

Abstract
We have previously shown that 2-acetylcyclopentanone (2-ACP), an enolate-forming 1,3-dicarbonyl compound, provides protection in cell culture and animal models of oxidative stress. The pathophysiology of ischemia-reperfusion injury (IRI) involves oxidative stress, and, therefore, we determined the ability of 2-ACP to prevent this injury in a rat liver model. IRI was induced by clamping the portal vasculature for 45 minutes (ischemia phase), followed by recirculation for 180 minutes (reperfusion phase). This sequence was associated with substantial derangement of plasma liver enzyme activities, histopathological indices, and markers of oxidative stress. The 2-ACP (0.80–2.40 mmol/kg), administered by intraperitoneal injection 10 minutes prior to reperfusion, provided dose-dependent cytoprotection, as indicated by normalization of the IRI-altered liver histologic and biochemical parameters. The 2-ACP (2.40 mmol/kg) was also hepatoprotective when injected before clamping the circulation (ischemia phase). In contrast, an equimolar dose of N-acetylcysteine (2.40 mmol/kg) was not hepatoprotective when administered prior to reperfusion. Our studies to date suggest that during reperfusion the enolate nucleophile of 2-ACP limits the consequences of mitochondrial-based oxidative stress through scavenging unsaturated aldehyde electrophiles (e.g., acrolein) and chelation of metal ions that catalyze the free radical-generating Fenton reaction. The ability of 2-ACP to reduce IRI when injected prior to ischemia most likely reflects the short duration of this experimental phase (45 minutes) and favorable pharmacokinetics that maintain effective 2-ACP liver concentrations during subsequent reperfusion. These results provide evidence that 2-ACP or an analog might be useful in treating IRI and other conditions that have oxidative stress as a common molecular etiology.
Introduction
Significant hepatocyte damage occurs when blood flow to the liver is transiently interrupted (ischemia phase) and then subsequently restored (reperfusion phase). This sequence of warm ischemia-reperfusion injury (IRI) can occur either purposefully (e.g., during liver transplantation) or as part of disease pathogenesis (e.g., sinusoidal obstruction, Budd-Chiari syndrome). The pathophysiology of warm IRI involves oxidative stress, mitochondrial dysfunction, Kupffer cell activation, and increased proinflammatory cytokine signaling. These deleterious processes are initiated during the ischemic phase and subsequently exacerbated as a function of cellular reoxygenation during the reperfusion phase (Klune and Tsung, 2010). Based on this pathogenic complexity, it has been difficult to identify primary mediators or mechanisms that might be relevant therapeutic targets. Nonetheless, oxidative stress appears to be a significant component of IRI (Papadopoulos et al., 2013), and the use of antioxidant compounds that decrease cellular levels of reactive oxygen species (ROS) is an obvious pharmacotherapeutic strategy. However, clinical trials provided only mixed support for antioxidant therapy (Jaeschke and Woolbright, 2012; Papadopoulos et al., 2013).
There is substantial evidence that the toxic consequences of oxidative stress are mediated by acrolein, 4-hydroxy-2-nonenal (HNE), and other unsaturated aldehydes generated during membrane lipid peroxidation (Uchida, 2003; Wood et al., 2006; Ellis, 2007; Yoshida et al., 2009). As reactive electrophiles, these aldehydes cause cytotoxicity by depleting cellular glutathione (GSH) levels and by reacting with functionally critical nucleophilic amino acid residues of proteins (Fritz and Petersen, 2013; LoPachin and Gavin, 2014). Therefore, as an alternative approach to antioxidants, nucleophilic chemicals that form irreversible adducts with these electrophiles might arrest oxidative damage and mitigate hepatocyte demise during reperfusion. Indeed, the thiol-based nucleophile, N-acetylcysteine (NAC), can reduce hepatocyte damage in animal models of IRI (Jegatheeswaran and Siriwardena, 2011). In previous studies, we showed that acetylacetone, 2-acetylcyclopentanone (2-ACP), and other 1,3-dicarbonyl compounds (Fig. 1A) provided direct protection in cell culture models of oxidative stress (H2O2) and electrophile (acrolein)-induced injury (LoPachin et al., 2011). These compounds ionize in aqueous solutions to form nucleophilic enolate anions (Fig. 1B). More recently, we demonstrated that 2-ACP prevented hepatotoxicity in a mouse model of acetaminophen overdose (Zhang et al., 2013). Whereas the idea that enolate-forming 1,3-dicarbonyl compounds, such as 2-ACP, might be cytoprotective is unprecedented, the concept stems from the recognition that curcumin (Fig. 1A), a phytopolyphenol with well documented cytoprotective capability (Begum et al., 2008), also possesses an enolizable 1,3-dicarbonyl function (pKa = 8.0; LoPachin et al., 2011). Furthermore, the chemistry of 1,3-dicarbonyl compounds is well known (Loudon, 2002; Bug and Mayr, 2003), and mechanistic studies have shown that the nucleophilic enolate of 2-ACP (Fig. 1B) mediates cytoprotection by inhibiting oxidative stress at multiple sites (LoPachin et al., 2011, 2012; Zhang et al., 2013). Because oxidative stress is a major pathogenic component of IRI, in the present study we determined the relative hepatoprotective efficacies of 2-ACP and NAC in a rat model of warm liver IRI.
Fig. 1.

Line structures and ionization of 1,3-dicarbonyl compounds. (A) Line structures for acetylacetone, 2-ACP, and curcumin. (B) Schematic diagram showing that loss of a proton from either the central (“α”) carbon in the diketo tautomer of 2-ACP or the hydroxyl group of the enol isomer yields the same resonance-stabilized enolate anion.
Materials and Methods
Reagents.
All chemicals, reagents, and experimental protectants were of the highest grade commercially available and were purchased from Sigma-Aldrich (St. Louis, MO). Bovine heart mitochondrial preparations were purchased from BioVision (Milpitas, CA).
Animals and Treatments.
