

Abstract
Objective(s):
Morphine dependence (MD) potently protects heart against ischemia reperfusion (IR) injury through specific signaling mechanisms, which are different from the pathways involved in acute morphine treatment or classical preconditioning. Since opioid receptor density changes post cerebral ischemia strongly correlated with brain histological damage, in the present study, we tried to elucidate the possible role of opioid receptors in IR injury among morphine-dependent mice.
Materials and Methods:
Accordingly, incremental doses (10 mg/kg/day to 30 mg/kg/day) of morphine sulphate were subcutaneously administered for 5 days before global brain ischemia induction through bilateral common carotid artery occlusion. Animals were received naloxone (5 mg/kg) or L-NAME (20 mg/kg) 30 min after the last morphine dose. Twenty four hr after the ischemia induction, Retention trial of passive avoidance test and western blot analysis were done. histological analysis (TUNEL and NISSL staining) performed 72 hr after ischemia.
Results:
MD improved post ischemia memory performance (P<0.01) and neuronal survival (P<0.001) and decreased apoptosis (P<0.05) in region I of hippocampus (CA1 region) in mouse. Treatment with naloxone or L-NAME abolished all MD aforementioned effects.
Conclusion:
Results of the present study suggested that opioid receptors activation in the early hr post ischemia is crucial for MD-induced hippocampus tolerance against IR injury. Opioid receptor-dependent balance of NO production was another key factor in MD-induced protection. Further studies are required to determine the effect of MD on opioid receptor changes after ischemia and its correlation with MD-induced protection.
Keywords: Apoptosis, Global brain ischemia, Hippocampus, Memory, Morphine, NO
Introduction
Brain ischemia is becoming a main cause of morbidity and mortality world-wide (1), (2). Global brain ischemia induces neuronal cell death, especially in vulnerable CA1 pyramidal cells (3). Therefore, innovative treatment strategies to protect the brain against ischemic injury will improve clinical outcomes. One of the important approaches is exploitation of endogenous ability of brain to “condition” itself against severe ischemia (4). In this context, ischemic preconditioning (IPC) is defined as brief episodes of ischemia which can protect brain from subsequent severe ischemic insult (5-6). Detailed mechanistic study of this phenomenon has indicated that different pharmacological compounds can mimic IPC and induce an equally effective form of neuroprotection through activation of similar signalling pathways (7-8). In addition, it is possible to directly stimulate related end-effectors or activate general processes by affecting the metabolism, protein synthesis, ATP-sensitive potassium channels (KATP) and other involved targets (9-10). The main advantage of pharmacological preconditioning in comparison with IPC-like interventions is its clinical feasibility (11).
Pharmacological preconditioning can afford strong protection of CA1 neurons against ischemic hallenge (12). Several in vivo and in vitro studies have been conducted to evaluate the effect of morphine and other opioid receptor agonists in limiting the harmful effects of ischemia-reperfusion injury. Morphine, in an in vitro model of anoxia-reoxygenation, markedly improved mitochondrial respiratory activity and inhibited alterations in mitochondrial membrane fluidity and lipoperoxidation and blocked the enhanced release of cytochrome c and subsequent neural cell apoptosis (13-14). Treatment with delta- opioid receptor agonist or specific delta-opioid receptor-1 agonist reduced IR-induced neural damage. Morphine reduced cerebral infarct size and prevented short-term memory impairment. These effects are actually abolished by pre-ischemic treatment with KATP channel blocker (glibenclamide) or naloxone (15-16).
Previously, Peart and Gross showed that 5 days pre-treatment with implanted morphine pellets could produce a profound increase in cardiac tissue tolerance against ischemia, even during 24 hr of morphine withdrawal period (17). Similarly, chronic morphine treatment was effective in heart preconditioning among aged mice while acute opioid treatment had no effect (18). Further evaluation suggested that morphine dependence acts through specific signalling pathways, which are different from mechanisms in acute morphine treatment or classical preconditioning (19). Conversely, our recent studies on rat kidney confirmed opioid receptors and nitric oxide (NO) involvement in morphine-dependence induction of protection through the same pathways of classical preconditioning (20, 21).
Although acute morphine protection against ischemic brain injury is a dose-dependent and well studied phenomenon (22), chronic use of morphine to prevent neuronal injury following cerebral ischemia has not been investigated up to now. Moreover, different opioid receptor subtypes endure changes post ischemia. This finding correlates with histological damage to the brain tissue (23-25). Based on the aforementioned data, we tried to examine the role of opioid receptors in MD-induced neuroprotection. Recent study on kidney ischemia model has shown that MD protects kidney through balancing post ischemia NO levels (21), therefore here, possible involvement of NO pathway was also evaluated.
