alone is employed as a local treatment. After radiotherapy, it might be possible to surgically excise previously inoperable tumors together with a safe margin.
The author thinks that radiotherapy is not necessary after adequate wide excision in cases involving patients that respond well to preoperative chemotherapy. However, the author considers that radiotherapy should be added after inadequate excision and in cases involving poor responders to chemotherapy or excessively narrow surgical margins.
Among patients who exhibited pulmonary metastasis at diagnosis, the patients who received total lung irradiation were found to have a better prognosis than those who did not undergo such treatment [63, 64]. A dose of 14–18 Gy is recommended for total lung irradiation [63]. However, clinicians should be aware that more than 50 % of ES patients have functional lung abnormalities [64].
In recent years, carbon ion beam treatment has prevailed in Japan [65], and it is often used to achieve local control of surgically inaccessible tumors due to anatomical reasons. However, when a tumor can be surgically excised without causing a reduction in function or quality of life, surgery should be selected.
Reconstructive surgery
It is very important to prevent surgical site infections in ES patients: if an infection occurs, postoperative chemotherapy must be postponed until the infection has been completely eradicated. In recent years, the dose intensity of chemotherapy has been increasing, which could lead to an increase in the infection rate. Orthopedic surgeons should not select reconstruction methods that are associated with high infection rates. In recent years, liquid nitrogen-treated recycled bone has been developed and has since been used frequently [66]. This method is reported to induce a cryo-immunological response [67] so it might be expected to result in a higher survival rate than other reconstruction methods.
The pelvis is the most frequent site for ES and patients with pelvic ES have a poor prognosis [68]. Fibula strut grafts are available for pelvic reconstruction after iliac excision (P1). Non-vascularized fibula grafts seem to be sufficient for pelvic reconstruction because bone union can be expected to occur within a few months after their implantation [69]. The author is currently examining the utility of hip joint transposition in cases involving large tumors of the iliac wing that affect the acetabulum [70, 71], and so far no postoperative surgical site infections have occurred in any patient. In a study of cases in which local control of pelvic ES was obtained using internal hemipelvectomy, the patients reported that they had a good postoperative quality of life [72].
The prompt administration of intensive chemotherapy is important in ES. Most ES patients are aged between 10 and 20 years of age. Although the reconstruction of skeletal defects using tumor prostheses might be an established treatment after tumor excision, if available biological reconstruction methods seem to be more suitable than tumor prostheses for cases involving children.
Metastasis and secondary malignancies
A total of 34 % of ES patients exhibit metastasis at diagnosis [27]. Patients with lung metastasis alone, display a better prognosis than patients with bone or bone marrow metastases. As the survival rate of ES has increased, cases of late relapse, e.g., relapses that occur more than 10 years after treatment, are being reported (Fig. 10). No standard treatment strategy for relapsed ES has been established. There have been several trials of the ICE regimen (IFO, carboplatin, and ETO) [73], topotecan + CPM [74], and the irinotecan (CPT-11) plus temozolomide regimen [75].
Fig. 10.
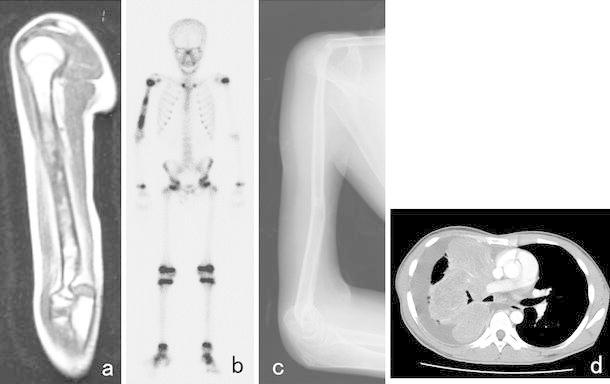
A 10-year-old boy with ES of the right humerus. He had a pain in his right upper arm. A plain X-ray showed a limited periosteal reaction. a T1-weighted MRI depicted an area of heterogeneous low signal intensity in the humerus. b A bone scan showed abnormally high isotope uptake in the right humerus. After the diagnosis of ES had been confirmed by open biopsy, preoperative chemotherapy composed of DOX, CPM, methotrexate, VCR, IFO, and ACT-D was administered. c After the preoperative chemotherapy, the patient underwent wide excision of the tumor in his right humerus. A sling procedure involving a vascularized fibula graft was performed for reconstruction, and postoperative chemotherapy was initiated. At 13 years after surgery, he visited our hospital due to dyspnea and coughing. A chest X-ray showed a white region in his right lung, and d CT depicted a large amount of pleural effusion and atelectasis due to the pulmonary metastasis of ES
In the EICESS92 study, which examined 647 cases, the frequency of secondary malignancies was 0.9 % among all cases and 1.2 % among patients that were with treated with ETO. On the other hand, none of the patients that were treated without ETO developed secondary malignancies [76]. In the Italian Sarcoma Group Experience study (1983–2006), which involved 543 ES patients that exhibited a median follow-up period of 86 months, secondary malignancies developed in 15 patients (3 %). Among the latter patients, the median interval between the diagnosis of the primary and secondary malignancies was 84 months. In another study, the cumulative frequency of secondary malignancies was 3.4 % after 10 years, 4.7 % after 20 years, and 5 % after 25 years [77]. In the follow-up period, tumor relapses, secondary malignancies, and late complications such as leg length discrepancies or scoliosis after thoracic surgery or radiation can all have important impacts on survival and quality of life (Fig. 11).
