

Abstract
5-Benzylglycinyl-amiloride (UCD38B) is the parent molecule of a class of anticancer small molecules that kill proliferative and nonproliferative high-grade glioma cells by programmed necrosis. UCD38B intracellularly triggers endocytosis, causing 40–50% of endosomes containing proteins of the urokinase plasminogen activator system (uPAS) to relocate to perinuclear mitochondrial regions. Endosomal “mis-trafficking” caused by UCD38B in human glioma cells corresponds to mitochondrial depolarization with the release and nuclear translocation of apoptotis-inducing factor (AIF) followed by irreversible caspase-independent cell demise. High-content quantification of immunocytochemical colocalization studies identified that UCD38B treatment increased endocytosis of the urokinase plasminogen activator (uPA), its receptor (uPAR), and plasminogen activator inhibitor-1 (PAI-1) into the early and late endosomes by 4- to 5-fold prior to AIF nuclear translocation and subsequent glioma demise. PAI-1 was found to comparably relocate with a subset of early and late endosomes in four different human glioma cell lines after UCD38B treatment, followed by caspase-independent, nonapoptotic cell death. Following UCD38B treatment, the receptor guidance protein LRP-1, which is required for endosomal recycling of the uPA receptor to the plasmalemma, remained abnormally associated with PAI-1 in early and late endosomes. The resultant aberrant endosomal recycling increased the total cellular content of the uPA–PAI-1 protein complex. Reversible inhibition of cellular endocytosis demonstrated that UCD38B bypasses the plasmalemmal uPAS complex and directly acts intracellularly to alter uPAS endocytotic trafficking. UCD38B represents a class of small molecules whose anticancer cytotoxicity is a consequence of causing the mis-trafficking of early and late endosomes containing uPAS cargo and leading to AIF-mediated necrotic cell death.
Introduction
High-grade gliomas (HGGs) are rapidly proliferative, highly infiltrative, and predominantly fatal primary brain cancers with hypovascularized infiltrative borders and characterized by the spontaneous formation of avascular necrotic tumor domains. Within the hypoxic-ischemic regions, HGGs demonstrate increased expression of proteins belonging to the urokinase plasminogen activator system (uPAS) (Harbeck et al., 2013). The major components of the uPAS are the urokinase-type plasminogen activator (uPA), tissue-type plasminogen activator, plasminogen activator inhibitor-1 (PAI-1) and plasminogen activator inhibitor-2, and the uPA receptor (uPAR). uPAS proteins play an important role in events leading to cancer cell infiltration, angiogenesis, and metastasis. uPA is a serine protease synthesized as pro-uPA that is secreted and becomes activated when bound to its cell surface receptor uPAR (Blasi et al., 1987). Activated uPA catalyzes the transformation of plasminogen into plasmin (Ellis et al., 1989). Plasmin is an extracellular serine protease capable of degrading proteins of the extracellular matrix and basement membranes (Andreasen et al., 1997). Plasminogen activator inhibitors are antiproteases belonging to the SERPIN super family that inhibit the enzymatic activities of uPA and tissue-type plasminogen activator. PAI-1 binds to the active site of uPA, generating a uPA–PAI-1 protein complex that is bound to the plasmalemmal uPAR receptor (uPAR::uPA–PAI-1). Enzymatic inhibition of secreted and receptor-bound uPA by PAI-1 impedes degradation of the extracellular matrix and fibrinolysis. Despite its enzymatic inhibition of uPA, elevated PAI-1 expression in several cancer cell types, notably high-grade glioma and breast cancers, strongly corresponds with enhanced tumor growth, infiltration, angiogenesis, and metastasis (Schmitt et al., 1997; Bajou et al., 2004). Previously, small molecules and antibodies designed to inhibit secreted and plasmalemmal uPA have been investigated as anticancer agents but are predominantly cytostatic, preventing cancer migration and angiogenesis (Setyono-Han et al., 2005; Ulisse et al., 2009). These plasmalemmal uPA inhibitors fundamentally differ from the anticancer cytotoxicity and intracellular mechanisms described for 5-benzylglycinyl-amiloride (UCD38B) and its pepidomimetic congeners.
The intracellular functions of uPA–PAI-1 are protean and poorly understood. Enzyme-linked immunosorbant assay (ELISA) can quantify protein complexes of uPA–PAI-1, and increased complex expression has been reported to strongly correlate with cancer recurrence and metastasis in lymph node–negative breast cancer (Harbeck et al., 2013). A summary of endocytotic trafficking of uPAS proteins is depicted in Fig. 1. PAI-1 binds to the active site of uPA, and the latter is bound to its plasmalemmal receptor (uPAR). PAI-1 regulates cancer cell invasion and detachment by controlling endocytic recycling of the uPAR::uPA–PAI-1 complex (Czekay et al., 2003; Cortese et al., 2008). Clathrin-mediated endocytic internalization of this tertiary uPAS complex requires additional binding by the endocytic guiding receptor protein, low density lipoprotein receptor–related protein-1 (LRP-1) (Herz et al., 1988, 1992). The resultant quaternary complex is internalized via clathrin-coated pits and transported to early endosomes and late endosomes, where uPA–PAI-1 becomes dissociated from uPAR. The uPA–PAI-1 complex then undergoes degradation in the lysosomes (Olson et al., 1992; Conese et al., 1995). uPAR and LRP-1 become dissociated and are recycled back from the endosomal compartment to the cell surface (Fig. 1).
Fig. 1.