Material and Methods
Animals and experimental protocol
Eighty male BALB/c mice (prepared from Razi institute, Karaj, Iran) weighting 25-30 g maintained on a standard laboratory conditions with the free access to water tap and chow. Experimental procedures confirmed by ethics committee of Tehran University of Medical Sciences and National institute of health (NIH) guide for the care and use of laboratory animals. Animals equally divided into 8 groups as follows:
- Sham: 5 days subcutaneous (SC) normal saline administration without bilateral common carotid arteries occlusion (BCCO)
- IR (Ischemia Reperfusion): 5 days normal saline administration (SC), 30 min global brain ischemia
- MD+Sham: 5 days morphine administration (SC) prior to sham operation
- MD+IR: 5 days morphine administration (SC) followed by 30 min global brain ischemia
- Nal+IR: 5 days normal saline administration (SC) followed by 30 min global brain ischemia, Naloxone (non-selective opioid receptor antagonist) (5 mg/kg) injection 30 min before surgical procedure
- MD+ Nal +IR: 5 days morphine administration (SC) followed by 30 min global brain ischemia, naloxone (5 mg/kg) injection 30 min after the last morphine dose
- L-NAME+IR: 5 days normal saline administration (SC) followed by 30 min global brain ischemia, L-NAME (50 mg/kg) injection 30 min before surgical procedure
- MD+L-NAME+IR: 5 days morphine administer-ation (SC) dose followed by 30 min global brain ischemia L-NAME (50 mg/kg) injection 30 min after the last morphine dose
In all groups global brain ischemia followed by 24 hr (n=6) or 72 hr (n=4) reperfusion period.
Drugs and reagent
The following drugs were used: morphine sulfate (Tolid-daru, Tehran, Iran), naloxone (Temad, Tehran, Iran), ketamine (Temad, Tehran, Iran) and xylazine (Temad, Tehran, Iran). Morphine was dissolved in saline; all drug solutions were prepared immediately before administration. Ketamine and xylazine were administrated intraperitoneally (IP). Morphine and naloxone were administered subcutaneously (SC).
Morphine dependence
Mice were morphine dependent by subcutaneous morphine sulphate administration for 5 consecutive days. Briefly during the first 4 days morphine was injected twice a day (day 1&2: 10 m/kg, day 3&4: 15 mg/kg; 8 am and 8 pm). The last morphine dose (30 mg/kg) was injected 4 hr before ischemia induction. This method has already been validated in rat model of morphine dependence (20). Naloxone 5 mg/kg or L-NAME (50 mg/kg) were administered 30 min after the last morphine dose and 3.5 hr before ischemia induction.
Global brain ischemia reperfusion
Four hr after the last morphine injection, mice were lightly anesthetized using mixture of ketamine (50 mg/kg) and xylazine (10 mg/kg). A midline incision was made along the neck to expose the left and right common carotid arteries followed by vagus nerve release from the surrounding tissues. Both common carotid arteries were carefully dissected away from the vagus and encircled with 6-0 suture to enable later occlusion with microsurgery clips. When animals were semiconscious, bilateral carotid arteries were occluded by an arterial clamp for global cerebral ischemia induction which was confirmed by animal loss of consciousness, eyeball whitening and mydriasis. Half an hr later, clamps were removed and the incision was closed in layers. Mice in each group were subjected to 24 hr reperfusion for protein expression and enzymatic activity assessments (n=6) or 72 hr for histological evaluations (n=4). Body temperature was monitored during the surgical procedure using a rectal probe and maintained at 36 ± 0.5°C (26).
Behavioural experiment
Passive avoidance test (PAT)
Passive avoidance test was done using the shuttle box consisting two compartments of light and dark with a guillotine door between. The floor was covered with stainless steel grids placed at 1 cm intervals. Intermittent electric shocks (50 Hz, 1 sec, 0.5 mA intensity) were delivered to the black compartment grid floor by an insulated stimulator. First, animals were habituated to the experimental conditions. Each animal was then gently placed in the light compartment equipped with an electric bulb. After 5 sec, the guillotine door was retracted and the animal was allowed to enter the dark compartment. Animals that waited more than 100 sec to cross were excluded from the study. Once the animal crossed with all four paws to the next compartment, the guillotine door was closed and the animal returned to the home cage after 10 sec (habituation trial). The acquisition trial was performed 30 min after the habituation trial. The animal was placed in the light compartment and 5 sec later the guillotine door was opened. As soon as the animal crossed to the dark compartment, the door was closed and a foot shock (50 Hz, 1 sec, 0.5 mA) was immediately induced. Retention trial was performed 24 hr after acquisition trial and ischemic lesion without applying electric shock. The time that animal spent in the light compartment before crossing to the dark compartment was measured as latency time and compared between different groups.