Fig. 11.
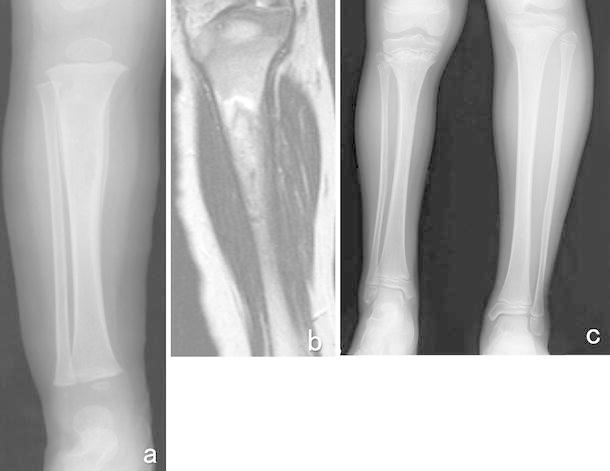
A 1-year-old boy suddenly suffered a pain in his right lower leg without any obvious cause. a A plain X-ray showed a radiolucent lesion and widening of the diaphysis of his right tibia. A periosteal reaction was also observed. b T2-weighted imaging demonstrated an area of high signal intensity in the tibia. An open biopsy resulted in a diagnosis of ES of the tibia. After preoperative chemotherapy composed of 6 courses of VIDE according to the protocol outlined in the Euro-EWING 99 study, radiotherapy involving 55.8 Gy was performed as a local therapy. After the radiotherapy, one course of the VAI regimen and 7 courses of the VAC regimen were administered. When the patient was 9.5 years old, CT detected lymph node swelling in his neck. A biopsy of the lymph node demonstrated metastasis from papillary thyroid cancer. The patient underwent excision of his right thyroid lobe and the swollen lymph node. c He displayed a 5-cm leg length discrepancy at 10 years after the local therapy
Further improving the cure rate
Increasing the dose intensity of anti-tumor drugs, e.g., via their bi-weekly administration, might lead to improvements in patient prognosis. New drugs such as pazopanib [78], eribulin [79], and trabectedin [80, 81] are becoming available and might lead to further increases in patient survival. Of course, controlling treatment complications and the early detection and prevention of secondary malignancies are mandatory. In Japan, single institutional trials are not sufficient for producing scientific evidence, as they would involve too few patients; however, the multi-institutional study being conducted by the JESS group is expected to produce high quality data.
Summary
The prognosis of ES patients has improved in recent years. To diagnose ES, molecular techniques that aim to detect characteristic fusion genes are commonly used in combination with conventional histological and immunohistochemical techniques. As for chemotherapy, several new lines of evidence have been obtained in large-scale studies conducted in Europe and North America. The use of multidisciplinary treatment teams composed of specialists from various fields such as pediatric oncologists, medical oncologists, radiation oncologists, and orthopedic surgeons is necessary to further improve patient prognosis. After treatment, ES patients must be carefully followed up for tumor relapses, growth-related musculoskeletal complications, and secondary malignancies.
Acknowledgments
The author thanks DR. Hiroyuki Yanai for his comments about the pathology of ES and Ms. Eri Sakaguchi for preparing this manuscript. The author is deeply grateful to the members of the tumor group of the Department of Orthopaedic Surgery, Okayama University Hospital (Dr. Toshiyuki Kunisada, Dr. Ken Takeda, Dr. Tomohiro Fujiwara, Dr. Joe Hasei, and Ms. Aki Yoshida), for their help during the preparation of this manuscript.