uPAS in glioma cells. uPA binds to the plasmalemmal receptor uPAR and converts plasminogen to plasmin for extracellular matrix degradation. PAI-1 binds to the active site of uPA, which is bound to uPAR to form a uPAR::uPA–PAI-1 complex. The endocytic receptor protein LRP-1 mediates internalization of the uPAS complex into early endosomes and late endosomes. uPA–PAI-1 is dissociated from uPAR and LRP-1 in late endosomes to undergo degradation in the lysosomes. Dissociated uPAR and LRP-1 are recycled back from the endosomal compartment to the cell surface.
Previously, we reported that 5-benzylglycinyl-amiloride (UCD38B), a cell permeant, competitive enzymatic inhibitor of uPA, kills proliferating and nonproliferating glioma cells by a novel caspase-independent, programmed necrotic cell death mechanism (Pasupuleti et al., 2013). The current investigation significantly advances these initial observations, as we now identify that UCD38B causes apoptosis-inducing factor (AIF)–mediated necrotic glioma cell death by redirecting a portion of early and late endosomes containing uPAS protein cargo with the guiding endosomal LRP-1 receptor protein to perinuclear mitochondrial regions. Surprisingly, we found that cell-permeant UCD38B bypasses the plasmalemmal uPAS receptor complex and intracellularly activates endosomal “mis-trafficking”. The drug-induced endosomal juxtaposition to the mitochondrial regions temporally corresponds with mitochondrial dilation and depolarization, which is associated with mitochondrial release and nuclear translocation of AIF.
Materials and Methods
Drugs and Antibodies.
UCD38B [5-benzylglycinyl-3-amino-6-chloro-N-(diaminomethylene) pyrazine-2-carboxamide] and UCD74A [5-glycinyl-3-amino-6-chloro-N-(diaminomethylene) pyrazine-2-carboxamide] were synthesized as described previously (Palandoken et al., 2005; Massey et al., 2012). Drug stocks of 250 mM were constituted in dimethylsulfoxide (DMSO) stored at −20°C and reconstituted for each experiment at a final concentration of 250 μM.
Receptor associated protein (RAP) from Millipore (Billerica, MA) was added to glioma cells at 1 μM for 1 hour at 37°C prior to UCD38B or UCD74A treatment.
Human anti–PAI-1 mouse monoclonal, human anti–LRP-1 goat polyclonal (1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), human anti–EEA-1, human anti–LAMP-1 rabbit polyclonal (1:100 dilution; Abcam, Cambridge, MA), and human anti-uPA mouse monoclonal and human anti-uPAR mouse monoclonal (1:100 dilution; American Diagnostica, Stamford, CT) were used as primary antibodies. Goat anti-mouse secondary antibody (1:20,000; Licor Biosciences, Lincoln, NE) was used for protein visualization on immunoblots.
Cell Culture.
U87MG, LN229, U118MG, and U138MG human glioma cell lines were derived from high-grade glial cancers and obtained from the American Type Culture Collection (Manassas, VA). Cells were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY). Dulbecco’s modified Eagle’s medium or minimum essential medium (for U138MG) was supplemented with 10% fetal bovine serum (Hyclone Laboratories, Rockford, IL), 1% l-glutamine (200 mM; Gibco), 100 U/ml penicillin, and 100 mg/ml streptomycin (Gibco).
Cell Death Assay.
A lactate dehydrogenase (LDH) assay measured the release of cytosolic LDH from dead and dying glioma cells. Ten thousand cells per well were plated in a 96-well microtiter plate and treated with the drugs for 24 hours at 37°C, 5% CO2. The LDH assay was performed according to the manufacturer’s protocol (Clontech Laboratories, Mountain View, CA). Absorbance was read at 492 nm using a SpectraMax M3 with SoftmaxPro software (LifeSciSoft, Kingston, NY).
For the Trypan blue exclusion assay, cells were harvested by gently rinsing with phosphate-buffered saline (PBS). Trypan blue was added to the cell suspension (10% v/v), and after 3 minutes, an aliquot was transferred to a hemacytometer for manual cell counting.
Immunofluorescence Confocal Microscopy.
Human U87MG glioma cells were cultured in chamber slides. Cells were treated with UCD38B or UCD74A for 30, 60, and 120 minutes at 250 μM at 37°C. Mitochondria were visualized by staining with MitoTracker or JC-1 (Molecular Probes, Eugene, OR) for 30 minutes prior to brief fixation. Cells were washed with PBS and fixed with 4% paraformaldehyde in PBS at room temperature for 15 minutes, followed by washing with PBS and permeabilization with 80% methanol in PBS at −20°C for 15 minutes. Blocking of nonspecific antibody binding sites was performed using 3% nonfat dry milk/1% bovine serum albumin (BSA) in PBS for 1 hour at room temperature. Cells were incubated with a primary antibody in 0.1% BSA/PBS at 4°C overnight at 1:100 dilution. A secondary antibody against mouse or rabbit, conjugated with Alexa Fluor 488, 594, and 647 (Molecular Probes) was used at 1:500 dilution in 0.1% BSA/PBS and incubated for 1 hour at room temperature. After the PBS wash, the slides were mounted with mounting media containing 4′,6-diamidino-2-phenylindole (Molecular Probes). Images were captured using a 40× objective with a spinning disk confocal microscope (Olympus IX81; Olympus, Center Valley, PA). UCD38B and UCD74A are comparable in quantum yield when visualized by fluorescent microscopy as previously described (Palandoken et al., 2005).
