

Anticytokine autoantibodies are an emerging mechanism of immunodeficiency. We identified anti–granulocyte macrophage colony-stimulating factor autoantibodies in 5 adults with disseminated/extrapulmonary nocardiosis. Their presence might increase risk for pulmonary alveolar proteinosis or other opportunistic infections, and may direct therapy.
Keywords: adult-onset immunodeficiency, anticytokine autoantibodies, nocardiosis, GM-CSF, opportunistic infection
Abstract
Background. Nocardia species cause infections in both immunocompromised and otherwise immunocompetent patients, although the mechanisms defining susceptibility in the latter group are elusive. Anticytokine autoantibodies are an emerging cause of pathogen-specific susceptibility in previously healthy human immunodeficiency virus-uninfected adults, including anti–granulocyte macrophage colony-stimulating factor (GM-CSF) autoantibodies with cryptococcal meningitis.
Methods. Plasma from patients with disseminated/extrapulmonary nocardiosis and healthy controls was screened for anticytokine autoantibodies using a particle-based approach. Autoantibody function was assessed by intranuclear staining for GM-CSF–induced STAT5 phosphorylation in normal cells incubated with either patient or normal plasma. GM-CSF–mediated cellular activation by Nocardia was assessed by staining for intracellular cytokine production and intranuclear STAT5 phosphorylation.
Results. We identified neutralizing anti–GM-CSF autoantibodies in 5 of 7 patients studied with central nervous system nocardiosis and in no healthy controls (n = 14). GM-CSF production was induced by Nocardia in vitro, suggesting a causative role for anti–GM-CSF autoantibodies in Nocardia susceptibility and dissemination.
Conclusions. In previously healthy adults with otherwise unexplained disseminated/extrapulmonary Nocardia infections, anti–GM-CSF autoantibodies should be considered. Their presence may suggest that these patients may be at risk for later development of pulmonary alveolar proteinosis or other opportunistic infections, and that patients may benefit from therapeutic GM-CSF administration.
Nocardiosis is an opportunistic infection that generally occurs in immunocompromised patients, especially in those with phagocyte defects that are induced by systemic corticosteroids or seen in chronic granulomatous disease [1]. However, there remain some patients with nocardiosis for whom no defect has been found. Nocardia's proclivity for central nervous system involvement has been long appreciated [2], although the mechanisms underlying this remain obscure.
Anticytokine autoantibodies are an emerging cause of adult-onset immunodeficiency [3]. Neutralizing anti–interferon gamma (IFN-γ) autoantibodies have been identified in the context of severe disseminated opportunistic infections [4]; anti–interleukin (IL) 17A, anti–IL-17F, and anti–IL-22 autoantibodies in association with and chronic mucocutaneous candidiasis [5, 6]; anti–IL-6 autoantibodies in the setting of staphylococcal skin infections [7]; and recently, anti–granulocyte macrophage colony-stimulating factor (GM-CSF) autoantibodies in association with Cryptococcus gattii meningitis [8].
Anti–GM-CSF autoantibodies were first recognized as etiologic in most cases of pulmonary alveolar proteinosis (PAP), a rare lung disorder characterized by the accumulation of proteinaceous material within the alveoli [9] due to defective GM-CSF–dependent surfactant clearance by pulmonary macrophages [10]. It is now appreciated that these autoantibodies may also contribute to infection susceptibility in the absence of PAP [8, 11]. Abrogation of GM-CSF signaling, either by gene knockout in mice or neutralizing autoantibodies, impacts other phagocytic activities [12, 13] and increases susceptibility to infection with opportunists typically controlled by phagocytes, such as Nocardia species, Histoplasma, and Cryptococcus [9, 14–16]. These observations implicate a direct role for GM-CSF in host defense against these opportunists.
The recognition of anti–GM-CSF autoantibodies in previously healthy patients with cryptococcal meningitis [8] prompted us to evaluate the plasma of healthy, human immunodeficiency virus (HIV)-uninfected and otherwise immunocompetent adults with disseminated/extrapulmonary nocardiosis.