This study was supported in part by the Health and Labor Sciences Research Expenses Commission; an Applied Research for Innovative Treatment of Cancer grant from the Ministry of Health, Labor, and Welfare (H26-084, H26-062); and a Grant-in-Aid for Scientific Research (B) from The Ministry of Education, Culture, Sports, Science and Technology (MEXT) (Grant Number 25293323)
Conflict of interest
The author declares that he has no conflict of interest.
References
- 1.Jaffe R, Santamaria M, Yunis EJ, Tannery NH, Agostini RM, Jr, Medina J, Goodman M. The neuroectodermal tumor of bone. Am J Surg Pathol. 1984;8(12):885–898. doi: 10.1097/00000478-198412000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Carlei F, Polak JM, Ceccamea A, Marangos PJ, Dahl D, Cocchia D, Michetti F, Lezoche E, Speranza V. Neuronal and glial markers in tumours of neuroblastic origin. Virchows Arch A Pathol Anat Histopathol. 1984;404(3):313–324. doi: 10.1007/BF00694896. [DOI] [PubMed] [Google Scholar]
- 3.Hashimoto H, Enjoji M, Nakajima T, Kiryu H, Daimaru Y. Malignant neuroepithelioma (peripheral neuroblastoma). A clinicopathologic study of 15 cases. Am J Surg Pathol. 1983;7(4):309–318. doi: 10.1097/00000478-198306000-00002. [DOI] [PubMed] [Google Scholar]
- 4.de Alava E, Lessnick SL, Sorensen PH. Ewing sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumours of soft tissue and bone. Lyon: WHO Press; 2013. pp. 305–309. [Google Scholar]
- 5.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, Thomas G. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359(6391):162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 6.Rosen G, Juergens H, Caparros B, Nirenberg A, Huvos AG, Marcove RC. Combination chemotherapy (T-6) in the multidisciplinary treatment of Ewing's sarcoma. Natl Cancer Inst Monogr. 1981;(56):289–99. [PubMed]
- 7.Rosen G. Current management of Ewing’s sarcoma. Prog Clin Cancer. 1982:267–82. [PubMed]
- 8.Nesbit ME, Jr, Gehan EA, Burgert EO, Jr, Vietti TJ, Cangir A, Tefft M, Evans R, Thomas P, Askin FB, Kissane JM, Pritchard DJ, Herrmann J, Neff J, Makley JT, Gilula L. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol. 1990;8(10):1664–1674. doi: 10.1200/JCO.1990.8.10.1664. [DOI] [PubMed] [Google Scholar]
- 9.Burgert EO, Jr, Nesbit ME, Garnsey LA, Gehan EA, Herrmann J, Vietti TJ, Cangir A, Tefft M, Evans R, Thomas P. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone: intergroup study IESS-II. J Clin Oncol. 1990;8(9):1514–1524. doi: 10.1200/JCO.1990.8.9.1514. [DOI] [PubMed] [Google Scholar]
- 10.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers PA, Donaldson SS, Moore S, Rausen AR, Vietti TJ, Miser JS. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 11.Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, Marcus K, Sailer S, Healey JH, Dormans JP, Weiss AR. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Childrenʼs Oncology Group. J Clin Oncol. 2012;30(33):4148–4154. doi: 10.1200/JCO.2011.41.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jürgens H, Exner U, Gadner H, Harms D, Michaelis J, Sauer R, Treuner J, Voute T, Winkelmann W, Winkler K, Göbel U. Multidisciplinary treatment of primary Ewing’s sarcoma of bone. A 6-year experience of a European Cooperative Trial. Cancer. 1988;61(1):23–32. doi: 10.1002/1097-0142(19880101)61:1<23::AID-CNCR2820610106>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 13.Paulussen M, Ahrens S, Dunst J, Winkelmann W, Exner GU, Kotz R, Amann G, Dockhorn-Dworniczak B, Harms D, Muller-Weihrich S, Welte K, Kornhuber B, Janka-Schaub G, Gobel U, Treuner J, Voute PA, Zoubek A, Gadner H, Jürgens H. Localized Ewing tumor of bone: final results of the cooperative Ewing’s Sarcoma Study CESS 86. J Clin Oncol. 2001;19(6):1818–1829. doi: 10.1200/JCO.2001.19.6.1818. [DOI] [PubMed] [Google Scholar]