All aspects of animal use in this study were in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Montefiore Medical Center Animal Care Committee. Adult male rats (Sprague-Dawley, 275–300 g; Taconic Farms, Germantown, NY) were used in this study. Rats were housed individually in polycarbonate boxes. Filtered drinking water and Purina Rodent Laboratory Chow (Purina Mills, St. Louis, MO) were available ad libitum. The animal room was maintained at approximately 22°C and 50% humidity with a 12-hour light/dark cycle.
IRI Model: Surgical Procedure.
Rats were fasted overnight with access to water, and experiments were begun the next morning. On the day of the experiment, rats were anesthetized (urethane, 1000 mg/kg i.p.), and the abdomen was shaved and swabbed with ethanol (70%), followed by a betadine preparation. A transverse abdominal incision (2.5 cm) was made, and the liver was elevated to expose the hepatic vasculature. Experimental IRI was induced according to a modification of Abe et al. (2009). Specifically, two atraumatic microclips (Roboz Surgical Instrument, Gaithersburg, MD) were used to cross-clamp the portal vein, hepatic artery, and bile duct above the right lateral lobe branch. This produced ischemia in the median and left lateral lobes (involving ∼70% of the liver), which was visually evident due to blanching of the lobes. The abdominal incision was closed, and body temperature was maintained at 37°C by a thermostatically controlled heating pad. Following the ischemic period, the clamps were removed and circulation was restored. The abdomen was closed for the duration of the reperfusion phase. Immediately following this period, animals were euthanized by bilateral pneumothorax, and blood was collected for subsequent determinations of liver-specific transaminase activities in plasma. Livers were excised, weighed, and frozen in liquid nitrogen for later biochemical analyses. Selected livers were fixed in ice-cold 10% phosphate-buffered formalin for histologic examination.
IRI Model: Experimental Approach.
Rats were randomly grouped (n = 10–15) according to the experimental treatment to be received. Preliminary studies showed that 45 minutes of ischemia followed by 180 minutes of reperfusion produced substantial liver damage, as indicated by both biochemical and histologic indices (see Results). As a general experimental protocol, putative hepatoprotectants (0.80–2.40 mmol/kg i.p.) were dissolved in dimethylsulfoxide (1%)/phosphate-buffered saline (PBS) and were injected 10 minutes prior to the induction of either ischemia or reperfusion. Vehicle control rats (n = 6) were anesthetized and received injections of dimethylsulfoxide-PBS (3 ml/g) in parallel with animals receiving hepatoprotectant. To control for the surgical procedure, sham-operated animals received a laparotomy. At 45 minutes postlaparotomy, the incision was closed and animals were euthanized 180 minutes later. Analyses of the respective blood and tissue samples for the vehicle- and sham-operated controls indicated no group mean statistical difference, and therefore, these control groups were combined and designated as sham/vehicle control.
Hepatoxicity Parameters and Histopathological Analyses.
To assess IRI-induced hepatocyte damage, the appearance of liver enzymes, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) in plasma was measured. In addition, plasma levels of lactate dehydrogenase (LDH) were determined as a general measure of cell damage. Cardiac blood was collected in heparin-coated tubes (1.5 ml; BD Biosciences, Franklin Lakes, NJ), and plasma samples were obtained by centrifugation (14,000g for 5 minutes). Samples were subsequently analyzed by an automated analyzer (Hitachi Modular Automated Clinical Chemistry Analyzer; Roche Diagnostics, Indianapolis, IN) and expressed as IU/ml plasma.
As indices of hepatocyte oxidative stress, unsaturated aldehyde products of lipid peroxidation and soluble thiol status were determined in liver homogenates prepared from the different experimental groups. To measure tissue concentrations of the unsaturated aldehydes, HNE, and malondialdehyde (MDA), frozen livers were pulverized and samples (2 g) were added to 5.5 ml radioimmunoprecipitation assay buffer (DeSeau et al., 1987) containing protease inhibitor cocktail and butylated hydroxytoluene (5 mM). Tissue samples were homogenized in a Dounce tissue grinder (10 strokes), and the homogenate was centrifuged at 500g (4°C) for 15 minutes to remove cellular debris. The supernatant was retained (S1); the pellet was washed once in radioimmunoprecipitation assay buffer (4.5 ml); and the supernatant (S2) was combined with S1. Total (free and protein-bound) unsaturated aldehydes were determined by the spectrophotometric method of Gerard-Monnier et al. (1998), as modified by Zhang et al. (2013). Briefly, an aliquot (200 μl) of the combined supernatant was added to 650 μl 1-methyl-2-phenylindole in an acetonitrile/methanol (3:1) mixture. The reaction was started by adding 150 μl 12 N hydrochloric acid. Absorbance (586 nm) was measured after incubation of the reaction mixture at 45°C for 60 minutes. The absorbance values used to determine MDA concentrations were based on a standard curve for 1,1,3,3-tetramethoxypropane as a source of MDA. To determine the respective HNE concentrations, parallel samples (200 μl) were added to the 1-methyl-2-phenylindole mixture, and the reaction was started by adding 150 μl methanesulfonic acid (37%) containing 100 μM Fe(III). Absorbance (586 nm) was measured after incubation at 45°C for 60 minutes. The final absorbance readings are linear functions of both the HNE and MDA concentrations, and, therefore, the HNE content can be derived by subtracting the previously determined MDA concentration from the combined unsaturated aldehyde content.