MATERIALS AND METHODS
Subjects
From September 2011 through July 2014, we screened 7 adult patients with disseminated/extrapulmonary nocardiosis who were otherwise immunologically normal under National Institutes of Health (NIH) institutional review board (IRB)–approved protocols 93-I-0119 or 95-I-0066. Patients were either evaluated at NIH or identified through a literature search of the MEDLINE database (National Library of Medicine, Bethesda, Maryland) using the keywords CNS, extrapulmonary or disseminated, and Nocardia or nocardiosis. We reviewed published cases of disseminated/extrapulmonary nocardiosis for any predisposing risk factor (eg, HIV, systemic corticosteroid administration, or other significant disease-associated or iatrogenic immunosuppression), contacted the corresponding author for any case meeting these criteria published within the last 2 years, and worked with them to characterize their patients, both clinically and in the laboratory, under our NIH IRB–approved protocols. Normal blood from healthy controls was obtained from the NIH blood bank under appropriate IRB-approved protocols. Plasma and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation [8].
Anticytokine Autoantibody Screening
Plasmas were screened for anticytokine autoantibodies using a particle-based approach [17]. Immunoglobulin isotypes and immunoglobulin G (IgG) subclasses were determined for anti–GM-CSF specific autoantibodies using the same strategy.
Detection of GM-CSF-Induced STAT5 Phosphorylation
Patient and normal PBMCs (1 × 106) were cultured in complete Roswell Park Memorial Institute (RPMI) 1640 media consisting of 2 mM glutamine, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 100 U/mL penicillin, and 100 µg/mL streptomycin with 10% patient or normal plasma. Cultures were left unstimulated or stimulated with GM-CSF (10 ng/mL; R&D Systems) for 30 minutes at 37°C. Monocytes were identified by CD14 (BD Pharmingen) surface staining before being fixed and permeabilized for intranuclear staining with anti-pSTAT5 (Y694) antibody (BD Biosciences) [8]. Data were collected using FACSCalibur (BD Biosciences), analyzed using FlowJo software (Treestar), and graphed with Prism5 software (GraphPad).
Preparation of Nocardia
Nocardia cyriacigeorgica strain GU2-H (NCGU2-H) [18] was selected as it is a model organism to study Nocardia pathogenesis. NCGU2-H was grown in Middlebrook 7h9 broth to mid–log phase at 37°C with mild rotational agitation (150 rpm). The culture was collected and spun down for 10 minutes at 3000g, and the pellet was washed once with Hank's balanced salt solution and twice with RPMI media 1640 (1×) with no additions (no antibiotics or serum). After resuspension in RPMI 1640 media (1×), the bacterial concentration was assessed spectrophotometrically by optical density at 580 nm (approximately 5 × 107 colony-forming units [CFUs]/mL). CFUs were confirmed by plating serial dilutions on solid media. For experiments with heat-killed bacteria, NCGU2-H was incubated for 30 minutes at 95°C.
Nocardia-Induced Intracellular GM-CSF and STAT5 Phosphorylation
Monocytes were isolated from patient or normal PBMCs using CD14 MicroBeads (Miltenyi) per the manufacturer's instructions. PBMCs or purified monocytes were cultured in antibiotic-free RPMI media with 10% patient or normal plasma. For the detection of intracellular GM-CSF, cultures were left unstimulated or incubated with heat-killed or live Nocardia (multiplicity of infection of 3) for 15 minutes, and then treated with brefeldin A (10 µg/mL). Cells were stained for CD14, CD3, CD4, GM-CSF, and tumor necrosis factor alpha (TNF-α) (BD Pharmingen) using BD Cytofix/Cytoperm kit (BD Biosciences). Data were collected using FACSCanto (BD Biosciences).
Phosphorylated STAT5 (pSTAT5) was detected in cells cultured in antibiotic-free media with 10% patient or normal plasma and incubated with heat-killed Nocardia for the indicated amount of time. Cells were stained for CD14 and intranuclear pSTAT5 as described above.
RESULTS
Initially we screened patients 1–3 with proven CNS nocardiosis (Table 1) who were referred to NIH for suspected immunodeficiency. Upon recognition that all 3 were positive for anti–GM-CSF autoantibodies, we identified 2 additional cases through systematic literature review (patients 4 and 5, Table 1) [19, 20]. We also screened all past samples stored in our laboratory from patients with proven or probable disseminated/extrapulmonary nocardiosis (patients 6 and 7, Table 1) [21].
Table 1.