- 14.Paulussen M, Craft AW, Lewis I, Hackshaw A, Douglas C, Dunst J, Schuck A, Winkelmann W, Kohler G, Poremba C, Zoubek A, Ladenstein R, van den Berg H, Hunold A, Cassoni A, Spooner D, Grimer R, Whelan J, McTiernan A, Jürgens H. European Intergroup Cooperative Ewing’s Sarcoma S. Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment—cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol. 2008;26(27):4385–4393. doi: 10.1200/JCO.2008.16.5720. [DOI] [PubMed] [Google Scholar]
- 15.Cripe TP. Ewing sarcoma: an eponym window to history. Sarcoma. 2011;2011:457532. doi: 10.1155/2011/457532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Deley MC, Paulussen M, Lewis I, Brennan B, Ranft A, Whelan J, Le Teuff G, Michon J, Ladenstein R, Marec-Berard P, van den Berg H, Hjorth L, Wheatley K, Judson I, Juergens H, Craft A, Oberlin O, Dirksen U. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol. 2014;32(23):2440–2448. doi: 10.1200/JCO.2013.54.4833. [DOI] [PubMed] [Google Scholar]
- 17.Lücke A. Beiträge zur Geschwulstlehre. Virchows Arch Pathol Anat. 1866;35:524–539. doi: 10.1007/BF01960751. [DOI] [Google Scholar]
- 18.Huvos AG. Ewing’s sarcoma. In: Huvos AG, editor. Bone tumor diagnosis, treatment, and prognosis. Philadelphia: WB Saunders; 1991. pp. 523–552. [Google Scholar]
- 19.Stou AP. A tumor of the ulnar nerve. Proc NY Pathol Soc. 1918;18:2–12. [Google Scholar]
- 20.Stou AP, Murray MD. Neuroephithelioma of the radial nerve with a study of its behavior in vitro. Rev Can Biol. 1942;1:651–659. [Google Scholar]
- 21.Ewing J. Diffuse endothelioma of bone. Proc NY Path Soc. 1921;21:17–24. [Google Scholar]
- 22.Angervall L, Enzinger FM. Extraskeletal neoplasm resembling Ewing’s sarcoma. Cancer. 1975;36(1):240–251. doi: 10.1002/1097-0142(197507)36:1<240::AID-CNCR2820360127>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Nesbitt KA, Vidone RA. Primitive neuroectodermal tumor (neuroblastoma) arising in sciatic nerve of a child. Cancer. 1976;37(3):1562–1570. doi: 10.1002/1097-0142(197603)37:3<1562::AID-CNCR2820370346>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 24.Askin FB, Rosai J, Sibley RK, Dehner LP, McAlister WH. Malignant small cell tumor of the thoracopulmonary region in childhood: a distinctive clinicopathologic entity of uncertain histogenesis. Cancer. 1979;43(6):2438–2451. doi: 10.1002/1097-0142(197906)43:6<2438::AID-CNCR2820430640>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir G. Chromosomal translocation (11;22) in cell lines of Ewing’s sarcoma. C R Seances Acad Sci III. 1983;296(23):1101–1103. [PubMed] [Google Scholar]
- 26.Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, Volk C, Thiery JP, Olschwang S, Philip I, Berger MP, Philip T, Lenoir GM, Mazabraud A. Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12) Cancer Genet Cytogenet. 1988;32(2):229–238. doi: 10.1016/0165-4608(88)90285-3. [DOI] [PubMed] [Google Scholar]
- 27.JOA Musculoskeletal Tumor Committee. Bone Tumor Registry in Japan. 2011.
- 28.JOA Musculoskeletal Tumor Committee. Soft Tissue Tumor Registry in Japan. 2011.
- 29.Li FP, Tu JT, Liu FS, Shiang EL. Rarity of Ewing’s sarcoma in China. Lancet. 1980;1(8180):1255. doi: 10.1016/S0140-6736(80)91719-5. [DOI] [PubMed] [Google Scholar]
- 30.Jensen RD, Drake RM. Rarity of Ewing’s tumour in negroes. Lancet. 1970;1(7650):777. doi: 10.1016/S0140-6736(70)91002-0. [DOI] [PubMed] [Google Scholar]
- 31.Fraumeni JF, Jr, Glass AG. Rarity of Ewing’s sarcoma among US negro children. Lancet. 1970;1(7642):366–367. doi: 10.1016/S0140-6736(70)90754-3. [DOI] [PubMed] [Google Scholar]
- 32.Gurney JG, Swensen AR, Bulterys M. Malignant bone tumors. In: Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, editors. Cancer incidence and survival among children and adolescents: United States SEER Program 1975–1995. Bethesda: National Cancer Institute, SEER Program; 1999. NIH Pub No 99-4649.