GSH and oxidized glutathione (GSSG) levels were determined by the method of Giustarini et al. (2013). To analyze GSSG, rat livers were homogenized in Tris buffer (50 mM) containing serine (2 mM), boric acid (20 mM), acivicin (0.020 mM), and N-ethylmaleimide (31 mM) and then deproteinized by trichloroacetic acid (TCA) precipitation. Liver homogenates were centrifuged at 14,000g (2 minutes), and the supernatant (S1) was retained. Unreacted N-ethylmaleimide was extracted from S1 using 3 volumes of dichloromethane, followed by centrifugation at 14,000g (30 seconds). The resulting supernatant (S2) was collected and was added (20 µl) to a cuvette containing PBS (925 µl); 20 mM 5,5′-dithiobis(2-nitrobenzoic acid) (5 µl), 4.8 mM NADPH (20 µl), and 20 IU ml−1 glutathione reductase (20 µl). As a blank, TCA (20 µl) was added to cuvettes in place of the S2 aliquot. Spectrophotometric absorbance was recorded at 412 nm (1 minute), and the sample slope or blank (TCA) slope was calculated. GSSG (0.10 µM) was subsequently added to the cuvette, and the absorbance was recorded at 412 nm for 1 minute. GSSG concentrations in liver homogenates (GSSGL) were calculated using the following algorithm: GSSGL = S × GSSGc/St × 49.5 (sample dilution factor) × 2 (dilution due to acidification)/protein concentration (mg/ml), where S = slope sample − slope blank, GSSGc = is the final concentration of standard GSSG in the cuvette (0.10 µM), and St = (slope sample + GSSG) − slope sample. Results are expressed as nmol/mg liver protein (±S.E.M.). To measure total GSH, S1 (10 μl) or GSH (thiol source) standards (4–64 μM) were added to a cuvette containing PBS (945 µl), 20 mM 5,5′-dithiobis(2-nitrobenzoic acid) (5 µl), 4.8 mM NADPH (20 µl), and 20 IU ml−1 glutathione reductase (20 µl). Absorbance was recorded at 412 nm (1 minute), and total GSH was expressed as nmol/mg liver protein (±S.E.M.).
For histologic assessment, liver samples were excised from rats in all experimental groups at the conclusion of the respective reperfusion phases. Tissue samples were fixed in ice-cold 10% buffered formalin solution, paraffin embedded, sectioned (5 μm), and stained with H&E. Sections from individual animals (n = 3 per experimental group) were blinded by code, and 5–8 fields on each slide were randomly selected and evaluated at the light-microscopy level for evidence of hepatocellular injury using standard morphologic criteria (e.g., necrosis, loss of architecture, vacuolization, karyolysis, apoptosis). The extent of cellular injury was determined semiquantitatively by assigning a severity score on a scale of 0–4, as described by He et al. (2009), where 0 = absent; 1 = mild; 2 = moderate; 3 = severe; and 4 = complete hepatocyte destruction. This scoring system was used to compare the relative extent of liver damage associated with the IRI plus vehicle and IRI plus ACP experimental groups.
Effects of 2-ACP and Malonate on Succinate Dehydrogenase Activity Determined In Vitro.
Succinate dehydrogenase (SDH; complex II) activity was measured spectrophotometrically in mitochondrial preparations as the rate of succinate-dependent reduction of 2,6-dichlorophenolindophenol (DCPIP; Wojtovich and Brookes, 2008). Briefly, SDH activity was determined in standard assay buffer containing Tris-SO4 (10 mM), succinate (5 mM), DCPIP (40 µM), KCN (1 mM), sucrose (250 mM), MgSO4 (2 mM), and K2SO4 (1 mM) at pH 7.4 and 25°C. The reaction was initiated by addition of mitochondrial protein (100 µg ml−1). The rate of DCPIP reduction was followed at 600 nm (ε = 21 × 103 M−1 cm−1; blue to colorless) for 60 minutes using a Molecular Devices SpectraMax Plus 384 plate reader (Sunnyvale, CA). To assess the effects of inhibitors on SDH, enzyme activities (µmol min−1 mg protein−1) were determined (n = 3–4 measurements per experimental group) in graded concentrations of sodium malonate dibasic (50–500 µM) or 2-ACP (500–1000 µM), and results were compared with control rates. The concentration of 1,3-dicarbonyl that inhibited 50% of enzyme activity (IC50 value) was calculated by the Cheng-Prusoff equation (Prism 3.0; GraphPad Software, San Diego, CA).
Calculations of Hard and Soft, Acids and Bases Quantum Mechanical Parameters.
To calculate hard and soft, acids and bases (HSAB) parameters for the electrophiles and nucleophiles involved in this study, the respective energies of the highest occupied molecular orbital (EHOMO) and the lowest unoccupied molecular orbital (ELUMO) were derived using Spartan14 (version 1.1.1) software (Wavefunction, Irvine, CA). For each chemical structure, ground state equilibrium geometries were calculated with Density Functional B3LYP 6-31G* in water starting from 6-31G* geometries. Global (whole-molecule) hardness (η) was calculated as η = (ELUMO − EHOMO)/2, and softness (σ) was calculated as the inverse of hardness or σ = 1/η. An electrophilicity index (ω) was calculated as ω = µ2/2η, here µ is chemical potential of the electrophile and µ = (ELUMO + EHOMO)/2. The nucleophilicity index (ω−) was calculated as ω− = ηA(µA − µB)2/2(ηA + ηB)2, where A = reacting nucleophile and B = reacting electrophile (see LoPachin et al., 2012 for more detailed discussion).
Statistical Analyses.
All statistical analyses were conducted using Prism 6.0 (GraphPad Software) with minimal significance set at the 0.05 level of probability. For analysis of liver enzymes in plasma and oxidative stress parameters, statistically significant differences between group mean data were determined by a Bonferroni test for multiple comparisons.
Results
Characterization of the IRI Model.
The ischemia-reperfusion sequence used in these studies caused substantial hepatocyte injury, as evidenced by elevations in plasma ALT, a liver-specific enzyme. Figure 2 shows that mean (±S.E.M.) ALT plasma levels rose from a control value of 80 ± 10 U/I to 7360 ± 390 U/I after 180 minutes of postischemia reperfusion. Severe IRI-induced parenchymal cell damage was substantiated by commensurate increases in plasma levels of AST and LDH (Fig. 2; Bessems et al., 2006). These enzyme alterations coincided with histopathological analyses that indicated moderate to severe liver cell death (Fig. 3). Measurements of liver homogenates also indicated significantly elevated indices of cellular oxidative stress, that is, unsaturated aldehyde accumulation and GSH loss (Fig. 4). The IRI model used in this study produces substantial liver damage and was therefore used to determine the relative hepatoprotective abilities of experimental protectants.
Fig. 2.