Quantification of Colocalized Immunocytological Fluorescent Biomarkers.
Quantification of double-labeled immunocytological markers that were colocalized was performed by imaging and analyzing cells using an automated high-content imaging system, ImageXpress Micro XL System (Molecular Devices, Sunnyvale, CA). High-content imaging was performed on four wells per treatment, with 36 sites per well, using a Custom Module software of MetaXpress 5.0 that was developed specifically for cytologic colocalization studies. A minimum of 10 cells per site was included, and sites without cells were excluded when calculating the mean and standard deviation analysis.
Electron Microscopy.
U87MG glioma cells were plated in Laboratory-Tek Permanox chamber eight-well slides from Electron Microscopy Sciences (Hatfield, PA). Cells were treated with the drugs UCD38B, UCD74A, or vehicle control at 250 μM for 1 hour. Cells were fixed in Karnovsky’s fixative (2.5% glutaraldehyde and 2.0% paraformaldehyde) for 1 hour. Slides were prepared as described previously (Pasupuleti et al., 2013). Cells were imaged in the Philips 120 BioTwin electron microscope at 80 kV equipped with a Gatan MegaScan model 794/20 digital camera.
Protein Fractionation and Immunoblotting.
Protein extracts were made in radioimmunoprecipitation assay lysis buffer (Cell Signaling, Danvers, MA), and 50 μg of protein was loaded on SDS-PAGE. For mitochondrial enriched fractions, cells were harvested, washed with 1× PBS, and centrifuged 1000 rpm for 5 minutes. Cells were then resuspended in mitochondrial isolation buffer (250 mM sucrose, 10 mM Tris-HCl, pH 7.4, and 0.1 mM EGTA) and homogenized using a Dounce homogenizer (Sigma-Aldrich, St. Louis, MO). Samples were centrifuged at 1000 rpm for 10 minutes to pellet the nuclei. Supernatant was collected in a new tube and centrifuged at 15,000 rpm for 20 minutes to collect the pellet (mitochondrial enriched) and supernatant (cytosol enriched) fractions. The pellet was solubilized in lysis buffer and centrifuged to pellet the debris. Protein samples were size fractionated through 12% SDS-PAGE and transferred onto a nitrocellulose membrane by immunoblotting. Blocking buffer (Licor Biosciences) diluted 1:1 in 1× PBS was used for 1 hour at room temperature to prevent nonspecific binding sites. Primary antibody incubation was performed overnight at 4°C. The secondary antibody incubation was carried out at room temperature for 1 hour. Goat anti-mouse, goat anti-rabbit, and donkey anti-goat secondary antibodies from Licor Biosciences were used at 1:10,000 dilution. Bands were visualized and quantified using an Odyssey infrared imager (Licor Biosciences).
uPA and PAI-1 Complex ELISA.
The human uPA–PAI-1 protein ELISA assay kit was purchased from Molecular Innovations (Novi, MI), with a human uPA–PAI-1 complex provided in a lyophilized form for standardization. It was reconstituted in 3% BSA/Tris-buffered saline for a final concentration of 200 ng/ml. The uPA–PAI-1 complex standard was prepared by serial dilution to obtain the 0–100 ng/ml range, and a linear standard curve was generated following the manufacturer instructions. U87MG cells were treated with UCD38B or UCD74A at 250 μM for 45 minutes, 1 hour, or 2 hours. DMSO (0.1% v/v) was used as vehicle. Protein extracts were made, and the protein concentration was determined by a BCA protein assay. A 96-well plate precoated with the uPA antibody was provided in the kit. The standard and protein were added to the 96-well plate. The plate was incubated at room temperature on a shaker for 30 minutes. The samples were aspirated and washed with the washing buffer included in the kit. A polyclonal PAI-1 antibody was added and incubated again at room temperature for 30 minutes on a shaker. A horseradish peroxidase–linked secondary antibody was added to the wells following the wash. Substrate solution from the kit was added to the wells after the wash and incubated for 5 minutes. The reaction was stopped by adding 1 N H2SO4, the plate was gently agitated, and the absorbance was read at 450 nm using SpectraMax M3 with SoftmaxPro software (LifeSciSoft).
Statistical Analysis.
Data are presented as mean ± standard deviation, and statistical significance was identified using one-way analysis of variance and Bonferroni’s nonparametric, multiple pairwise comparisons using SigmaStat software version 2.00 (SPSS Science Inc., Chicago, IL).
Results
UCD38B Initiates Intracellular Relocation of uPA, PAI-1, and uPAR to Mitochondrial Regions in U87MG Glioma Cells.