Clinical and Laboratory Features
| Patient No. | Age/Sex/Ethnicity | Anti–GM-CSF Autoantibody Status | Nocardia Speciation and Sites | Other Infections | Comorbidities/ Other Autoimmunity | Absolute CD4 Count | Absolute Neutrophil Count | DHR Analysis | IgG Level | Follow- up | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 44/M/US white | Positive | N. paucivorans (brain) | None | None | 849 cells/µL | 2840 cells/µL | Normal | 483 mg/dL | Resolved; Stable on TMP-SMX prophylaxis | |
| 2 | 73/M/white Portuguese | Positive | Nocardia unspeciated (cutaneous) | Pulmonary aspergillosis | None | 221 cells/µL | 8870 cells/µL | Not done | 2240 mg/dL | Stable on TMP-SMX, AMC, and GM-CSF | |
| 3 | 61/M/US white | Positive | N. farcinica (brain) | None | None | 397 cells/µL | 4770 cells/µL | Normal | 704 mg/dL | Stable on TMP-SMX, MXF, and GM-CSF | |
| 4 | 50/M/US white | Positive | N. paucivorans (brain) | None | None | 707 cells/µL | 1900 cells/µL | Normal | 818 mg/dL | Complete recovery after 12 mo antibiotic therapy. Off antibiotics for >1 y | Previously reported [19] |
| 5 | 52/F/African American | Positive | N. asteroides (lung, brain) | Disseminated cryptococcosis | Type II diabetes mellitus (hemoglobin A1C 8.6%), prior necrotizing pancreatitis | 403 cells/µL | 3190 cells/µL | Not done | 965 mg/dL | Off antibiotics, no signs of PAP to date | Previously reported [20] |
| 6 | 18/M/US white | Negative | N. transvalensis (CNS) | None | Developed panhypopituitarism from infection | 726 cells/µL | 2900 cells/µL | Normal | 424 mg/dL | Resolved | Previously reported [21]; mild hypo- gammaglobulinemia likely steroid induced |
| 7 | 54/F/US white | Negative | N. beijingensis (lung, presumed CNS) | Chronic pulmonary infections: MAC, Aspergillus, Stenotrophomonas, Serratia | Bronchiectasis | 842 cells/µL | 2110 cells/µL | Normal | 1160 mg/dL | Unknown |
Abbreviations: AMC, amoxicillin/clavulanate; CNS, central nervous system; DHR, dihydrorhodamine; GM-CSF, granulocyte macrophage colony-stimulating factor; IgG, immunoglobulin G; MAC, Mycobacterium avium complex; MXF, moxifloxacin; PAP, pulmonary alveolar proteinosis; TMP-SMX, trimethoprim-sulfamethoxazole.
Case 1
An otherwise healthy 44-year-old HIV-uninfected US white man presented in September 2011 with 3 weeks of headaches, forgetfulness, blurry vision, and confusion upon return from 9 months in Iraq. Magnetic resonance imaging (MRI) of the brain revealed 2 complex ring-enhancing masses with surrounding edema and midline shift. Brain biopsies showed necrosis with neutrophilic infiltrate and periodic acid-Schiff (PAS)–positive filamentous organisms; culture grew Nocardia paucivorans. Lymphocyte phenotyping and neutrophil counts were normal. Dihydrorhodamine oxidation testing was normal, excluding chronic granulomatous disease. Initial intravenous antibiotic therapy included trimethoprim-sulfamethoxazole (TMP-SMX), imipenem-cilastatin, and amikacin. The patient had progressive somnolence and vomiting with an increase in lesion size, prompting ultrasound-guided drainage. Imipenem-cilastatin was stopped due to isolate resistance, and intravenous TMP-SMX and amikacin were continued for 8 weeks. The patient then received oral TMP-SMX and linezolid for 8 weeks followed by TMP-SMX alone with full functional and radiographic recovery.
We screened the patient for anticytokine autoantibodies 7 months after his diagnosis of Nocardia and found neutralizing autoantibodies against GM-CSF. Banked sera collected over the 20 years preceding infection showed development of anti–GM-CSF autoantibodies about 10 years prior to developing CNS nocardiosis.
Computed tomography (CT) of the chest at initial presentation showed subtle ground glass opacities, possibly consistent with PAP. However, normal exercise capacity and absence of respiratory complaints have deflected further diagnostics.