Dose-dependent effects of 2-ACP administered intraperitoneally 10 minutes prior to unclamping the portal circulation (reperfusion phase) on IRI-induced plasma appearance of ALT, AST, and LDH in rat (n = 10–15/group). The 2-ACP at the 0.80 mmol/kg dose was not effective relative to IRI + vehicle (data not shown). Data are expressed as mean activity ± S.E.M., and joining lines indicate statistically significant differences in treatment groups at **P < 0.01 level.
Fig. 3.

Representative photomicrographs illustrating parenchymal status in rat liver from control (A), IRI (B), and IRI + 2-ACP (C; 2.40 mmol/kg i.p.) experimental groups. Tissue was excised at the conclusion of the 180-minute reperfusion period, fixed, and then stained with H&E. The arrows on (B) (IRI group) denote the zonal distribution of severe, confluent hepatocyte necrosis involving the periportal regions. The perivenular regions remained viable. Magnification 100× (400× inset), and the scale bar equals 100 µm.
Fig. 4.

Effects of 2-ACP (2.40 mmol/kg i.p.) administered 10 minutes prior to unclamping the portal circulation (reperfusion phase) on (A) IRI-induced changes in MDA, HNE, and (B) GSH and GSSG concentrations in rat liver homogenates (n = 10/group). Data are expressed as mean percentage of control ± S.E.M. Joining lines indicate statistically significant differences in treatment groups at *P < 0.05, **P < 0.01, and ***P < 0.001 levels.
The 2-ACP Administered Prior to Reperfusion Is Hepatoprotective.
Initial studies determined the ability of 2-ACP to prevent the expression of injury during the reperfusion phase. Results indicate that 2-ACP (0.80–2.40 mmol/kg) injected 10 minutes before unclamping the hepatic circulation produced significant dose-dependent reductions in ALT, AST, and LDH plasma levels reflecting hepatocyte protection (Fig. 2). The changes in plasma biomarkers corresponded to histopathological alterations noted in the different experimental groups. Thus, all tissue sections from the control group exhibited unremarkable liver histology, that is, no evidence of ischemia or hepatocyte injury (histologic score = 0; Fig. 3A). In contrast, tissue sections from the IRI plus vehicle group collected after 180 minutes of reperfusion exhibited severe, confluent hepatocyte necrosis involving all of zone 1 and focally extending into zone 2. Eosinophilic acellular/proteinaceous material comprised the areas of zonal necrosis. Also evident was severe lobular hepatocyte apoptosis and scattered foci of spotty necrosis (Fig. 3B). Correspondingly, histologic assessment revealed moderate-to-severe levels of hepatocyte injury, that is, a mean (±S.E.M.) score of 2.91 ± 0.21. However, when 2-ACP (2.40 mmol/kg) was injected 10 minutes before unclamping the hepatic circulation, this level of severe IRI-induced hepatocyte damage was significantly reduced. Histologic analyses of tissue slides from the IRI plus ACP group indicated mild ischemic changes with rare hepatocyte necrosis/apoptosis and no areas of confluent necrosis (Fig. 3C). Commensurate with these relatively mild cytologic changes, the respective histologic score (1.27 ± 0.17) was significantly lower than that of the IRI plus vehicle group.
As indices of cellular oxidative stress, unsaturated aldehydes (MDA and HNE), and GSH, GSSG concentrations were measured in liver homogenates prepared from the different experimental groups. Commensurate with histologic evidence of liver cell death (Fig. 3), the IRI plus vehicle group was associated with relatively large increases in mean concentrations of total MDA and HNE (Fig. 4A). However, these data primarily reflect changes in adduction of membrane-bound proteins because severe loss of cellular integrity during 180 minutes of reperfusion (Fig. 3) will permit egress of free aldehyde and the aldehyde adducts of soluble proteins. IRI was also associated with substantial reductions in GSH, which most likely represents loss due to electrophile scavenging (free radicals, unsaturated aldehydes), oxidation to GSSG, and cell lysis. The observed reduction in GSSG is consistent with several IRI-induced changes, that is, increased ATPase-mediated transport, elevated membrane permeability with subsequent efflux, formation of protein mixed disulfides, and the irretrievable loss of GSH substrate (Shivakumar et al., 1995). In accordance with histologic evidence of hepatocyte preservation and decreased lysis (Fig. 3), 2-ACP administered prior to reperfusion reduced the elevated aldehyde concentrations associated with IRI and limited corresponding GSH/GSSG perturbations (Fig. 4).
NAC Administered Prior to Reperfusion Is Not Hepatoprotective.
NAC (2.40 mmol/kg i.p.) was administered prior to re-establishing hepatic circulation. In contrast to 2-ACP, an equimolar dose of NAC did not affect IRI-induced changes in the plasma levels of ALT, AST, or LDH (Fig. 5).
Fig. 5.

Effects of 2-ACP (2.40 mmol/kg i.p.) or equimolar NAC administered 10 minutes prior to unclamping the portal circulation (reperfusion phase) on IRI-induced plasma appearance of ALT, AST, and LDH in rat (n = 15/group). Data are expressed as mean activity ± S.E.M., and joining lines indicate statistically significant differences at the ***P < 0.001 level.
Hepatoprotective Effects of 2-ACP Administered Prior to Ischemia.
As illustrated in Fig. 6, administration of 2-ACP by intraperitoneal injection 10 minutes before induction of liver ischemia significantly decreased (55–65%) the IRI-induced derangement of plasma enzymes. The same level of hepatoprotection can therefore be achieved when administered before either reperfusion (Fig. 2) or ischemia (Fig. 6).
Fig. 6.

Effects of 2-ACP (2.40 mmol/kg i.p.) administered 10 minutes prior to clamping the portal circulation (ischemia phase) on IRI-induced plasma appearance of ALT, AST, and LDH in rat (n = 15/group). Data are expressed as mean activity ± S.E.M., and joining lines indicate statistically significant differences in treatment groups at the ***P < 0.01 level.
The 2-ACP Does Not Inhibit Succinate Dehydrogenase Activity.