Histological analyses
Three days after global brain ischemia, animals were deeply anesthetized and transcardially perfused with 20 ml PBS (0.1 molar, pH 7.4) followed by 50 ml of chilled paraformaldehyde (4%) in PBS. Brains were then removed and post-fixed in 4% paraformaldehyde overnight. After dehydration in graded concentration of ethanol and butanol, brains were first embedded in paraffin and then cut into 7 µm- thick serial sections. Neuronal cell loss was assessed by histological examination of the dorsal hippocampus (CA1 region) of brain sections stained with Cresyl Violet. Intact pyramidal neurons in CA1 region was counted under a light microscope (Olympus, Hamburg, Germany) at 400× magnification. Four sections were counted in area of 53500 µm2 in each mouse.
To detect DNA fragmentation in degenerating neurons and find an index for apoptotic cell death, sections were processed for TUNEL nuclear staining using an in situ cell death detection kit (Roche Molecular Biochemicals kit, Germany). Briefly, sections were deparaffinised and treated with proteinase K solution. To block endogenous peroxidase activity, sections were incubated in 3% H2O2 for 10 min, re-incubated in TUNEL Reaction Buffer for 10 min, and then incubated in TUNEL Reaction Mixture for 1 hr at 37-40°C in humidified chamber. Finally, the tissue sections were incubated with 0.05% 3, 3-diaminobanzidine (DAB) substrate for 1-2 min to visualize apoptotic cells and counterstained with Gill’s hematoxylin for 30 sec (27). Apoptotic cells were observed under light microscope with 400X magnification (Olympus, Hamburg, Germany). Positive Controls were provided through incubating the sections with DNAase (3000U/ml in 50 mM Tris-HCl, pH 7.5, 1 mg/ml BSA) for 10 min at 15°C to induce DNA strand breaks prior to labelling procedure. Negative Controls were also prepared through incubating the sections only with label solution (without terminal transferase) instead of TUNEL reaction mixture (27).
Western blot analysis
To measure Bcl-2 and Bax proteins expression, animals were deeply anesthetized 24 hr post global brain ischemia. Brain was immediately removed from the skull, and hippocampus was dissected. The hippocampus was rapidly frozen by immersion in liquid nitrogen before being sealed into vials and stored at -70°C. Tissue samples were lysed in RIPA lysis buffer (#9806, Cell Signalling Technology, Italy) on ice for 30 min and centrifuged at 13000 g for 20 min at 4°C. Protein concentration was determined by spectrophotometric method. Loading buffer was added to the samples and boiled for 5 min at 95°C. Then, they were run on 10% sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membrane. Membranes were blocked with 5% milk in Tris-buffered saline with Tween (TBST) for 1 hr and incubated with primary antibodies including β-actin [Abcam; β-actin (ab8226), as loading control, Cell Signalling Technology, Italy], anti-Bax [#2772, Rabbit polyclonal antibody for BAX, Cell Signalling Technology, Italy] and anti-Bcl-2 [#2876, Rabbit polyclonal antibody for Bcl-2, Cell Signalling Technology, Italy] at 4°C overnight. Membranes were then washed and incubated with the HRP-conjugated anti-mouse secondary antibody [#7072, Cell Signalling Technology, Italy] for 1 hr at room temperature in the dark and scanned for analysis of the relative level of each protein expression to β-actin expression.
Statistical analysis
Results were expressed as mean ± SEM. Analysis was performed using SPSS statistical software, version 16.0. All data were analyzed by One-way Analysis of variance (ANOVA), followed by post hoc Tukey’s test for between-group comparisons. P<0.05 considered to be statistically significant.
Results
Effect of morphine dependence on memory performance
Passive avoidance test was done for all experimental groups to determine the effects of ischemic injury and interventions on learning and memory performance. As shown in Figure 1, mean latency time in the retention trial significantly decreased after IR (57.33±15.3; P<0.01 vs Sham). Morphine dependence did not change it in sham operated animals but it increased latency time post ischemia (147±20.03, P<0.05 vs IR). Naloxone by itself had no effect on post ischemia memory performance, but in morphine dependent group it decreased latency time (60.15±18.26, P<0.05 vs MD+IR). Latency time in L-NAME treated group significantly decreased compared with MD+IR group (78.03±10.57, P<0.01 vs. MD+IR).
Figure 1.