Case 2
An otherwise healthy 73-year-old HIV-uninfected Portuguese white man presented in April 2012 with 4 weeks of fever, cough, hemoptysis, and asthenia. Chest CT demonstrated pneumonia, prompting treatment with imipenem/cilastatin. A few days later he developed several erythematous, elevated cutaneous lesions with ulceration, and central necrosis on his limbs, thorax and abdomen; culture of a biopsied lesion grew Nocardia species. Amikacin was added to imipenem-cilastatin and at 4 weeks of therapy, the cutaneous lesions had resolved and the pneumonia had improved, but new cavitation was present. He remained febrile with elevated inflammatory markers. CT-guided biopsy of the cavity demonstrated bronchial invasion with fungal elements from which Aspergillus fumigatus was cultured. Voriconazole was added to intravenous treatment for Nocardia and the patient was discharged after 6 weeks on voriconazole, TMP-SMX, and amoxicillin-clavulanate.
In August 2012, he developed fever, vomiting, and altered consciousness. Brain MRI showed several scattered cerebral lesions with extensive adjacent edema, suggesting progression of his Nocardia infection. Despite reinitiation of intravenous antibiotics, he had further neurological decline. Repeat brain MRI showed an increase in lesion size and edema. Brain biopsy demonstrated structures consistent with Nocardia, although cultures were negative. Lung biopsy was positive for Nocardia species by polymerase chain reaction. Neutrophil counts were normal and lymphocyte phenotyping revealed mild CD4+ lymphopenia (221 cells/µL) in the setting of active infection.
Seven months after initial presentation, neutralizing anti–GM-CSF autoantibodies were sought (Figure 1A). Neither CT of the chest nor bronchoalveolar lavage with biopsy suggested PAP. Because of continued deterioration, he received recombinant GM-CSF 250 µg subcutaneously thrice weekly, with gradual clinical and radiologic improvement. Six months after initiation of GM-CSF, the patient had dramatic clinical improvement and had returned to almost autonomous life. For this reason, GM-CSF was stopped. One month later he presented with recrudescent hemoptysis, vomiting, weight loss, and pleurisy. Chest CT showed lesions consistent with invasive aspergillosis, and brain MRI demonstrated enlarging lesions with increased edema. Voriconazole, imipenem-cilastatin, amikacin, and GM-CSF were reinitiated, again with clinical improvement at the time of this writing (Supplementary Figure 1).
Figure 1.
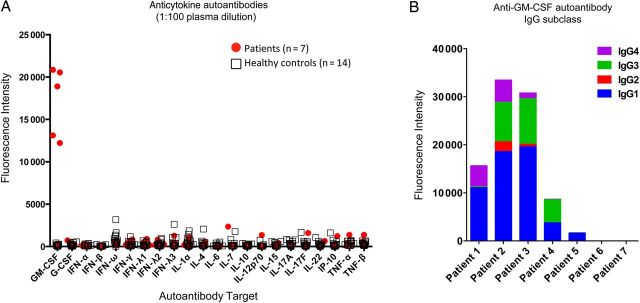
Laboratory evaluation of patients with disseminated/extrapulmonary nocardiosis. A, Multiplex screen for anticytokine autoantibodies against granulocyte macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), interferon (IFN)–α, IFN-β, IFN-ω, IFN-γ, IFN-λ1, IFN-λ2, IFN-λ3, interleukin (IL)–1α, IL-4, IL-6, IL-7, IL-10, IL-12p70, IL-15, IL-17A, IL-17F, IL-22, Interferon-gamma-inducible protein 10 (IP-10), tumor necrosis factor (TNF)–α, and TNF-β in 7 patients with disseminated/extrapulmonary nocardiosis and healthy controls (n = 14). B, Evaluation of anti–GM-CSF immunoglobulin G (IgG) subclass in patient plasma.