Figure 7 shows that malonate, a well described competitive inhibitor of mitochondrial SDH, produced concentration-dependent (50–500 µM) enzyme inhibition with an IC50 of 147.4 ± 9.5 µM. These data are consistent with previous determinations of malonate inhibition of SDH in rat mitochondria (IC50 = 42 µM; Wojtovich and Brookes, 2008) and in bovine heart submitochondrial particles (IC50 = 96 ± 1.3 µM; Jones and Hirst, 2013). Figure 7 also shows that incubation of mitochondrial proteins with a broad concentration range of 2-ACP (500–1000 µM) did not significantly affect SDH activity.
Fig. 7.

Concentration-dependent effects of malonate (50–500 µM) and 2-ACP (500–1000 µM) on the succinate dehydrogenase activity in bovine heart mitochondrial preparations. Data are expressed as mean percentage of control ± S.E.M. The calculated IC50 for malonate = 147.4 ± 9.5 µM. The 2-ACP did not produce concentration-dependent changes in SDH activity, and therefore an IC50 could not be calculated.
HSAB Calculations of Unsaturated Aldehyde Electrophilicity and Hepatoprotectant Nucleophilicity.
Table 1 presents the respective values of the HSAB parameters, softness (σ) and electrophilicity (ω), determined for acrolein and other unsaturated aldehydes that participate in oxidative stress injury. Results indicate that these aldehydes are soft, relatively strong electrophiles. For comparative purposes, we included the HSAB parameters for N-acetyl-p-benzoquinonimine, the very powerful quinone imine metabolite of acetaminophen (Tylenol; McNeil-PPC, Inc., Fort Washington, PA), and for acrylamide a very weak electrophile that produces cumulative neurotoxicity (see LoPachin and Gavin, 2014, for details). Table 2 presents the respective values of the HSAB parameters, softness (σ) and nucleophilicity (ω−), for GSH (endogenous thiol) and cytoprotective nucleophiles. The data show that 2-ACP, GSH, and NAC are soft, relatively nucleophilic compounds. Also presented are acidity constants expressed as pKa values and the corresponding percentage of each nucleophile in the anionic state (% ionization) at cytophysiological conditions. As indicated in Table 2, the thiolate anions are stronger nucleophiles, but 2-ACP is more acidic (lower pKa), and therefore, a greater proportion of this substance will exist in the ionized nucleophilic enolate state at physiologic pH.
TABLE 1.
HSAB parameters for unsaturated aldehydes
Softness (σ) and electrophilicity (ω) for the selected compounds were calculated as described in Materials and Methods. Nonaldehydes N-acetyl-p-benzoquinoimine (NAPQI) and acrylamide are included for comparative purposes.
| Electrophile | Structure | Softness (σ × 103 ev−1) | Electrophilicity (ω, ev) |
|---|---|---|---|
| NAPQI |  |
499 | 6.83 |
| Acrolein |  |
371 | 3.82 |
| HNE |  |
377 | 3.80 |
| Crotonaldehyde |  |
367 | 3.56 |
| Malondialdehyde | 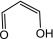 |
384 | 3.39 |
| Acrylamide |  |
315 | 2.62 |
TABLE 2.
HSAB and ionization parameters for thiolate and enolate nucleophiles
Softness (σ) and nucleophilicity (ω−; acrolein as the reacting electrophile) for the selected compounds were calculated as described in Materials and Methods.
| Softness (σ × 103 ev−1) | Nucleophilicity (ω− × 103 ev) | pKa | % Anion (pH = 7.4) | |
|---|---|---|---|---|
| 2-ACP (enolate) | 418 | 204 | 7.8 | 28.5 |
| GSH (thiolate) | 427 | 239 | 8.6 | 5.9 |
| NAC (thiolate) | 367 | 316 | 9.5 | 0.8 |
Discussion
Previous research has indicated that 2-ACP and related 1,3-dicarbonyl compounds were effective cytoprotectants in cell culture and animal models of oxidative stress injury (LoPachin et al., 2011; Zhang et al., 2013). Oxidative stress also plays a prominent role in IRI, presumably through ischemic inhibition of mitochondrial SDH activity, which leads to disruption of electron transport during reperfusion and the generation of ROS (Papadopoulos et al., 2013; Chouchani et al., 2014). Accordingly, we found that 2-ACP injected prior to the reperfusion phase (Fig. 2) provided dose-dependent hepatoprotection against IRI. A similar level of protection was achieved when 2-ACP was administered prior to induction of ischemia (Fig. 6). Although pathogenic processes initiated during the ischemic episode might be targets for protection, it is more likely that 2-ACP selectively inhibits reperfusion-based mechanisms (see ahead). Because the ischemic period is short (45 minutes), and the presumed plasma half-life of 2-ACP is sufficiently long (∼12 hours; Ballantyne and Cawley, 2001), the observed hepatoprotective ability of 2-ACP injected prior to ischemia is probably due to maintenance of effective liver concentrations during reperfusion. Regardless of the injection sequence, the efficacy of 2-ACP hepatoprotection is clearly indicated by histologic preservation in the IRI plus ACP group (Fig. 3, C versus B) and by significant reductions in hepatocyte indices of oxidative stress (Fig. 4).
It is noteworthy that both 2-ACP and curcumin are enolate-forming 1,3-dicarbonyl compounds (Fig. 1). The chemical reactions of anionic enolates are well known (Loudon, 2002; Bug and Mayr, 2003; Eames, 2009), and the relevance of these reactions to general cytoprotection has been demonstrated (Vajragupta et al., 2005; Weber et al., 2006; Begum et al., 2008; LoPachin et al., 2011, 2012). Thus, 2-ACP is not a strong antioxidant (LoPachin et al., 2011), but will ionize readily in biologic buffers to yield a nucleophilic enolate (Fig. 1B). The enolate carbanion generated by this process can form 1,4-Michael adducts with acrolein, HNE, and other electrophilic unsaturated aldehydes that participate in oxidative stress–induced injury (LoPachin et al., 2011). Similarly, curcumin and other phytopolyphenols (e.g., phloretin, resveratrol) can ionize to form nucleophilic enolate sites that scavenge electrophilic aldehydes (Awasthi et al., 1996; Zhu et al., 2009, 2012).