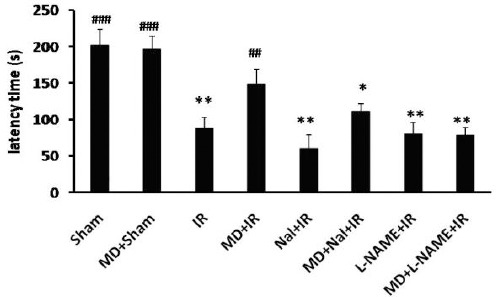
Latency time in PAT (Mean±SE, n=10) in different groups. Ischemia reperfusion (IR), morphine dependence (MD), Naloxone (Nal). One-way ANOVA followed by post-hoc Tucky test to compare the mean latency time between groups. ## P<0.01 and ### P<0.001 vs. IR group and *P<0.05 and ** P<0.01 vs. MD+IR group
Histological assessment
TUNEL staining was applied 72 hr post ischemia to examine DNA fragmentation in neurons undergoing post ischemia apoptosis. Three sections from each hippocampus and from each section, one visual field was observed under light microscope (400X). The ratio of apoptotic to the total cell number in the visual field represented as the apoptotic cell percentage. TUNEL positive cells were detected very rarely in the hippocampal sections of sham operated groups (Figure 2 A). IR induced a dramatic increase in the number of dark brown TUNEL-positive CA1 neurons (75.6 ± 4.1%, P<0.001 vs. Sham, Figure 2 B and H). Morphine dependence significantly decreased DNA fragmentation post IR (39±2.3%, P<0.05 vs IR, Figure 2 C and H). Naloxone by itself had no effect on neuronal cell apoptosis post ischemia, but it abolished morphine dependence protective effect on decreased neuronal cell apoptosis (67.9±2.6%, P< 0.05 vs MD+IR, Figure 2 D, E and H). L-NAME pre-treatment decreased morphine dependence neuroprotective effect (62.5 ± 3.2%, P< 0.05 vs. MD+IR, Figure 2 F, G and H).
Figure 2.
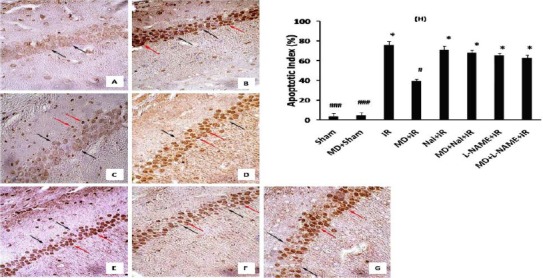
Effects of morphine dependency on ischemia-induced apoptosis of hippocampal CA1 neurons. Representative pictures of sham (A), IR (B), MD+IR (C), Nal+ IR (D), MD+ Nal+ IR (E), L-NAME+IR (F) and MD+L-NAME+IR (G) 72 hr after ischemia (400×). Black arrows are indicating intact cells and red arrows indicating apoptotic cells. Data has been expressed as (Mean± SEM). One-way ANOVA followed by Ttucky test was applied to compare the mean percentage of apoptosis between groups. # P<0.05 and ### P<0.001 vs. IR group and *P<0.05 vs. MD+IR group (H)
After 72 hr reperfusion, brains were fixed with paraformaldehyde (4%). Coronal sections were prepared from paraffinized brains and were stained with Cresyl Violet. Then the viable neurons in CA1 region were counted. Pyramidal cells of CA1 region in sham group had normal arrangement, round shape, transparent conformation and intact nuclei (Figure 3 A). In contrast, 72 hr post ischemia, most CA1 pyramidal cells were shrunken (viable cells ratio = 20.5 ± 2.2%, P<0.001, Figure 3 B and H), however MD increased viable cell number (58.6 ± 3.8%, P<0.01 vs. IR, Figure 3 C and H). Naloxone (25.2 ± 4.6%, P< 0.01 vs. MD+IR, Figure 3 D, E and H) and L-NAME (24.4 ± 3.8%, P< 0.01 vs. MD+IR, Figure 3 F, G and H) decreased viable cell number in morphine dependent mice.
Figure 3.