Case 3
An otherwise healthy 61-year-old HIV-uninfected white man presented in May 2013 with 5 months of progressive left upper arm discoordination, headache, seizures, and visual disturbance. Brain MRI showed a right frontoparietal mass with surrounding edema and at least 5 additional tiny enhancing foci. Chest CT showed an 8-mm nodule in the left lower lobe. Right parietal craniotomy with evacuation of the brain abscess found gram-positive, weakly acid-fast positive and PAS-positive filamentous organisms; culture grew Nocardia farcinica. Immunoglobulin levels, lymphocyte phenotyping, neutrophil counts, and dihydrorhodamine testing were normal. The patient was treated with 8 weeks of intravenous antimicrobials (imipenem-cilastatin followed by amikacin) and 2 oral agents (TMP-SMX with moxifloxacin or linezolid), based on in vitro susceptibility testing; TMP-SMX and moxifloxacin were continued thereafter. Anti–GM-CSF autoantibodies were identified 12 months after initial presentation (Supplementary Figure 2).
In April 2014, he again had neurologic decline despite ongoing treatment with moxifloxacin and TMP-SMX. Amikacin and linezolid were added, and brain MRI showed increased enhancement at the previous surgical site, prompting repeat evacuation. Biopsy revealed florid necrotizing granulomatous inflammation, although all cultures and stains were negative. Adjuvant subcutaneous GM-CSF 250 µg thrice weekly led to improved cognition and MRI findings 2 months later.
Additional Cases
Two additional cases were reported elsewhere in detail [19, 20] without a known diagnosis until we contacted the authors. One was a 50-year-old man who presented with systemic and neurological symptoms and was identified on brain biopsy to have N. paucivorans on stain and culture. He was successfully treated over 12 months with TMP-SMX, imipenem, and moxifloxacin. The other was a 52-year-old African American woman who presented with cryptococcal meningitis. Upon review of medical records, she had been treated 6 years earlier for brain biopsy–proven N. farcinica. The 2 additional cases identified at NIH were found through medical records review and included an 18-year-old man with culture-positive Nocardia transvalensis of the central nervous system and a 54-year-old woman with chronic bronchiectasis and multiple pulmonary infections since the age of 22, including Nocardia beijingensis, which was presumed the source of 2 temporal lesions seen by MRI (Table 1).
Anticytokine Autoantibody Testing
Plasmas of patients 1–5 demonstrated only anti–GM-CSF autoantibodies in a multiplex assay for anticytokine autoantibodies (Figure 1A). They were exclusively of IgG isotype (not shown) and predominantly of IgG1 subclass (Figure 1B). Patients 6 and 7 were negative for all autoantibodies screened (Figure 1A).
In normal PBMCs, only anti–GM-CSF autoantibody-positive patient plasma inhibited GM-CSF–induced STAT5 phosphorylation (Figure 2A), whereas patient PBMCs washed of autologous plasma demonstrated normal pSTAT5 induction (Figure 2B; PBMCs were not available for patient 7).
Figure 2.

In vitro evaluation of patient plasma and cells. A, Normal peripheral blood mononuclear cells (PBMCs) were incubated in the presence of normal or patient plasma and left unstimulated or stimulated with granulocyte macrophage colony-stimulating factor (GM-CSF). Intracellular staining for phosphorylated STAT5 (pSTAT5) was measured by flow cytometry, gating on CD14+ monocytes. B, Washed normal or patient PBMCs were left unstimulated or stimulated with GM-CSF. A stimulation index was calculated using the ratio of the geometric mean fluorescence of pSTAT5 staining for stimulated to unstimulated conditions for each person (PBMCs not available for patient 7).
GM-CSF Pathway Activation in Response to Nocardia
Heat-killed and live Nocardia-induced intracellular GM-CSF and TNF-α in 2 hours and was even more prominent after 6 hours in normal monocytes (Figure 3A), but not in normal lymphocytes (not shown). Exogenous GM-CSF induced STAT5 phosphorylation in 5 minutes, with maximal signal after 20 minutes. Nocardia-induced STAT5 phosphorylation was only evident after 1.5 hours and peaked at 2.5 hours in normal monocytes (Figure 3B).
Figure 3.