- 33.Patterson FR, Basra SK. Ewing’s sarcoma. In: Schwartz HS, editor. Orthopaedic knowledge update: musculoskeletal tumors 2. Rosemont: American Academy of Orthopaedic Surgeons; 2007. pp. 175–183. [Google Scholar]
- 34.Gyorke T, Zajic T, Lange A, Schafer O, Moser E, Mako E, Brink I. Impact of FDG PET for staging of Ewing sarcomas and primitive neuroectodermal tumours. Nucl Med Commun. 2006;27(1):17–24. doi: 10.1097/01.mnm.0000186608.12895.69. [DOI] [PubMed] [Google Scholar]
- 35.Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67(7):1886–1893. doi: 10.1002/1097-0142(19910401)67:7<1886::AID-CNCR2820670712>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 36.Olsen SH, Thomas DG, Lucas DR. Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol. 2006;19(5):659–668. doi: 10.1038/modpathol.3800569. [DOI] [PubMed] [Google Scholar]
- 37.Nascimento AG, Unii KK, Pritchard DJ, Cooper KL, Dahlin DC. A clinicopathologic study of 20 cases of large-cell (atypical) Ewing’s sarcoma of bone. Am J Surg Pathol. 1980;4(1):29–36. doi: 10.1097/00000478-198004010-00003. [DOI] [PubMed] [Google Scholar]
- 38.Ozaki T, Nakagawa Y, Yoshida A, Numoto K, Sugihara S, Kunisada T, Hamazaki S, Inoue H. Amplification of MYCL in atypical Ewing tumor analysis of metaphase and microarray comparative genomic hybridization. Cancer Genomic Proteomic. 2004;1:275–282. [PubMed] [Google Scholar]
- 39.Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, Dei Tos AP, Weiss SW. Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol. 2005;29(8):1025–1033. [PubMed] [Google Scholar]
- 40.Giovannini M, Biegel JA, Serra M, Wang JY, Wei YH, Nycum L, Emanuel BS, Evans GA. EWS-erg and EWS-Fli1 fusion transcripts in Ewing’s sarcoma and primitive neuroectodermal tumors with variant translocations. J Clin Invest. 1994;94(2):489–496. doi: 10.1172/JCI117360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor. ERG. Nat Genet. 1994;6(2):146–151. doi: 10.1038/ng0294-146. [DOI] [PubMed] [Google Scholar]
- 42.Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, Shapiro DN. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene. 1995;10(6):1229–1234. [PubMed] [Google Scholar]
- 43.Kaneko Y, Yoshida K, Handa M, Toyoda Y, Nishihira H, Tanaka Y, Sasaki Y, Ishida S, Higashino F, Fujinaga K. Fusion of an ETS-family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Gene Chromosome Cancer. 1996;15(2):115–121. doi: 10.1002/(SICI)1098-2264(199602)15:2<115::AID-GCC6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 44.Urano F, Umezawa A, Hong W, Kikuchi H, Hata J. A novel chimera gene between EWS and E1A-F, encoding the adenovirus E1A enhancer-binding protein, in extraosseous Ewing’s sarcoma. Biochem Biophys Res Commun. 1996;219(2):608–612. doi: 10.1006/bbrc.1996.0281. [DOI] [PubMed] [Google Scholar]
- 45.Peter M, Couturier J, Pacquement H, Michon J, Thomas G, Magdelenat H, Delattre O. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene. 1997;14(10):1159–1164. doi: 10.1038/sj.onc.1200933. [DOI] [PubMed] [Google Scholar]
- 46.Ozaki T, Paulussen M, Poremba C, Brinkschmidt C, Rerin J, Ahrens S, Hoffmann C, Hillmann A, Wai D, Schäf er KL, Böcker W, Jürgens H, Winkelmann W, Dockhorn-Dworniczak B. Genetic imbalances revealed by comparative genomic hybridization in Ewing tumors. Genes Chromosomes Cancer. 2001;32(2):164–171. doi: 10.1002/gcc.1178. [DOI] [PubMed] [Google Scholar]
- 47.Kunisada T, Yoshida A, Morimoto Y, Itani T, Yanai H, Sugihara S, Ozaki T. Fusion gene assay in the treatment of bone and soft tissue tumors. Proceedings of the 43rd Annual Musculoskeletal Tumor Meeting of the Japanese Orthopaedic Association. 2010;84:S977 (in Japanese).