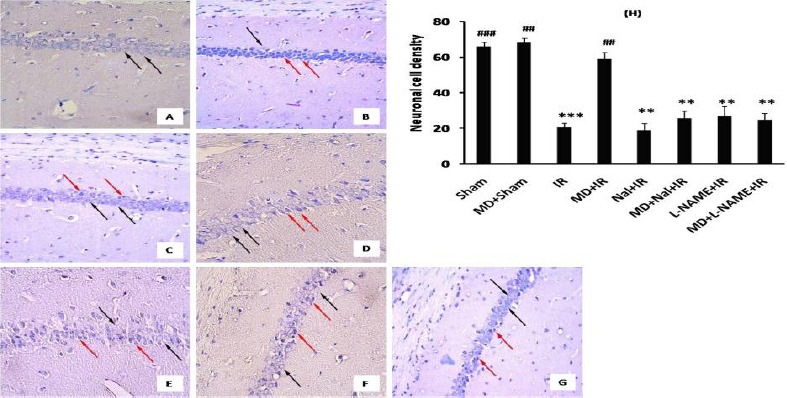
Effect of morphine dependency on ischemia-induced neuronal cell loss. Representative pictures of sham (A), IR (B), MD+IR (C), Nal+ IR (D), MD+ Nal+ IR (E), L-NAME+IR (F) and MD+L-NAME+IR (G) 72 hr after ischemia (400×). Black arrows are indicating intact cells and red arrows indicating necrotic cells. Data has been expressed as the number of counted live hippocampal CA1 neurons (Mean± SEM). Statistical analysis for cell density was done using one-way ANOVA followed by Tucky test (Mean± SEM) (H). ## P<0.01 and ### P<0.001 vs. IR group and **P<0.01 and *** P<0.001 vs. MD+IR group
Bcl-2 and bax protein expression
Protein expression has been normalized to β-actin expression in each sample and showed as protein expression to β-actin expression ratio. Expression of pro-apoptotic protein Bax and anti-apoptotic protein Bcl-2 significantly increased 24 hr post ischemia (P<0.05 vs Sham, Figure 4). Morphine dependence decreased the expression of Bax protein and increased Bcl-2 protein expression 24 hr post ischemia (P<0.05 vs. IR, Figure 4). Noloxone administration before ischemia blocked the effect of morphine dependence on Bax and Bcl-2 protein expression (P<0.05 and P<0.01 vs. MD+IR, respectively, Figure 4). L-NAME by itself could not change the expression of Bax or Bcl-2 post ischemia, but abolished the effect of morphine dependence on expression of Bax and Bcl-2 (P<0.05 vs. MD+IR, Figure 4).
Figure 4.
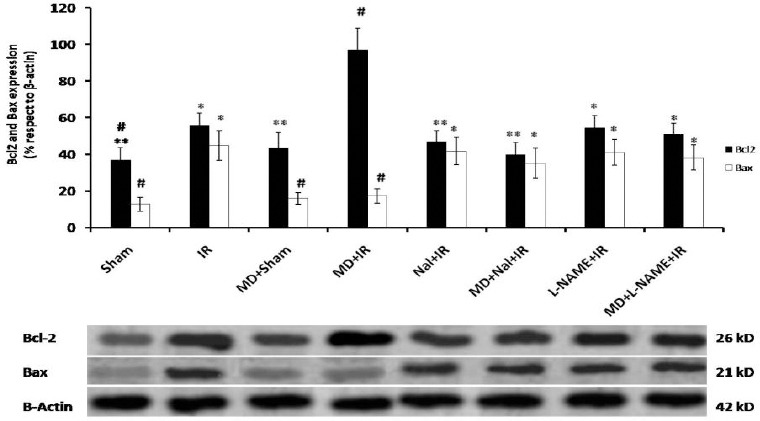
Expression of pro-apoptotic protein Bax and anti-apoptotic protein Bcl-2 in samples prepared from hippocampal tissue after treatment. The results show the ratio of Bax or Bcl-2 protein expression to β-actin expression (upper panel). Data are shown as Mean ± SEM. Ischemia reperfusion (IR), morphine dependence (MD), Naloxone (Nal). One-way ANOVA followed by post-hoc Tucky test was applied to compare the mean rate of protein expression between groups. # P<0.05 and ## P<0.01 vs. IR group. *P<0.05 and **P<0.01 vs. MD+IR group
Discussion
Finding new molecular mechanisms to increase vital organs tolerance against ischemia is the point of interest among basic scientists and clinicians. The first ideas of tissue preconditioning through induction of morphine dependence in animals comes from the studies on heart that showed morphine dependence potentiated protection via specific molecular mechanisms that are different from the classical preconditioning pathways(17-19).
In the present study, we set out to clarify the morphine dependence potential role in brain tolerance against ischemia and contribution of opioid receptors and NO in MD-induced ischemia protection. Results of the behavioural studies confirmed the effect of morphine dependence on preserving memory performance after global brain ischemia. Biochemical and histological experiments showed that MD decreased oxidative stress, apoptotic cell death in CA1 region and increased cell survival after ischemia. These effects were abolished in naloxone or L-NAME treated groups and suggested opioid receptors and NO contributions in MD induced protection. Same results have been obtained in kidney model of ischemia (21).
Morphine dependence improves post ischemia memory
According to Motamedi and et al, morphine dependence effects on rat learning and memory is a task-dependent phenomenon and cannot significantly affect passive avoidance test outcomes (30). In the present study, memory performance in morphine dependent mice is similar to the sham group, which confirms that the present protocol of dependence does not affect memory in normal mice. However, it affects post ischemia memory formation through increasing latency time in PAT (Figure 1). Biochemical and histological evaluations show that the PAT results in MD mice are correlated with improved tissue damage in this group.