Nocardia-induced activation of the granulocyte macrophage colony-stimulating factor (GM-CSF) signaling pathway. A, Normal peripheral blood mononuclear cells (PBMCs) were incubated with media alone or heat-killed or live Nocardia (multiplicity of infection 3) in the presence of brefeldin A for 2 or 6 hours and stained for intracellular GM-CSF and tumor necrosis factor (TNF)–α. B, Time course for STAT5 phosphorylation (pSTAT5) after stimulation with GM-CSF or Nocardia in PBMCs at the indicated time points gating on CD14+ monocytes or CD4+ lymphocytes. C, Normal purified monocytes were incubated with normal plasma, normal plasma spiked with neutralizing commercial anti–GM-CSF monoclonal antibody, or patient plasma and evaluated for Nocardia-induced intracellular GM-CSF following brefeldin A treatment (top panel) or stained for pSTAT5) (bottom panel). D, Purified patient monocytes were evaluated for Nocardia-induced intracellular GM-CSF in the presence of normal or patient plasma and brefeldin A.
Nocardia-Induced GM-CSF Signaling Is Independent of Lymphocytes
Purified monocytes were incubated with normal plasma, normal plasma spiked with neutralizing commercial anti–GM-CSF monoclonal antibody, or patient plasma, and evaluated for Nocardia-induced intracellular GM-CSF (Figure 3C, top panel). Nocardia-induced pSTAT5 was seen in purified monocytes incubated with normal plasma but prevented by anti–GM-CSF antibodies (Figure 3C, bottom panel) and tracked with the presence of Nocardia-induced GM-CSF production.
As expected, purified patient monocytes produced GM-CSF in response to Nocardia similarly to normal cells, irrespective of extracellular GM-CSF blockade by patient plasma (Figure 3D).
DISCUSSION
We describe 7 patients with disseminated/extrapulmonary nocardiosis, 5 of whom had neutralizing anti–GM-CSF autoantibodies. Of those 5, one patient also had invasive aspergillosis, also indicative of underlying phagocyte dysfunction. Despite the known causal relationship between GM-CSF autoantibodies and PAP, none of the patients carried this diagnosis initially, although 2 had mild characteristic radiographic changes that were identified in retrospect. Archived sera from our first patient demonstrated anti–GM-CSF autoantibodies that predated clinical expression of disease, indicating that the autoantibodies were not merely a reactive phenomenon and may have had a causal role. Although patient 1 was also noted to have slightly low levels of IgG (Table 1), the levels were only mildly depressed, and hypogammaglobulinemia is not generally associated with Nocardia susceptibilitiy [22]. Interestingly, patient 5 was screened for GM-CSF autoantibodies based on her diagnosis of CNS cryptococcosis; upon further evaluation, it was discovered that she had a history of Nocardia brain abscess.
Consistent with both anti–GM-CSF autoantibody-associated cryptococcosis or PAP and anti–IFN-γ–associated immunodeficiency, the patients with anti–GM-CSF autoantibody-associated disseminated/extrapulmonary nocardiosis presented in adulthood without other underlying comorbidities, including other forms of autoimmunity. Of the 2 patients who were negative for anti–GM-CSF autoantibodies, one presented as a teenager, increasing the likelihood of a yet-unidentified genetic defect. The other had an extensive history of pulmonary disease, including severe bronchiectasis with chronic polymicrobial infections, and did not have proven extrapulmonary nocardiosis; improvement of her brain lesions involved multiple interventions, including TMP-SMX administration.
Nocardia-induced GM-CSF appears to be a critical part of the host response to infection with this pathogen. GM-CSF–induced STAT5 phosphorylation occurs rapidly and in response to concentrations of exogenous GM-CSF as low as 1 pg/mL [8], whereas Nocardia-induced pSTAT5 was appreciated after 1.5 hours and peaked at 2.5 hours, consistent with the time frame required for protein synthesis (Figure 3B). Abrogation of GM-CSF release by a neutralizing autoantibody apparently impedes the events necessary to control Nocardia. Heat-killed and live bacteria had comparable effects on cytokine production (Figure 3A), demonstrating that GM-CSF induction does not require bacterial replication or metabolism and that inducing factors are heat stable. Blocking extracellular transport of cytokines with brefeldin A demonstrates that the Nocardia-induced production of GM-CSF and TNF-α are not GM-CSF dependent (Figure 3A).