- 48.Zoubek A, Dockhorn-Dworniczak B, Delattre O, Christiansen H, Niggli F, Gatterer-Menz I, Smith TL, Jürgens H, Gadner H, Kovar H. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol. 1996;14(4):1245–1251. doi: 10.1200/JCO.1996.14.4.1245. [DOI] [PubMed] [Google Scholar]
- 49.de Alava E, Kawai A, Healey JH, Fligman I, Meyers PA, Huvos AG, Gerald WL, Jhanwar SC, Argani P, Antonescu CR, Pardo-Mindan FJ, Ginsberg J, Womer R, Lawlor ER, Wunder J, Andrulis I, Sorensen PH, Barr FG, Ladanyi M. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol. 1998;16(4):1248–1255. doi: 10.1200/JCO.1998.16.4.1248. [DOI] [PubMed] [Google Scholar]
- 50.Ginsberg JP, de Alava E, Ladanyi M, Wexler LH, Kovar H, Paulussen M, Zoubek A, Dockhorn-Dworniczak B, Juergens H, Wunder JS, Andrulis IL, Malik R, Sorensen PH, Womer RB, Barr FG. EWS-FLI1 and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing’s sarcoma. J Clin Oncol. 1999;17(6):1809–1814. doi: 10.1200/JCO.1999.17.6.1809. [DOI] [PubMed] [Google Scholar]
- 51.Le Deley MC, Delattre O, Schaefer KL, Burchill SA, Koehler G, Hogendoorn PC, Lion T, Poremba C, Marandet J, Ballet S, Pierron G, Brownhill SC, Nesslbock M, Ranft A, Dirksen U, Oberlin O, Lewis IJ, Craft AW, Jürgens H, Kovar H. Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-EWING 99 trial. J Clin Oncol. 2010;28(12):1982–1988. doi: 10.1200/JCO.2009.23.3585. [DOI] [PubMed] [Google Scholar]
- 52.Obata H, Ueda T, Kawai A, Ishii T, Ozaki T, Abe S, Tanaka K, Tsuchiya H, Matsumine A, Yabe H. Japanese Musculoskeletal Oncology G. Clinical outcome of patients with Ewing sarcoma family of tumors of bone in Japan: the Japanese Musculoskeletal Oncology Group cooperative study. Cancer. 2007;109(4):767–775. doi: 10.1002/cncr.22481. [DOI] [PubMed] [Google Scholar]
- 53.Wexler LH, DeLaney TF, Tsokos M, Avila N, Steinberg SM, Weaver-McClure L, Jacobson J, Jarosinski P, Hijazi YM, Balis FM, Horowitz ME. Ifosfamide and etoposide plus vincristine, doxorubicin, and cyclophosphamide for newly diagnosed Ewing’s sarcoma family of tumors. Cancer. 1996;78(4):901–911. doi: 10.1002/(SICI)1097-0142(19960815)78:4<901::AID-CNCR30>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 54.Granowetter L, Womer R, Devidas M, Krailo M, Wang C, Bernstein M, Marina N, Leavey P, Gebhardt M, Healey J, Shamberger RC, Goorin A, Miser J, Meyer J, Arndt CA, Sailer S, Marcus K, Perlman E, Dickman P, Grier HE. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children’s Oncology Group Study. J Clin Oncol. 2009;27(15):2536–2541. doi: 10.1200/JCO.2008.19.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyers PA, Krailo MD, Ladanyi M, Chan KW, Sailer SL, Dickman PS, Baker DL, Davis JH, Gerbing RB, Grovas A, Herzog CE, Lindsley KL, Liu-Mares W, Nachman JB, Sieger L, Wadman J, Gorlick RG. High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing’s sarcoma does not improve prognosis. J Clin Oncol. 2001;19(11):2812–2820. doi: 10.1200/JCO.2001.19.11.2812. [DOI] [PubMed] [Google Scholar]
- 56.Ladenstein R, Potschger U, Le Deley MC, Whelan J, Paulussen M, Oberlin O, van den Berg H, Dirksen U, Hjorth L, Michon J, Lewis I, Craft A, Jürgens H. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol. 2010;28(20):3284–3291. doi: 10.1200/JCO.2009.22.9864. [DOI] [PubMed] [Google Scholar]