Previously, we determined that the cell-permeant UCD38B, a competitive enzymatic inhibitor of uPA, caused relocation of a portion of intracellular uPA to a mitochondrial enriched subcellular fraction after 120 minutes (Pasupuleti et al., 2013). Immunocytochemistry localized this uPA migration to perinuclear mitochondrial regions using confocal microscopy (Pasupuleti et al., 2013). PAI-1 binds to the active site of uPA that is bound to uPAR, forming plasmalemmal and intracellular uPAR::uPA–PAI-1 complexes. Therefore, we investigated the intracellular localization of PAI-1 and uPAR in U87MG glioma cells following 120-minute treatment with UCD38B or its cell-impermeant homolog UCD74A. Mitochondria were visualized using MitoTracker, and nuclei were stained with 4′,6-diamidino-2-phenylindole to localize the intracellular distribution of PAI-1 and uPAR following treatment. PAI-1 was distributed homogeneously in the cytoplasm and plasmalemmal regions of untreated cells. Following 120-minute treatment of UCD38B, a portion of PAI-1 and uPAR was localized to perinuclear mitochondrial regions (Fig. 2A; Fig. 3A). In contrast, no alteration in intracellular PAI-1 or uPAR localization was identified following treatment with UCD74A. For biochemical verification, cytosolic and mitochondrial fractions were separated by subcellular fractionation following glioma treatment for 120 minutes. Immunoblotting was performed to visualize PAI-1 and uPAR using either an anti–PAI-1 or anti-uPAR monoclonal antibody, respectively. Actin and voltage-dependent anion channel protein represented the cytosolic and mitochondrial fractions, respectively. Relative levels of PAI-1 or uPAR expression in each fraction were normalized to their respective protein markers. Like uPA, PAI-1 and uPAR were enriched in the mitochondrial fractions when treated with UCD38B (Fig. 2, B and C; Fig. 3, B and C). As a control, neither PAI-1 nor uPAR were enriched in mitochondrial fractions following incubation with cell-impermeant UCD74A.
Fig. 2.

Localization of PAI-1 in U87MG glioma cell. Intracellular localization of PAI-1 was visualized by confocal microscopy with an anti–PAI-1 antibody in U87MG cells that had been treated for 120 minutes with 250 μM of UCD38B, UCD74A, or vehicle (0.1% v/v DMSO). (A) Alexa 488 was used as a secondary antibody. MitoTracker and 4′,6-diamidino-2-phenylindole (DAPI) were used to visualize the mitochondria and nucleus, respectively, and the scale bar is 50 μm. Cytosolic (C) and mitochondrial (M) enriched fractions were separated by SDS-PAGE. PAI-1 was visualized on an immunoblot with Actin and voltage-dependent anion channel as cytosolic and mitochondrial marker proteins, respectively (B). Densitometry of PAI-1 expression, normalized to marker proteins. (C) is presented as the mean ± S.D. of n = 3 with ***P < 0.001.
Fig. 3.

Localization of uPAR in U87MG glioma cells. U87MG cells were treated for 120 minutes with 250 μM of UCD38B, UCD74A, or vehicle (0.1% v/v DMSO). (A) Anti-uPAR antibody was used to detect uPAR by immunofluorescence confocal microscopy. Alexa 488 was used as a secondary antibody. MitoTracker and 4′,6-diamidino-2-phenylindole (DAPI) were used to visualize the mitochondria and nucleus, respectively. Scale bar is 50 μm. uPAR levels in cytosolic (C) and mitochondrial (M) enriched fractions were detected on immunoblots (B) with Actin and voltage-dependent anion channel used as cytosolic and mitochondrial marker proteins, respectively. Normalized protein levels of uPAR in cytosolic and mitochondrial fractions were determined by densitometry (C) as the mean ± S.D. of n = 3 with ***P < 0.001.
Electron Microscopy Reveals an Increase in Endosomal and Lysosomal Formation in U87MG Glioma Cells Following UCD38B Treatment.
Electron micrographs were obtained following 60-minute exposure to UCD38B, preceding the mitochondrial depolarization observed in live U87MG glioma cells following UCD38B treatment (Pasupuleti et al., 2013). In vehicle-treated control cells, cellular morphology remained unchanged (Fig. 4A). By contrast, U87MG glioma cells treated with UCD38B demonstrated an increase in the formation of endosomes and lysosomes, coinciding with the appearance of intracellular vacuolization (Fig. 4B). In glioma cells treated with UCD38B, electron microscopy demonstrated that endosomes and lysosomes lay in close proximity to dilated mitochondria with deformed cristae (Fig. 4C). These morphologic observations indicated that UCD38B treatment increased endocytosis, with an augmentation of endosomes and lysosomes lying in close proximity to dilated mitochondria.
Fig. 4.

Electron microscopy reveals endocytosis in U87MG glioma cells. Time-matched, vehicle-treated (control) U87MG glioma cells. (A) U87MG glioma cells were treated with UCD38B at 250 μM for 60 minutes, and cellular changes were identified by electron microscopy. Arrows indicate increase in cytoplasmic vacuolization (B) and lysosome formation in UCD38B-treated cells (C). Scale bar is 1 μm.
PAI-1 Relocation to Early and Late Endosomes Is Increased Following UCD38B Treatment in U87MG Glioma Cells.
Electron microscopy demonstrated that UCD38B treatment within 60 minutes initiated a marked increase in endocytosis associated with endosomal relocation to mitochondrial regions. Therefore, we investigated the effect of UCD38B on endosomal trafficking of uPA, PAI-1, and uPAR in U87MG glioma cells. Colocalization of uPAS proteins with early and late endosomes was determined by immunofluorescence confocal microscopy. EEA-1 and LAMP-1 antibodies were used to identify early and late endosomal proteins, respectively. U87MG glioma cells were treated with drug vehicle or UCD74A for 60 minutes or UCD38B for 15 and 60 minutes. PAI-1 did not demonstrate significant colocalization with early or late endosomes following 15-minute exposure with UCD38B. However, after 60 minutes, PAI-1 immunostaining was found to colocalize with endosomes labeled with either EEA-1 or LAMP-1 antibodies (Fig. 5, B and D). No alteration of intracellular PAI-1 colocalization with early or late endosomes was observed in glioma cells following 60-minute exposure to either drug vehicle or UCD74A (Fig. 5, A and C). U87MG glioma cells were reimaged using the ImageXpress high-content imaging system to quantify intracellular colocalization. Following UCD38B treatment for 60 minutes, PAI-1 colocalized with 40 and 54% of EEA-1 and LAMP-1 immunofluorescent markers for early and late endosomes, respectively. This magnitude of endosomal colocalization of PAI-1 in UCD38B-treated cells was highly significant when compared with vehicle- or UCD74A-treated U87MG glioma cells (Fig. 5E).