In cell culture models of acrolein toxicity, the rank order of cytoprotection for a series of enolate-forming 1,3-dicarbonyl compounds was found to be directly related to the respective reaction rates for acrolein adduct formation (LoPachin et al., 2011). These rates are determined by the following: 1) enolate nucleophilicity, which influences the second order rate constant (k) for the adduct reaction, and 2) the acidity of the parent dicarbonyl compound (pKa), which governs the concentration of the reacting enolate nucleophile. Calculations of HSAB parameters indicate that acrolein and related unsaturated aldehydes are soft, relatively strong electrophiles (Table 1) that react preferentially with comparably soft nucleophiles. Experimental evidence indicates that the toxicologically relevant soft nucleophilic targets of the unsaturated aldehydes are sulfhydryl thiolate groups on specific cysteine residues that modulate the function of critically important enzymes and proteins (reviewed in LoPachin et al., 2009a; Fritz and Petersen, 2011, 2013; LoPachin and Gavin, 2014). HSAB calculations (Table 2) show that the 2-ACP enolate is a soft, strong carbanion nucleophile, and, because the parent compound (2-ACP) is relatively acidic (pKa = 7.8), a significant concentration of this anion will exist at physiologic pH. These data suggest that IRI hepatoprotection is mediated by the soft enolate of 2-ACP, which can act as a surrogate nucleophile target for acrolein and other soft electrophilic aldehydes generated during reperfusion (see detailed discussions in LoPachin et al., 2012; LoPachin and Gavin, 2014). The aldehyde-enolate interaction is a kinetically favored soft–soft reaction that is preferable to soft–hard interactions, for example, the reactions of acrolein with hard nucleophilic nitrogen groups on carnosine (histidine analog) or N-acetyl lysine (LoPachin et al., 2007, 2009b).
As indicated above, aldehyde scavenging by the enolate is dependent not only on softness and nucleophilicity, but also on the acidity of the parent compound. Thus, nucleophilicity (ω−; Table 2) of the 2-ACP enolate is comparable to that of the respective thiolate forms of NAC and GSH. However, due to a lower pKa value, the 2-ACP enolate will be present in significantly higher concentrations than either thiolate at physiologic conditions. This limited ionization at cellular conditions might explain the inability of NAC to prevent IRI in the present study (Fig. 5), because the relatively low concentration of the active species (thiolate) would consequentially limit electrophile scavenging. In fact, there is evidence that NAC cytoprotection involves an indirect mechanism that is not based on electrophile scavenging, for example, increased GSH synthesis or mitochondrial bioenergetics (Zwingmann and Bilodeau, 2006; Jaeschke and Woolbright, 2012).
The bidentate enolate of 2-ACP can also arrest oxidative stress by chelating iron [Fe(III)] and copper [Cu(II)] metal ions (Jiao et al., 2006; Eames, 2009; LoPachin et al., 2011) that catalyze the free radical–generating Fenton reaction. Indeed, the metal-chelating abilities of 1,3-dicarbonyl enolates have been recognized for more than a century (Eames, 2009). Recent cell culture studies of H2O2 toxicity demonstrated that metal chelation was an important criterion for 1,3-dicarbonyl cytoprotection (LoPachin et al., 2011). In these studies, the relative chelating abilities for the dicarbonyl series were quantified by direct in chemico measurements (LoPachin et al., 2011). Thus, compounds with rigid structures that precluded bidentate metal ion coordination such as 1,3-cyclopentanedione were ineffective despite having the ability to form enolates. However, 2-ACP and other β-diketone compounds with flexible structures that accommodate metal chelation were highly protective. Metal ion chelation by the enolate of curcumin has also been identified as a cytoprotective trait of this phytopolyphenol (Bernabe-Pineda et al., 2004; Jiao et al., 2006).
Malonic acid is a 1,3-dicarbonyl compound that provides cytoprotection in several models of in vivo and in vitro IRI. However, cytoprotection is not based on enolate nucleophilicity, because this acid exists as the dianion (malonate) at physiologic conditions and further ionization would be extremely unfavorable if not precluded entirely. Instead, like other dicarboxylates, malonate is a succinate analog and is a competitive inhibitor of mitochondrial SDH. Substantial evidence now indicates that ischemia-induced reversal of this enzyme causes accumulation of succinate in mitochondria. During reoxygenation, SDH oxidation of the excess succinate drives ROS production through reverse electron transport involving complex I (reviewed in Chouchani et al., 2014). Malonate cytoprotection in IRI is presumably mediated by SDH inhibition, which prevents succinate accumulation and subsequent ROS production. The 2-ACP is also a 1,3-dicarbonyl compound, and therefore, the possibility exists that SDH inhibition might mediate corresponding hepatoprotection. However, we have demonstrated that, even at relatively high concentrations, 2-ACP is not an effective inhibitor of mitochondrial SDH activity (Fig. 7). As a dicarboxylic acid derivative, malonate can inhibit SDH by acting as a structural analog of the citric acid cycle intermediate succinate. In contrast, 2-ACP is a β-diketone and is therefore not a suitable substrate inhibitor for SDH. These data indicate that the observed hepatoprotective ability of 2-ACP does not involve inhibition of enzyme activity.