- 57.Dunst J, Sauer R, Burgers JM, Hawliczek R, Kurten R, Winkelmann W, Salzer-Kuntschik M, Muschenich M, Jürgens H. Radiation therapy as local treatment in Ewing’s sarcoma. Results of the Cooperative Ewing’s Sarcoma Studies CESS 81 and CESS 86. Cancer. 1991;67(11):2818–2825. doi: 10.1002/1097-0142(19910601)67:11<2818::AID-CNCR2820671118>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 58.Ozaki T, Hillmann A, Hoffmann C, Rube C, Blasius S, Dunst J, Jürgens H, Winkelmann W. Significance of surgical margin on the prognosis of patients with Ewing’s sarcoma. A report from the Cooperative Ewing’s Sarcoma Study. Cancer. 1996;78(4):892–900. doi: 10.1002/(SICI)1097-0142(19960815)78:4<892::AID-CNCR29>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 59.Bacci G, Toni A, Avella M, Manfrini M, Sudanese A, Ciaroni D, Boriani S, Emiliani E, Campanacci M. Long-term results in 144 localized Ewing’s sarcoma patients treated with combined therapy. Cancer. 1989;63(8):1477–1486. doi: 10.1002/1097-0142(19890415)63:8<1477::AID-CNCR2820630805>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 60.Wilkins RM, Pritchard DJ, Burgert EO., Jr Unni KK. Ewing’s sarcoma of bone. Experience with 140 patients. Cancer. 1986;58(11):2551–2555. doi: 10.1002/1097-0142(19861201)58:11<2551::AID-CNCR2820581132>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 61.Kawaguchi N, Ahmed AR, Matsumoto S, Manabe J, Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res. 2004;419:165–172. doi: 10.1097/00003086-200402000-00027. [DOI] [PubMed] [Google Scholar]
- 62.Schuck A, Ahrens S, Paulussen M, Kuhlen M, Konemann S, Rube C, Winkelmann W, Kotz R, Dunst J, Willich N, Jürgens H. Local therapy in localized Ewing tumors: results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2003;55(1):168–177. doi: 10.1016/S0360-3016(02)03797-5. [DOI] [PubMed] [Google Scholar]
- 63.Paulussen M, Ahrens S, Burdach S, Craft A, Dockhorn-Dworniczak B, Dunst J, Frohlich B, Winkelmann W, Zoubek A, Jürgens H. Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol. 1998;9(3):275–281. doi: 10.1023/A:1008208511815. [DOI] [PubMed] [Google Scholar]
- 64.Bolling T, Schuck A, Paulussen M, Dirksen U, Ranft A, Konemann S, Dunst J, Willich N, Jürgens H. Whole lung irradiation in patients with exclusively pulmonary metastases of Ewing tumors. Toxicity analysis and treatment results of the EICESS-92 trial. Strahlenther Onkol. 2008;184(4):193–197. doi: 10.1007/s00066-008-1810-x. [DOI] [PubMed] [Google Scholar]
- 65.Serizawa I, Kagei K, Kamada T, Imai R, Sugahara S, Okada T, Tsuji H, Ito H, Tsujii H. Carbon ion radiotherapy for unresectable retroperitoneal sarcomas. Int J Radiat Oncol Biol Phys. 2009;75(4):1105–1110. doi: 10.1016/j.ijrobp.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 66.Tsuchiya H, Wan SL, Sakayama K, Yamamoto N, Nishida H, Tomita K. Reconstruction using an autograft containing tumour treated by liquid nitrogen. J Bone Joint Surg Br. 2005;87(2):218–225. doi: 10.1302/0301-620X.87B2.15325. [DOI] [PubMed] [Google Scholar]
- 67.Murakami H, Demura S, Kato S, Nishida H, Yoshioka K, Hayashi H, Inoue K, Ota T, Shinmura K, Yokogawa N, Fang X, Tsuchiya H. Increase of IL-12 following reconstruction for total en bloc spondylectomy using frozen autografts treated with liquid nitrogen. PLoS One. 2013;8(5):e64818. doi: 10.1371/journal.pone.0064818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoffmann C, Ahrens S, Dunst J, Hillmann A, Winkelmann W, Craft A, Gobel U, Rube C, Voute PA, Harms D, Jürgens H. Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer. 1999;85(4):869–877. doi: 10.1002/(SICI)1097-0142(19990215)85:4<869::AID-CNCR14>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 69.Akiyama T, Clark JC, Miki Y, Choong PF. The non-vascularised fibular graft: a simple and successful method of reconstruction of the pelvic ring after internal hemipelvectomy. J Bone Joint Surg Br. 2010;92(7):999–1005. doi: 10.1302/0301-620X.92B7.23497. [DOI] [PubMed] [Google Scholar]