The immunologic effects of GM-CSF on Toll-like receptor activation, phagocytosis, bactericidal activity, oxidative burst, and cell adhesion in neutrophils and macrophages [12, 13] provide a biological rationale for how anti–GM-CSF autoantibodies [23] may contribute to Nocardia susceptibility. Poorly differentiated pulmonary macrophages, as seen in patients with anti–GM-CSF autoantibody-associated PAP, may result in an ineffective pulmonary barrier, thereby facilitating dissemination beyond the lung. Although one patient had cutaneous involvement, all patients demonstrated CNS disease. The presence of anti–GM-CSF autoantibodies in serum and cerebrospinal fluid [8] could also contribute to extrapulmonary disease and CNS involvement, respectively. The factors underlying the neurotropic tendencies of Nocardia remain elusive, and whether autoantibodies predispose to CNS infection specifically or a more general tendency toward dissemination will need to be further explored as more cases are identified.
PAP was first recognized in 1958 as a rare idiopathic lung disease characterized by alveolar accumulation of lipoproteinaceous material and associated respiratory compromise [9]. It was nearly 40 years later, when GM-CSF receptor null [24] and GM-CSF cytokine-null [25] mice developed PAP, that this pathway was recognized as being important in human PAP. Shortly thereafter, anti–GM-CSF autoantibodies [10] were identified and later proven causal [23, 26, 27] in about 90% of cases of human PAP. Typical of other autoimmune diseases, PAP varies in severity and may relapse, remit, or spontaneously resolve. Infectious complications of PAP were reported in the original descriptions and have been recognized in subsequent series as well, particularly with Nocardia [16, 28]. However, it remained unrecognized that anti–GM-CSF autoantibodies might lead to opportunistic infections in the absence of alveolar pathology [8].
Anti–GM-CSF autoantibodies have been recognized in healthy controls and intravenous immunoglobulin but at 10- to 1000-fold lower levels than in patients with PAP and almost exclusively as GM-CSF–bound complexes, leaving them with minimal residual biological activity [29]. It is unknown if those healthy controls are similar in character but only different in amount, or if they differ in both quality and quantity from pathogenic autoantibodies. It is also unclear what differentiates those who get infection from those who get PAP, and from those who get both. Infection susceptibility may occur at a different titer or after a different duration of autoantibodies than PAP. Additionally, unidentified genetic or autoimmune risk factors might influence the clinical phenotype.
Anti–GM-CSF autoantibodies should be sought in the context of unexplained opportunistic infections with C. gattii and Nocardia, particularly because their presence may inform therapy or possibly confer risk for the development of PAP. Inhaled and subcutaneous GM-CSF has been used with success in PAP, and 2 patients in this series appeared to respond to exogenous GM-CSF administration. These data suggest that pharmacologic doses of GM-CSF somehow overcome autoantibody blockade and might benefit patients who do not respond appropriately to antimicrobials. Rituximab has been used to target the pathogenic autoantibody-producing B cells in refractory cases [30, 31]. Nocardia induces GM-CSF production, but blockade of GM-CSF signaling apparently may permit dissemination. Anticytokine autoantibody-associated immunodeficiencies provide a unique window into understanding host–pathogen interactions and the critical signals that influence the outcomes of initial and subsequent encounters.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online (http://cid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Disclaimer. The content of this publication is the sole responsibility of the authors and does not necessarily reflect the views or policies of the National Institutes of Health (NIH) or the Department of Health and Human Services, the Department of Defense, or the Departments of the Army, Navy, or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the US government.
Financial support. This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, at the NIH.