Our results indicate that 2-ACP provided significant hepatoprotection in an animal model of IRI. The anionic enolate of this 1,3-dicarbonyl is a soft nucleophile that can form irreversible Michael adducts with acrolein, HNE, and other soft aldehyde electrophiles generated during the reperfusion phase of IRI. The 2-ACP can also reduce cellular oxidative stress through metal ion chelation and resulting inhibition of the free radical-generating Fenton reaction. Although these cytoprotective reactions of 2-ACP are consistent with the known chemistry of the 1,3-dicarbonyl compounds, it is possible that 2-ACP either directly or indirectly influences cellular stress responses (e.g., Nrf2/Keap1, Sirt1 pathways), as has been suggested for curcumin cytoprotection (Yang et al., 2009; Chung et al., 2010). The premise that enolate-forming 1,3-dicarbonyl compounds might be a possible therapeutic approach was based on the recognition that the heptadienone bridge of curcumin was also a 1,3-dicarbonyl (Fig. 1A). However, unlike most phytopolyphenols (Lambert et al., 2007; Halliwell, 2008), simple 1,3-diketones, such as 2-ACP, are chemically stable, relatively water-soluble compounds that are rapidly absorbed and have large volumes of distribution (Ballantyne and Cawley, 2001; LoPachin et al., 2011). Furthermore, the acute animal toxicity of these chemicals is low (LD50 > 800 mg/kg), and longitudinal dosing studies indicate a low incidence of systemic toxicity (e.g., 400–600 mg/kg per day × 60 days; Ballantyne and Cawley, 2001; B. Geohagen and R. LoPachin, unpublished data). The mechanism of cytoprotection, in conjunction with favorable pharmacokinetics and low toxicity, suggests that 2-ACP or an analog might be useful in treating warm IRI associated with stroke, myocardial infarction, and other organ failure. IRI-induced oxidative stress is also an underlying cause of allograft failure following transplantation. The preservation solutions used for donor organ resection and storage (e.g., UW, HTK, and Leeds solutions) uniformly contain antioxidants (e.g., allopurinol, carvedilol). Nonetheless, it is recognized that cell damage is still an issue (Park and Lee, 2008; Ben Mosbah et al., 2010), and therefore, the addition of a 1,3-dicarbonyl derivative could improve the cytopreservative capabilities of these solutions.
Acknowledgments
The authors thank Dr. Allan W. Wolkoff (Division of Gastroenterology and Liver Diseases, Albert Einstein College of Medicine) for helpful comments and guidance. The authors also thank Irene Ostrovsky (Chemistry Laboratory, Department of Pathology), Tewolde Yimer, and staff: Anie George, Faina Kremerman, Don Kutsyk, Kseniya Vayner, and Aleksandr Yakubov for analysis of rat plasma biochemistry.
Abbreviations
- 2-ACP
2-acetylcyclopentanone
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- DCPIP
2,6-dichlorophenolindophenol
- EHOMO
highest occupied molecular orbital energy
- ELUMO
lowest unoccupied molecular orbital energy
- GSH
glutathione
- GSSG
oxidized glutathione
- HNE
4-hydroxy-2-nonenal
- HSAB
hard and soft, acids and bases
- IRI
ischemia-reperfusion injury
- LDH
lactate dehydrogenase
- MDA
malondialdehyde
- NAC
N-acetylcysteine
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- TCA
trichloroacetic acid
Authorship Contributions
Participated in research design: LoPachin, Gavin, Korsharskyy.
Conducted experiments: Geohagen, Vydyanathan, Shaparin.
Contributed new reagents or analytic tools: Gavin.
Performed data analysis: Zhang, Liu, Bivin.
Wrote or contributed to the writing of the manuscript: LoPachin, Gavin, Korsharskyy, Vydyanathan.
Footnotes
This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grant R01-ES003830-26].
References
- Abe Y, Hines IN, Zibari G, Pavlick K, Gray L, Kitagawa Y, Grisham MB. (2009) Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic Biol Med 46:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi S, Srivatava SK, Piper JT, Singhal SS, Chaubey M, Awasthi YC. (1996) Curcumin protects against 4-hydroxy-2-trans-nonenal-induced cataract formation in rat lenses. Am J Clin Nutr 64:761–766. [DOI] [PubMed] [Google Scholar]
- Ballantyne B, Cawley TJ. (2001) 2,4-Pentanedione. J Appl Toxicol 21:165–171. [DOI] [PubMed] [Google Scholar]
- Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, et al. (2008) Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther 326:196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Mosbah I, Roselló-Catafau J, Alfany-Fernandez I, Rimola A, Parellada PP, Mitjavila MT, Lojek A, Ben Abdennebi H, Boillot O, Rodés J, et al. (2010) Addition of carvedilol to University Wisconsin solution improves rat steatotic and nonsteatotic liver preservation. Liver Transpl 16:163–171. [DOI] [PubMed] [Google Scholar]
- Bernabe-Pineda M, Ramirez-Sliva MT, Romero-Romo MA, Ganzalez-Vergara R, Rojas-Heernandex A. (2004) Spectrophotometric and electrochemical determination of the formation constants of the complexes curcumin-Fe(III)-water and curcumin-Fe(II)-water. Spectrochim Acta A Mol Biomol Spectrosc 60:1105–1113. [DOI] [PubMed] [Google Scholar]
- Bessems M, ’t Hart NA, Tolba R, Doorschodt BM, Leuvenink HGD, Ploeg RJ, Minor T, van Gulik TM. (2006) The isolated perfused rat liver: standardization of a time-honoured model. Lab Anim 40:236–246. [DOI] [PubMed] [Google Scholar]
- Bug T, Mayr H. (2003) Nucleophilic reactivities of carbanions in water: the unique behavior of the malodinitrile anion. J Am Chem Soc 125:12980–12986. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I. (2010) Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys 501:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSeau V, Rosen N, Bolen JB. (1987) Analysis of pp60c-src tyrosine kinase activity and phosphotyrosyl phosphatase activity in human colon carcinoma and normal human colon mucosal cells. J Cell Biochem 35:113–128. [DOI] [PubMed] [Google Scholar]
- Eames J. (2009) Acid-base properties of enols and enolates, in The Chemistry of Metal Enolates (Zablicky J. ed) pp 411–460, John Wiley & Sons, West Sussex, England. [Google Scholar]
- Ellis EM. (2007) Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther 115:13–24. [DOI] [PubMed] [Google Scholar]