Fig. 5.

Colocalization of PAI-1 with early and late endosomes in U87MG glioma cells. Colocalization of PAI-1 with early endosomes using EEA-1 as an early endosome marker protein was detected by immunofluorescent confocal microscopy. U87MG cells were treated with drug vehicle control (0.1% DMSO) or UCD74A (A) at 250 μM for 60 minutes. Glioma cells were exposed to UCD38B at 250 μM for 15 or 60 minutes (B). Colocalization of PAI-1 with late endosomes using LAMP-1 as a marker protein was visualized in glioma cells treated with vehicle or UCD74A for 60 minutes (C) or UCD38B (D) for 15 or 60 minutes. Scale bar is 50 μm. U87MG glioma cells were imaged and colocalization of immunofluorescent labels was quantified using the high-content imaging system ImageXpress and analyzed by a custom module using MetaXpress 5.0 software (E). Results are presented as the mean ± S.D. of n = 15 with ***P < 0.001.
uPA and uPAR Relocation to Early and Late Endosomes Is Increased Following UCD38B Treatment in U87MG Glioma Cells.
Analogously, uPA and uPAR were separately shown to colocalize with EEA-1 and LAMP-1 in glioma cells treated with UCD38B after 60 minutes but not at 15 minutes. (Fig. 6, B and D; Fig. 7, B and D). Treatment for 60 minutes with UCD74A or drug vehicle did not alter the minimal colocalization of uPA and uPAR with EEA-1 and LAMP-1 (Fig. 6, A and C; Fig. 7, A and C). High-content image analysis quantified that uPA colocalized with early and late endosomal markers 48 and 38%, respectively, in U87MG glioma cells following 60-minute treatment with UCD38B (Fig. 6E). Similarly, uPAR colocalized with 49 and 50% of early and late endosomes during this treatment interval (Fig. 7E). Colocalization of uPA and uPAR with early and late endosomes was significant in comparison with vehicle- and UCD74A-treated U87MG glioma cells (Fig. 6E; Fig. 7E). After 60 minutes, UCD38B induced a comparable, 4- to 5-fold increase in the colocalization of PAI-1, uPA, and uPAR with early and late endosomes (Fig. 6E; Fig. 7E), which indicated a concerted mechanism of intracellular trafficking.
Fig. 6.

Colocalization of uPA with early and late endosomes in U87MG glioma cells. Association of uPA with early endosomes using EEA-1 as a marker protein was analyzed using anti-uPA antibody. U87MG glioma cells were treated for 60 minutes with either drug vehicle, 250 μM of UCD74A (A), or 250 μM of UCD38B for 15 and 60 minutes (B). uPA and late endosome localization using LAMP-1 as a marker protein was examined in glioma cells treated with drug vehicle (control), UCD74A for 60 minutes (C), or UCD38B for 15 or 60 minutes (D). Scale bar is 50 μm. Quantification of uPA colocalization with endosomes was determined by imaging U87MG glioma cells using the high-content imaging system ImageXpress and analyzed by a custom module using MetaXpress 5.0 software (E). Results are presented as the mean ± S.D. of n = 15 with ***P < 0.001.
Fig. 7.

Colocalization of uPAR with early and late endosomes in U87MG glioma cells. Association of uPAR with early endosomes and late endosomes using EEA-1 and LAMP-1 as marker proteins, respectively, was analyzed by confocal microscopy. U87MG cells were treated with vehicle control, 250 μM of UCD74A for 60 minutes (A and C), or 250 μM of UCD3B for 15 or 60 minutes (B and D). Scale bar is 50 μm. Quantification of colocalization of uPAR with endosomes was quantified in U87MG glioma cells using the high-content imaging system ImageXpress and analyzed with a custom module using MetaXpress 5.0 software (E). Results are presented as the mean ± S.D. of n = 15 with ***P < 0.001.
PAI-1 Relocation to Early and Late Endosomes Is Increased Following UCD38B Treatment in Three Additional Human Glioma Cell Lines.
Drug-induced relocation of uPAS endosomes as a mechanism leading to AIF-mediated cancer cell necrosis has not been previously described. Therefore, it was important to determine whether endosomal relocation initiated by UCD38B was generalized to additional human glioblastoma cell lines. Because of its regulatory centrality in uPAS endosomal trafficking, PAI-1 was selected as the representative protein to investigate drug-induced endosomal colocalization in the human glioma cell lines LN229, U118MG, and U138MG. After 60 minutes of UCD38B treatment, confocal microscopy and high-content imaging again demonstrated that 3- to 5-fold more PAI-1 was colocalized with early and late endosomes in treated LN229 (Supplemental Fig. 1), U118MG (Supplemental Fig. 2) and U138MG (Supplemental Fig. 3) human glioma cells than in vehicle- or UCD74A-treated glioma cells.
LRP-1 and PAI-1 Demonstrate a Marked Increase in Relocation to Early and Late Endosomes Relocated to Mitochondrial Regions Following UCD38B Treatment in U87MG Gliomas.