- 70.Ozaki T, Hillmann A, Winkelmann W. Treatment outcome of pelvic sarcomas in young children: orthopaedic and oncologic analysis. J Pediatr Orthop. 1998;18(3):350–355. [PubMed] [Google Scholar]
- 71.Gebert C, Wessling M, Hoffmann C, Roedl R, Winkelmann W, Gosheger G, Hardes J. Hip transposition as a limb salvage procedure following the resection of periacetabular tumors. J Surg Oncol. 2011;103(3):269–275. doi: 10.1002/jso.21820. [DOI] [PubMed] [Google Scholar]
- 72.Rödl RW, Hoffmann C, Gosheger G, Leidinger B, Jürgens H, Winkelmann W. Ewing’s sarcoma of the pelvis: combined surgery and radiotherapy treatment. J Surg Oncol. 2003;83(3):154–160. doi: 10.1002/jso.10256. [DOI] [PubMed] [Google Scholar]
- 73.Van Winkle P, Angiolillo A, Krailo M, Cheung YK, Anderson B, Davenport V, Reaman G, Cairo MS. Ifosfamide, carboplatin, and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: the Children’s Cancer Group (CCG) experience. Pediatr Blood Cancer. 2005;44(4):338–347. doi: 10.1002/pbc.20227. [DOI] [PubMed] [Google Scholar]
- 74.Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jürgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. 2006;47(6):795–800. doi: 10.1002/pbc.20719. [DOI] [PubMed] [Google Scholar]
- 75.Wagner LM, McAllister N, Goldsby RE, Rausen AR, McNall-Knapp RY, McCarville MB, Albritton K. Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer. 2007;48(2):132–139. doi: 10.1002/pbc.20697. [DOI] [PubMed] [Google Scholar]
- 76.Paulussen M, Ahrens S, Lehnert M, Taeger D, Hense HW, Wagner A, Dunst J, Harms D, Reiter A, Henze G, Niemeyer C, Gobel U, Kremens B, Folsch UR, Aulitzky WE, Voute PA, Zoubek A, Jürgens H. Second malignancies after Ewing tumor treatment in 690 patients from a cooperative German/Austrian/Dutch study. Ann Oncol. 2001;12(11):1619–1630. doi: 10.1023/A:1013148730966. [DOI] [PubMed] [Google Scholar]
- 77.Longhi A, Ferrari S, Tamburini A, Luksch R, Fagioli F, Bacci G, Ferrari C. Late effects of chemotherapy and radiotherapy in osteosarcoma and Ewing sarcoma patients: the Italian Sarcoma Group Experience (1983–2006) Cancer. 2012;118(20):5050–5059. doi: 10.1002/cncr.27493. [DOI] [PubMed] [Google Scholar]
- 78.Keir ST, Morton CL, Wu J, Kurmasheva RT, Houghton PJ, Smith MA. Initial testing of the multitargeted kinase inhibitor pazopanib by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2012;59(3):586–588. doi: 10.1002/pbc.24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kolb EA, Gorlick R, Reynolds CP, Kang MH, Carol H, Lock R, Keir ST, Maris JM, Billups CA, Desjardins C, Kurmasheva RT, Houghton PJ, Smith MA. Initial testing (stage 1) of eribulin, a novel tubulin binding agent, by the pediatric preclinical testing program. Pediatr Blood Cancer. 2013;60(8):1325–1332. doi: 10.1002/pbc.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lau L, Supko JG, Blaney S, Hershon L, Seibel N, Krailo M, Qu W, Malkin D, Jimeno J, Bernstein M, Baruchel S. A phase I and pharmacokinetic study of ecteinascidin-743 (Yondelis) in children with refractory solid tumors. A Children’s Oncology Group study. Clin Cancer Res. 2005;11(2.1):672–7. [PubMed]
- 81.Baruchel S, Pappo A, Krailo M, Baker KS, Wu B, Villaluna D, Lee-Scott M, Adamson PC, Blaney SM. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children’s Oncology Group. Eur J Cancer. 2012;48(4):579–585. doi: 10.1016/j.ejca.2011.09.027. [DOI] [PubMed] [Google Scholar]