Potential conflicts of interest. All authors: No potential conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Dorman SE, Guide SV, Conville PS, et al. Nocardia infection in chronic granulomatous disease. Clin Infect Dis. 2002;35:390–4. doi: 10.1086/341416. [DOI] [PubMed] [Google Scholar]
- 2.Anagnostou T, Arvanitis M, Kourkoumpetis TK, et al. Nocardiosis of the central nervous system: experience from a general hospital and review of 84 cases from the literature. Medicine (Baltimore) 2014;93:19–32. doi: 10.1097/MD.0000000000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Browne SK. Anticytokine autoantibody-associated immunodeficiency. Annu Rev Immunol. 2014;32:635–57. doi: 10.1146/annurev-immunol-032713-120222. [DOI] [PubMed] [Google Scholar]
- 4.Browne SK, Burbelo PD, Chetchotisakd P, et al. Adult-onset immunodeficiency in Thailand and Taiwan. N Engl J Med. 2012;367:725–34. doi: 10.1056/NEJMoa1111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kisand K, Boe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puel A, Doffinger R, Natividad A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207:291–7. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puel A, Picard C, Lorrot M, et al. Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J Immunol. 2008;180:647–54. doi: 10.4049/jimmunol.180.1.647. [DOI] [PubMed] [Google Scholar]
- 8.Rosen LB, Freeman AF, Yang LM, et al. Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J Immunol. 2013;190:3959–66. doi: 10.4049/jimmunol.1202526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258:1123–42. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 10.Kitamura T, Tanaka N, Watanabe J, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999;190:875–80. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saijo T, Chen J, Chen SC, et al. Anti-granulocyte-macrophage colony-stimulating factor autoantibodies are a risk factor for central nervous system infection by Cryptococcus gattii in otherwise immunocompetent patients. MBio. 2014;5:e00912–4. doi: 10.1128/mBio.00912-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibata Y, Berclaz PY, Chroneos ZC, et al. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–67. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 13.Uchida K, Beck DC, Yamamoto T, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. 2007;356:567–79. doi: 10.1056/NEJMoa062505. [DOI] [PubMed] [Google Scholar]
- 14.Lee YC, Chew GT, Robinson BW. Pulmonary and meningeal cryptococcosis in pulmonary alveolar proteinosis. Aust N Z J Med. 1999;29:843–4. doi: 10.1111/j.1445-5994.1999.tb00803.x. [DOI] [PubMed] [Google Scholar]
- 15.Oerlemans WG, Jansen EN, Prevo RL, Eijsvogel MM. Primary cerebellar nocardiosis and alveolar proteinosis. Acta Neurol Scand. 1998;97:138–41. doi: 10.1111/j.1600-0404.1998.tb00623.x. [DOI] [PubMed] [Google Scholar]
- 16.Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect. 2012;65:173–9. doi: 10.1016/j.jinf.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Ding L, Mo A, Jutivorakool K, et al. Determination of human anticytokine autoantibody profiles using a particle-based approach. J Clin Immunol. 2012;32:238–45. doi: 10.1007/s10875-011-9621-8. [DOI] [PubMed] [Google Scholar]
- 18.Beaman BL, Maslan S. Effect of cyclophosphamide on experimental Nocardia asteroides infection in mice. Infect Immun. 1977;16:995–1004. doi: 10.1128/iai.16.3.995-1004.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammoud M, Kraft C, Pulst-Korenberg J, Chenoweth C, Gregg KS. Disseminated Nocardia paucivorans infection in an immunocompetent host. Infection. 2014;42:917–20. doi: 10.1007/s15010-014-0609-1. [DOI] [PubMed] [Google Scholar]
- 20.Henry TL, Newman S, Wickboldt AT, Seoane L. The new AIDS chameleon? Ochsner J. 2013;13:470. [Google Scholar]
- 21.Kranick SM, Zerbe CS. Case report from the NIH Clinical Center: CNS nocardiosis. J Neurovirol. 2013;19:505–7. doi: 10.1007/s13365-013-0193-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furst DE. Serum immunoglobulins and risk of infection: how low can you go? Semin Arthritis Rheum. 2009;39:18–29. doi: 10.1016/j.semarthrit.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135:223–35. doi: 10.1016/j.clim.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robb L, Drinkwater CC, Metcalf D, et al. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc Natl Acad Sci U S A. 1995;92:9565–9. doi: 10.1073/pnas.92.21.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dranoff G, Crawford AD, Sadelain M, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–6. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 26.Sakagami T, Beck D, Uchida K, et al. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. Am J Respir Crit Care Med. 2010;182:49–61. doi: 10.1164/rccm.201001-0008OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakagami T, Uchida K, Suzuki T, et al. Human GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. N Engl J Med. 2009;361:2679–81. doi: 10.1056/NEJMc0904077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215–35. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 29.Uchida K, Nakata K, Suzuki T, et al. Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood. 2009;113:2547–56. doi: 10.1182/blood-2009-05-155689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Browne SK, Zaman R, Sampaio EP, et al. Anti-CD20 (rituximab) therapy for anti-IFN-gamma autoantibody-associated nontuberculous mycobacterial infection. Blood. 2012;119:3933–9. doi: 10.1182/blood-2011-12-395707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kavuru MS, Malur A, Marshall I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J. 2011;38:1361–7. doi: 10.1183/09031936.00197710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.