LRP-1 is responsible for guiding clathrin-dependent internalization of the uPA–PAI-1 complex bound to transmembrane uPAR (Fig. 1). Therefore, it was important to identify whether uPAS endocytosis and endosomal trafficking guided by LRP-1 was altered by UCD38B treatment. Confocal microscopy demonstrated that LRP-1 remained abnormally colocalized with intracellular PAI-1 in U87MG glioma cells following 60-minute treatment with UCD38B, while no alteration was observed with UCD74A treatment (Fig. 8A). LRP-1 also remained abnormally colocalized with both EEA-1 (Fig. 8B) and LAMP-1 (Fig. 8C) in U87MG glioma cells treated with UCD38B. In glioma cell controls treated with vehicle or UCD74A, there was no change in LRP-1 localization.
Fig. 8.

Colocalization of PAI-1 and LRP-1 in U87MG glioma cells. U87MG cells were treated with either vehicle control, UCD74A, or UCD38B at 250 μM for 60 minutes. PAI-1 seen in green was immunostained with anti–PAI-1 primary and Alexa 488 secondary antibody. LRP-1 in red is detected by anti–LRP-1 primary and Alexa 594 secondary antibody (A). DAPI, 4′,6-diamidino-2-phenylindole . LRP-1 in green was associated with early endosomes immunostained with EEA-1 antibody (red) (B). Colocalization of LRP-1 with late endosomes was detected with LAMP-1 antibody (red) (C). Scale bar is 50 μm.
We have used JC-1 in live U87MG glioma cells and determined that mitochondrial depolarization occurred between 60 and 120 minutes following UCD38B treatment (Pasupuleti et al., 2013). Here, MitoTracker was employed to visualize mitochondria, and confocal fluorescent microscopy identified that 60-minute treatment of UCD38B caused early (Fig. 9A) and late (Fig. 9B) endosomes to become in close proximity with mitochondria, as compared with glioma cells treated with vehicle or UCD74A. ImageXpress high-content quantification demonstrated a 3.5-fold increase in endosomal coregistration with mitochondria (Fig. 9C). To verify that mitochondrial membrane depolarization was associated with this drug-induced colocalization of endosomes with mitochondria, we pretreated the glioma cells with JC-1 dye. JC-1 forms red aggregates and green monomers in cells having normally polarized mitochondria. The red aggregates are diminished or disappear with mitochondria depolarization or permeabilization. The late endosomal marker protein LAMP-1 (stained in blue) was selected as a marker of endosomal colocalization within the same glioma cells that had been preloaded with JC-1 (Fig. 9D). UCD38B treatment for 75 minutes in glioma cells caused the disappearance of JC-1 red aggregates, with only the retention of green monomers, indicating mitochondrial depolarization that had increased colocalization with late endosomes (blue) (Fig. 9D). In summary, these data indicated that UCD38B initiated retrafficking of early and late endosomes containing uPAS and LRP-1 proteins to mitochondria regions and associated with mitochondrial membrane depolarization, which was not observed in glioma cells treated with vehicle or UCD74A.
Fig. 9.

Close proximity of endosomes and mitochondria in UCD38B-treated U87MG glioma cells. U87MG cells were treated with either vehicle control, UCD74A, or UCD38B at 250 μM for 60 minutes. Translocation of early (EE) and late endosomes (LE) was verified by immunocytochemistry using EEA-1 and LAMP-1, respectively. Mitochondria are visualized in red using MitoTracker. Early endosomes were detected using anti–EEA-1 antibody (A). Late endosomes were detected by anti–LAMP-1 antibody (B). Colocalization of endosomes with mitochondria was quantified using ImageXpress and analyzed with a custom module using MetaXpress 5.0 software. Results are presented as the mean ± S.D. of n = 15 with ***P < 0.001 (C). U87MG cells were preloaded with JC-1 dye at 5 μM for 15 minutes and treated with either vehicle control, UCD38B, or UCD74A at 250 μM for 75 minutes. Glioma cells were then fixed for immunocytochemistry to immunostained for late endosomes using anti–LAMP-1 as primary and Alexa 647 as secondary antibody, respectively (D). Scale bar is 50 μm.
UCD38B Targets Intracellular uPAS in U87MG Glioma Cells Rather than Plasmalemmal Complex.
We then investigated whether UCD38B targeted the uPA–PAI-1–uPAR complex bound to LRP-1 on plasmalemma. LRP-1–mediated endocytosis was prevented using the LRP-1 inhibitor, RAP, but did not prevent UCD38B-induced glioma cell death, as measured by the LDH assay (data not shown). Endosomal trafficking can be reversibly inhibited by cooling glioma cells to 4°C and then restoring their temperature to 37°C. U87MG glioma cells were exposed to UCD38B or UCD74A at 4°C for 2, 6, and 24 hours, and did not undergo cell death when compared with time-matched controls at 37°C. UCD38B is autofluorescent and can be demonstrated to be intracellular using fluorescent microscopy of washed U87MG cells, as we previously described in contrast to autofluorescent cell-impermeant UC74A (Leon et al., 2013) (Table 1). UCD38B was observed to be intracellularly localized at 4°C after a 2-hour exposure (Fig. 10A). We then determined that internalized UCD38B that had entered the glioma cells could initiate endosomal mis-trafficking and subsequent cell death by washing the cells twice at 4°C and then shifting them to 37°C. Glioma cells were incubated at 4°C for 2, 6, and 24 hours. When these glioma cells were shifted to 37°C for 24 hours, intracellular UCD38B killed 40, 60, and 65% of glioma cells, respectively, as determined by the LDH assay (Fig. 10B) and verified by the Trypan blue exclusion assay (Supplemental Fig. 4). We verified that intracellular UCD38B caused PAI-1 relocation to mitochondrial regions only in glioma cells shifted to 37°C that subsequently died (Fig. 10C). Importantly, no cell death was detected in control glioma cells under the same conditions when treated with the cell-impermeant, autofluorescent congener UCD74A. Together, these data indicate that UCD38B bypasses the uPAS protein complex in the plasmalemma and directly targets intracellular uPAS proteins to initiate uPAS endosomal relocation that was followed by subsequent glioma cell demise.