Morphine dependence effects on post ischemia neuronal cell survival
CA1 neurons loss was seen 2-4 days post global cerebral ischemia, and lasted for weeks (3, 31). In the present study, hippocampus slices were prepared 3 days post ischemic insult, when the CA1 neuronal loss reached its peak. Morphine dependence significantly increased live neurons in CA1 region 72 hr post ischemia (Figure 3). In a recent study, Dong et al, showed that morphine treatment (1 mg/kg, 60 min before ischemia) decreased neuronal degeneration, number of apoptotic cells and caspase-3 expression in rats. Increasing morphine dose to 7 mg/kg strongly decreased pathological changes and number of apoptotic cells and caspase-3 expression (22, 32). Our results showed that TUNEL positive cells were markedly reduced in ischemic brains prepared from morphine dependent mice compared to vehicle-treated control mice (Figure 2). Apoptosis maximally occurs 72 hr post ischemia. This is due to the anti- and pro- apoptotic proteins changes in primary phases of reperfusion (31). Therefore we preferred to prepare samples for protein expression after 24 hr of reperfusion. Ischemia reperfusion increased the expression of Bax and Bcl-2. Increased Bcl-2 expression might be a compensatory mechanism against apoptosis. Morphine dependence decreased Bax expression and Bax/Bcl-2 ratio but increased Bcl-2 expression and CA1 neuronal survival 72 hr post ischemia.
Cell survival and death is strictly related to the oxidative state because it interacts with cellular macromolecules and signalling pathways. Reactive oxygen species (ROS) through macromolecules oxidation act as a trigger for apoptotic pathways (33). Increased ratio of Bax/Bcl-2 expression after ischemia reperfusion could be related to of ROS production, which reflected in increased MDA level, and decreased anti-oxidative mechanisms including SOD and catalase activities. These results are concomitant with limited neuronal cell death and improved Bax/Bcl-2 ratio in the morphine dependent group.
Role of opioid receptors
Single dose naloxone administration before ischemia affected all protective aspects of morphine dependence including behavioural test, neuronal cell loss apoptosis and oxidative state. Interestingly, these findings suggest that opioid receptors activation just before ischemia is crucial to express protective effects of chronic morphine use against ischemia. In cardiac models of ischemia, cardioprotective effects of δ1-opioid receptor agonist have been abolished by δ1-opioid antagonist administration either 30 min before or 48 hr after receptor activation (34). In kidney model of ischemia, naloxone administration just before ischemia also abolished protective effects of chronic morphine use (20, 21).
Boutinet et al, have previously reported early changes in different opioid receptors subtypes after focal cerebral ischemia in different sub-cortical regions in mice (24). Delta and mu opioid receptors density in the cortex of mice were decreased 6 h after middle cerebral artery occlusion (MCAO) which was correlated with later histological damage (25). Therefore opioid receptors activation in the early phases after ischemia seems to be crucial in ischemia injury outcome. In the present work, naloxone by itself had no effect on the ischemic injury but it affected varying degrees of protection afforded by chronic morphine use. This may suggest that reoccupation of opioid receptors in the first hr after ischemia is crucial for morphine dependence to be effective against hippocampus ischemic damage.
On the other hand, protective effects of chronic morphine use both in cardiac and renal tissues lasted even 24 hr after the last morphine dose (17, 21). In the present study, samples for biochemical and histological studies have been prepared 24 and 72 hr after the last morphine dose, and their results confirmed the morphine dependence persisting effect even after 3 days. Boutinet et al also found that opioid receptors underwent changes at later phases after ischemia. Utilizing the quantitative autoradiography method, they showed that mu, delta and kapa opioid receptor expressions have been decreased in the ipsilateral cortex one day after MCAO and stayed low up to 7 days after ischemia which is correlated with strong histological damage (23). Although we did not quantify opioid receptors density during reperfusion, these data strongly suggest that both acute and late signalling mechanisms involved in MD induced protection are opioid receptor dependent.
Balanced NO production as a key element in MD induced neuroprotection
NO role in brain ischemia reperfusion injury, like other organs is a matter of controversy. Some studies have proved its cytotoxicity while others reported NO as one of the key signalling factors in brain preconditioning against ischemia (35, 36). This controversy mainly relates to the enzymatic source and tissue level of NO (35, 37). In the present study, treatment with L-NAME in morphine dependent group suppressed post ischemia NO production and all mentioned morphine protective effects. So L-NAME increased apoptotic cell death and Bax/Bcl-2 ratio of CA1 region. These findings also focused on NO crucial role in protective effects of chronic morphine use against ischemia reperfusion injury.
Altogether, the results suggested that chronic morphine treatment before global cerebral ischemia decreased IR induced hippocampal tissue damage, apoptosis and memory deficits through the opioid receptors and NO dependent pathways.