- Fritz KS, Petersen DR. (2011) Exploring the biology of lipid peroxidation-derived protein carbonylation. Chem Res Toxicol 24:1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz KS, Petersen DR. (2013) An overview of the chemistry and biology of reactive aldehydes. Free Radic Biol Med 59:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard-Monnier D, Erdelmeier I, Régnard K, Moze-Henry N, Yadan JC, Chaudière J. (1998) Reactions of 1-methyl-2-phenylindole with malondialdehyde and 4-hydroxyalkenals: analytical applications to a colorimetric assay of lipid peroxidation. Chem Res Toxicol 11:1176–1183. [DOI] [PubMed] [Google Scholar]
- Giustarini D, Dalle-Donne I, Milzani A, Fanti P, Rossi R. (2013) Analysis of GSH and GSSG after derivatization with N-ethylmaleimide. Nat Protoc 8:1660–1669. [DOI] [PubMed] [Google Scholar]
- Halliwell B. (2008) Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch Biochem Biophys 476:107–112. [DOI] [PubMed] [Google Scholar]
- He S, Atkinson C, Qiao F, Cianflone K, Chen X, Tomlinson S. (2009) A complement-dependent balance between hepatic ischemia/reperfusion injury and liver regeneration in mice. J Clin Invest 119:2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Woolbright BL. (2012) Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev 26:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegatheeswaran S, Siriwardena AK. (2011) Experimental and clinical evidence for modification of hepatic ischaemia-reperfusion injury by N-acetylcysteine during major liver surgery. HPB 13:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Wilkinson J, 4th, Christine Pietsch E, Buss JL, Wang W, Planalp R, Torti FM, Torti SV. (2006) Iron chelation in the biological activity of curcumin. Free Radic Biol Med 40:1152–1160. [DOI] [PubMed] [Google Scholar]
- Jones AJY, Hirst J. (2013) A spectrophotometric coupled enzyme assay to measure the activity of succinate dehydrogenase. Anal Biochem 442:19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klune JR, Tsung A. (2010) Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. Surg Clin North Am 90:665–677. [DOI] [PubMed] [Google Scholar]
- Lambert JD, Sang S, Yang CS. (2007) Possible controversy over dietary polyphenols: benefits vs risks. Chem Res Toxicol 20:583–585. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Geohagen BC, Das S. (2007) Neurotoxic mechanisms of electrophilic type-2 alkenes: soft soft interactions described by quantum mechanical parameters. Toxicol Sci 98:561–570. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Petersen DR, Barber DS. (2009a) Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem Res Toxicol 22:1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopachin RM, Geohagen BC, Gavin T. (2009b) Synaptosomal toxicity and nucleophilic targets of 4-hydroxy-2-nonenal. Toxicol Sci 107:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Geohagen BC, Zhang L, Casper D, Lekhraj R, Barber DS. (2011) β-dicarbonyl enolates: a new class of neuroprotectants. J Neurochem 116:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopachin RM, Gavin T, Decaprio A, Barber DS. (2012) Application of the hard and soft, acids and bases (HSAB) theory to toxicant-target interactions. Chem Res Toxicol 25:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T. (2014) Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem Res Toxicol 27:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudon GM. (2002) Chemistry of enolate ions, enols and α,β-unsaturated carbonyl compounds, in Organic Chemistry, 4th ed, pp 997–1068, Oxford University Press, New York. [Google Scholar]
- Park SW, Lee SM. (2008) Antioxidant and prooxidant properties of ascorbic acid on hepatic dysfunction induced by cold ischemia/reperfusion. Eur J Pharmacol 580:401–406. [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Siempis T, Theodorakou E, Tsoulfas G. (2013) Hepatic ischemia and reperfusion injury and trauma: current concepts. Arch Trauma Res 2:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar BR, Kolluri SVR, Ravindranath V. (1995) Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. J Pharmacol Exp Ther 274:1167–1173. [PubMed] [Google Scholar]
- Uchida K. (2003) 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res 42:318–343. [DOI] [PubMed] [Google Scholar]
- Vajragupta O, Boonchoong P, Morris GM, Olson AJ. (2005) Active site binding modes of curcumin in HIV-1 protease and integrase. Bioorg Med Chem Lett 15:3364–3368. [DOI] [PubMed] [Google Scholar]
- Weber WM, Hunsaker LA, Gonzales AM, Heynekamp JJ, Orlando RA, Deck LM, Vander Jagt DL. (2006) TPA-induced up-regulation of activator protein-1 can be inhibited or enhanced by analogs of the natural product curcumin. Biochem Pharmacol 72:928–940. [DOI] [PubMed] [Google Scholar]
- Wojtovich AP, Brookes PS. (2008) The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim Biophys Acta 1777:882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood PL, Khan MA, Moskal JR, Todd KG, Tanay VAMI, Baker G. (2006) Aldehyde load in ischemia-reperfusion brain injury: neuroprotection by neutralization of reactive aldehydes with phenelzine. Brain Res 1122:184–190. [DOI] [PubMed] [Google Scholar]
- Yang C, Zhang X, Fan H, Liu Y. (2009) Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res 1282:133–141. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Tomitori H, Machi Y, Hagihara M, Higashi K, Goda H, Ohya T, Niitsu M, Kashiwagi K, Igarashi K. (2009) Acrolein toxicity: comparison with reactive oxygen species. Biochem Biophys Res Commun 378:313–318. [DOI] [PubMed] [Google Scholar]
- Zhang L, Gavin T, Geohagen BC, Liu Q, Downey KJ, LoPachin RM. (2013) Protective properties of 2-acetylcyclopentanone in a mouse model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther 346:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Zheng ZP, Cheng KW, Wu JJ, Zhang S, Tang YS, Sze KH, Chen F, Wang M. (2009) Natural polyphenols as direct trapping agents of lipdi peroxidation-derived acrolein and 4-hydroxy-trans-2-nonenal. Chem Res Toxicol 22:1721–1729. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Zhang N, Cheng MK, Lau CF, Chao J, Sun Z, Chang RCC, Chen F, Wang M. (2012) In vitro attenuation of acrolein-induced toxicity by phloretin, a phenolic compound from apple. Food Chem 135:1762–1768. [DOI] [PubMed] [Google Scholar]
- Zwingmann C, Bilodeau M. (2006) Metabolic insights into the hepatoprotective role of N-acetylcysteine in mouse liver. Hepatology 43:454–463. [DOI] [PubMed] [Google Scholar]