TABLE 1.
uPA–PAI-1 complex in glioma cells detected by ELISA
uPA–PAI-1 complex was determined by ELISA following drug treatment. U87MG glioma cells were treated with UCD38B for 45 minutes and 2 hours or with UCD74A for 2 hours. Protein extracts were made and 0.5 mg/ml of the total protein was used for detection of the uPA–PAI-1 complex. Results are presented as the mean ± S.D. of n = 3.
| U87MGs treated with drug |
uPA–PAI-1 complex |
|---|---|
| ng/ml | |
| Vehicle, 120 minutes | 4.1 ± 0.03 |
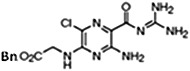 UCD38B, 45 minutes UCD38B, 45 minutes |
5.5 ± 0.3** |
| UCD38B, 120 minutes | 6.2 ± 0.1** |
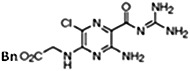 UCD74A, 120 minutes UCD74A, 120 minutes |
4.1 ± 0.06 |
P < 0.01.
Fig. 10.

Intracellular UCD38B cytotoxicity. U87MG glioma cells were treated with UCD38B or UCD74A for 2 hours at 4°C. The cells were rinsed with PBS and visualized under a fluorescent microscope. UCD38B was cell permeant at 4°C, as determined by intracellular fluorescence, and UCD74A was cell impermeant. Upper panel shows fluorescence, and lower panel shows bright field images at 40×. Scale bar is 50 μm (A). U87MG glioma cells were incubated for 2, 6, and 24 hours at 4°C. Glioma cells were washed with 1× PBS, replaced with fresh media with no drug and then transferred to 37°C for 24 hours. Cytotoxicity of intracellular UCD38B was determined by LDH assay (B). PAI-1 colocalization with mitochondria was examined by immunofluorescence. U87MG cells were exposed to UCD38B or UCD74A for 2 hours at 4°C, followed by a 37°C shift to induce glioma cell death. Mitochondria and PAI-1 were visualized using MitoTracker and anti–PAI-1 monoclonal antibody, respectively. 4′,6-diamidino-2-phenylindole (DAPI) was used to stain the nucleus. Scale bar is 50 μm (C).
uPA–PAI-1 Complexes Are Increased in U87MG Glioma Cells Following Treatment with UCD38B Treatment.
We postulated that UCD38B was disrupting normal endosomal transport of the quaternary protein complex comprised of uPAR, uPA, PAI-1, and LRP-1. Disruption of uPAS recycling by UCD38B would hamper recycling and the degradation of the quaternary uPAS complex, causing an increase in the cellular content of uPA–PAI-1 complexes. A commercially available ELISA kit was employed to quantitatively measure the amount of uPA–PAI-1 complexes in treated and control glioma cells. U87MG glioma cells were treated for 120 minutes with UCD38B, UCD74A, or drug vehicle. The amount of the uPA-PAI complex (in nanograms per milliliter) was quantified in 0.5 mg/ml of the total protein extracted from the treated glioma cells and normalized to a standard curve (Table 1). Intracellular levels of the uPA–PAI-1 complex were increased significantly following treatment with UCD38B, but not with UCD74A or vehicle control.
Discussion
HGGs, especially World Health Organization grade 4 glioblastomas, are highly aggressive primary cancers of the central nervous system, accounting for 78% of adult onset primary central nervous system malignancies (Dunn and Black, 2003; Buckner et al., 2007). The 5-year survival for patients with glioblastoma multiforme receiving conventional therapy is approximately 9%. Malignant gliomas recur in greater than 90% of cases despite radiation therapy, chemotherapy, or antiangiogenic agents, most notably bevacizumab.
5-benzylglycinyl-amiloride (UCD38B) is the parent molecule of a class of anticancer small molecules found to kill proliferative and nonproliferative glioma and breast cancer cell types (Leon et al., 2013; Pasupuleti et al., 2013). Previously, we demonstrated that UCD38B is a cell-permeant molecule that rapidly initiates a set of intracellular events causing mitochondrial depolarization with the release and nuclear translocation of AIF. Nuclear AIF mediates irreversible programmed necrotic cancer cell death, independent of caspase and poly ADP-ribose polymerase activation (Leon et al., 2013; Pasupuleti et al., 2013). The cell-impermeant congener of UCD38B, 5-glycinyl-amiloride (UCD74A), is a comparably competitive uPA inhibitor, inhibits cancer cell migration, and is cytostatic but not cytotoxic (Table 1) (Massey et al., 2012; Pasupuleti et al., 2013). Our initial reports describing the anticancer cytotoxicity of UCD38B against proliferating and nonproliferating well characterized human glioblastoma and human breast cancer cell lines produced several unanswered questions about its cellular mechanisms, which are better understood as a consequence of this investigation.