Conclusion
Considering the role of opioid receptors on neuroprotection, their post ischemic density and function changes and protective effects on tissue damage outcomes, recent data of morphine dependence-induced preconditioning may provide important insights on molecular mechanisms involved in pharmacological neuroprotection. Accordingly, chronic morphine use despite its minus social and cultural features can be considered as an alternative for neuroprotective implications. Moreover, based on morphine dependence protective role in cerebral ischemia, we should follow the safe and feasible ways of using this method to protect the brain from other acute and chronic neuronal disorders.
Acknowledgment
The results described in this paper were part of PhD student thesis. All authors appreciate Iran University of Medical Sciences, Tehran, Iran for funding all aspects of the present study.
References
- 1.Kinlay S. Changes in stroke epidemiology, prevention, and treatment. Circulation. 2011;124:494–496. doi: 10.1161/CIRCULATIONAHA.111.069633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanco M, Castillo J. Stroke in 2012: Major advances in the treatment of stroke. Nat Rev Neurol. 2013;2:68–70. doi: 10.1038/nrneurol.2012.274. [DOI] [PubMed] [Google Scholar]
- 3.Bendel O, Alkass K, Bueters T, von Euler M, von Euler G. Reproducible loss of CA1 neurons following carotid artery occlusion combined with halothane-induced hypotension. Brain Res. 2005;1033:135–142. doi: 10.1016/j.brainres.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara A, El Bejaoui S, Seyen S, Tirelli E, Plumier JC. The usefulness of operant conditioning procedures to assess long-lasting deficits following transient focal ischemia in mice. Behav Brain Res. 2009;205:525–534. doi: 10.1016/j.bbr.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 2007;38:680–685. doi: 10.1161/01.STR.0000251444.56487.4c. [DOI] [PubMed] [Google Scholar]
- 6.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- 9.Andoh T, Chock PB, Chiueh CC. Preconditioning-mediated neuroprotection: role of nitric oxide, cGMP, and new protein expression. Ann N Y Acad Sci. 2002;962:1–7. doi: 10.1111/j.1749-6632.2002.tb04051.x. [DOI] [PubMed] [Google Scholar]
- 10.Correia SC, Carvalho C, Cardoso S, Santos RX, Santos MS, Oliveira CR, et al. Mitochondrial preconditioning: a potential neuroprotective strategy. Front Aging Neurosci. 2010;2:211–218. doi: 10.3389/fnagi.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yellon DM, Dana A. The preconditioning phenomenon: A tool for the scientist or a clinical reality? Circ Res. 2000;87:543–550. doi: 10.1161/01.res.87.7.543. [DOI] [PubMed] [Google Scholar]
- 12.Thompson JW, Dave KR, Young JI, Perez-Pinzon MA. Ischemic preconditioning alters the epigenetic profile of the brain from ischemic intolerance to ischemic tolerance. Neurotherapeutics. 2013;10:789–797. doi: 10.1007/s13311-013-0202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peart JN, Gross ER, Gross GJ. Opioid-induced preconditioning: recent advances and future perspectives. Vascul Pharmacol. 2005;42:211–218. doi: 10.1016/j.vph.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 14.He X, Sandhu HK, Yang Y, Hua F, Belser N, Kim DH, Xia Y. Neuroprotection against hypoxia/ischemia: delta-opioid receptor-mediated cellular/molecular events. Cell Mol Life Sci. 2013;70:2291–2303. doi: 10.1007/s00018-012-1167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narita M, Kuzumaki N, Miyatake M, Sato F, Wachi H, Seyama Y, et al. Role of delta-opioid receptor function in neurogenesis and neuroprotection. J Neurochem. 2006;97:1494–1505. doi: 10.1111/j.1471-4159.2006.03849.x. [DOI] [PubMed] [Google Scholar]
- 16.Charron C, Messier C, Plamondon H. Neuroprotection and functional recovery conferred by administration of kappa- and delta 1-opioid agonists in a rat model of global ischemia. Physiol Behav. 2008;93:502–511. doi: 10.1016/j.physbeh.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Peart JN, Gross GL. Morphine-tolerant mice exhibit a profound and persistent cardioprotective phenotype. Circulation. 2004;109:1219–1222. doi: 10.1161/01.CIR.0000121422.85989.BD. [DOI] [PubMed] [Google Scholar]
- 18.Peart JN, Gross GL. Chronic exposure to morphine produces a marked cardioprotective phenotype in aged mouse hearts. Exp Gerontol. 2004;39:1021–1026. doi: 10.1016/j.exger.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 19.Peart JN, Gross GJ. Cardioprotective effects of acute and chronic opioid treatment are mediated via different signaling pathways. Am J Physiol Heart Circ Physiol. 2006;291:H1746–1753. doi: 10.1152/ajpheart.00233.2006. [DOI] [PubMed] [Google Scholar]
- 20.Habibey R, Pazoki-Toroudi H. Morphine dependence protects rat kidney against ischaemia-reperfusion injury. Clin Exp Pharmacol Physiol. 2008;35:1209–1214. doi: 10.1111/j.1440-1681.2008.04986.x. [DOI] [PubMed] [Google Scholar]
- 21.Habibey R, Ajami M, Ebrahimi SA, Hesami A, Babakoohi S, Pazoki-Toroudi H. Nitric oxide and renal protection in morphine-dependent rats. Free Radic Biol Med. 2010;49:1109–1118. doi: 10.1016/j.freeradbiomed.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 22.He Dong, Xiangyu Ji, Dong Wang, Yueyi Ren, Shiduan Wang, Jianfang Song. Effect of morphine preconditioning on neuronal apoptosis following cerebral ischemia/reperfusion injury. Neural Regen Res. 2010;5:1144–1149. [Google Scholar]
- 23.Boutin H, Catherine A, Mackenzie ET, Jauzac P, Dauphin F. Long-term alterations in mu, delta and kappa opioidergic receptors following middle cerebral artery occlusion in mice. Acta Neuropathol. 2007;114:491–500. doi: 10.1007/s00401-007-0269-7. [DOI] [PubMed] [Google Scholar]
- 24.Boutin H, Dauphin F, Jauzac P, MacKenzie ET. Exofocal alterations in opioidergic receptor densities following focal cerebral ischemia in the mouse. Exp Neurol. 2000;164:314–321. doi: 10.1006/exnr.2000.7400. [DOI] [PubMed] [Google Scholar]
- 25.Boutin H, Jauzac P, MacKenzie ET, Dauphin F. Potential use of early alterations in mu and delta opioid receptors as a predictive index for delayed brain ischemic damage. Neurobiol Dis. 2003;13:63–73. doi: 10.1016/s0969-9961(03)00033-0. [DOI] [PubMed] [Google Scholar]
- 26.Zare Mehrjerdi F, Aboutaleb N, Habibey R, Ajami M, Soleimani M, Arabian M, et al. Increased phosphorylation of mTOR is involved in remote ischemic preconditioning of hippocampus in mice. Brain Res. 2013;14(1526):94–101. doi: 10.1016/j.brainres.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 27.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86:271–278. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- 29.Honorio JE, Jr, Vasconcelos GS, Rodrigues FT, Sena Filho JG, Barbosa-Filho JM, Aguiar CC, et al. Monocrotaline: histological damage and oxidant activity in brain areas of mice. Oxid Med Cell Longev. 2012;2012:541–547. doi: 10.1155/2012/697541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motamedi F, Ghasemi M, Davoodi FG, Naghdi N. Comparison of learning and memory in morphine dependent rats using different behavioral models. Iran J Pharm Res. 2010;21:225–230. [Google Scholar]
- 31.Ajami M, Eghtesadi S, Razaz JM, Kalantari N, Habibey R, Nilforoushzadeh MA, et al. Expression of Bcl-2 and Bax after hippocampal ischemia in DHA +EPA treated rats. Neurol Sci. 2011;32:811–88. doi: 10.1007/s10072-011-0621-5. [DOI] [PubMed] [Google Scholar]
- 32.Rehni AK, Singh TG, Jaggi AS, Singh N. Pharmacological preconditioning of the brain: a possible interplay between opioid and calcitonin gene related peptide transduction systems. Pharmacol Rep. 2008;60:904–913. [PubMed] [Google Scholar]
- 33.Loh KP, Huang SH, De Silva R, Tan BK, Zhu YZ. Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res. 2006;3:327–37. doi: 10.2174/156720506778249515. [DOI] [PubMed] [Google Scholar]
- 34.Frassdorf J, Weber NC, Obal D, Toma O, Müllenheim J, Kojda G, et al. Morphine induces late cardioprotection in rat hearts in vivo: the involvement of opioid receptors and nuclear transcription factor kappaB. Anesth Analg. 2005;101:934–941. doi: 10.1213/01.ane.0000172130.70274.84. [DOI] [PubMed] [Google Scholar]
- 35.Huang PL. Nitric oxide and cerebral ischemic preconditioning. Cell Calcium. 2004;36:323–329. doi: 10.1016/j.ceca.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Strijbos PJ. Nitric oxide in cerebral ischemic neurodegeneration and excitotoxicity. Crit Rev Neurobiol. 1998;12:223–243. doi: 10.1615/critrevneurobiol.v12.i3.40. [DOI] [PubMed] [Google Scholar]
- 37.Goligorsky MS, Brodsky SV, Noiri E. Nitric oxide in acute renal failure: NOS versus NOS. Kidney Int. 2002;61:855–861. doi: 10.1046/j.1523-1755.2002.00233.x. [DOI] [PubMed] [Google Scholar]