One important question was why the anticancer cytotoxicity of UCD38B is independent of the cell cycle. Previously, we identified that UCD38B initiates relocation of uPA, and in this study, we identified that uPA, along with PAI-1, uPAR, and LRP-1, are coordinately relocated to mitochondrial regions by a concerted drug-induced mis-trafficking of uPAS endosomes. uPA, PAI-1, and uPAR colocalization with early and late endosomes is increased following UCD38B exposure, corroborated by subcellular fractionation and immunocytochemistry, and quantified by a high-content cytologic image analysis. Colocalization of uPAS proteins with early and late endosomes is increased 4- to 5-fold, and approximately 40–50% of uPAS endosomes are relocated to perinuclear mitochondrial domains. This unusual anticancer drug mechanism of action has not been previously reported, so it was important that these observations were generalized to the additional human glioma cell lines LN229, U118MG, and U138MG (Supplemental Figs. 1–3). PAI-1 was selected as a representative uPAS protein and demonstrated to colocalize with early and late endosomal markers following treatment with UCD38B. UCD38B initiates intracellular relocation of a subset of uPAS endosomes to perinuclear mitochondrial regions beginning at 60 minutes of UCD38B treatment, which coincides with mitochondrial depolarization in live U87MG glioma cells loaded with the fluorescent dye JC-1 that we previously reported (Pasupuleti et al., 2013). Mitochondrial depolarization is associated with the release and nuclear translocation of AIF, triggering a caspase-independent necrotic glioma cell death. This programmed AIF-mediated, caspase-independent glioma cell necrosis following UCD38B treatment is consistently observed in the additional human glioblastoma cell lines LN229, U118MG, and U138MG (Supplemental Figs. 5 and 6), which are capable of initiating high-grade glial tumors.
Endosomal relocation to adjacent mitochondrial domains is triggered by UCD38B within 60–120 minutes, corresponding with electron microscopic findings. Electron microscopy showed that UCD38B caused a marked increase in the appearance of vesicles and lysosomes in U87MG glioma cells following 60-minute treatment with UCD38B, which is consistent with enhanced endocytosis. Confocal microscopy and quantitative image analysis confirmed that UCD38B initiates a 3.5-fold increase in early and late endosomes colocalizing with mitochondrial regions and that 35% of endosomes appear to be relocated following drug treatment. Similar trafficking between endosomes and mitochondria has been reported following cell death caused by the vacuolating cytotoxin A toxin produced by Helicobacter pylori (Calore et al., 2010), tumor necrosis factor α (Garcia-Ruiz et al., 2002), and Fas (Ouasti et al., 2007).
AIF-mediated glioma necrosis produced by UCD38B is independent of canonical apoptosis (Leon et al., 2013; Pasupuleti et al., 2013) and appears to be distinct from ripoptosome-mediated necroptosis (Feoktistova et al., 2012). RIP-1 inhibition by necrostatin does not alter the anticancer cytotoxicity of UCD38B (Leon et al., 2013). Cell death induced by UCD38B was also unaffected by autophagic inhibition by the AMPK inhibitor compound C, the lysosomotropic agent chloroquine (Shintani and Klionsky, 2004; Meley et al., 2006), or by knockdown of the autophagy protein beclin-1 (Leon et al., 2013). The precise cause of mitochondria membrane depolarization associated with uPAS endosomal colocation initiated by UCD38B remains unclear. Mitochondrial membrane depolarization under these conditions can be triggered by different physiologic effectors, including calcium-induced calcium release, reduced concentrations of adenine nucleotides, inorganic phosphate, reactive oxygen species, changes in pH, reduced mitochondrial polarization (Crompton, 1999), and Bax (Marzo et al., 1998). Previously, we reported that calcium overload in glioma cells resulting from reversal of the sodium-calcium exchanger coupled with altered activation of sodium-proton exchange causes increased cytosolic free calcium to be stored within the mitochondria and endoplasmic reticulum (Harley et al., 2010). Calcium signaling released from the endoplasmic reticulum that is contacting mitochondrial domains has been described to affect the mode and extent of mitochondrial permeability induced by Bcl proteins (Mattson et al., 2000). The role of mitochondria in participating in programmed necrosis and necroptosis (Marshall and Baines, 2014) and the potential importance of these new cell death mechanisms caused by anticancer agents, including UCD38B, has recently been reviewed and is an active area of cell biologic investigation (Fulda, 2013).
LRP-1 is an endosomal guidance protein required for clathrin-mediated endocytosis and uPAS recycling (Cortese et al., 2008). Endocytotic internalization of uPAR bound to uPA–PAI-1 that is mediated by LRP-1 can be prevented by preincubating with the specific LRP-1 antagonist RAP (Taylor and Hooper, 2007). UCD38B antiglioma cytotoxicity was unaffected by preincubation with RAP. This surprising result was explained by reversibly inhibiting endosomal trafficking by cooling glioma cells to 4°C. Intracellular UCD38B, but not UCD74A, was capable of causing endosomal relocation and subsequent glioma demise when endosomal trafficking was re-established at 37°C. Reversible inhibition of endosomal trafficking demonstrated that cell-permeant UCD38B directly enters cancer cells and kills them by selectively mis-trafficking a subset of endosomes containing uPAS cargo.
Early endosomes are normally routed to the juxtanuclear endocytic recycling compartment from where the recycling endosomes originate (Maxfield and McGraw, 2004; Grant and Donaldson, 2009). Recycling endosomes transport uPAR and LRP-1 back to the plasma membrane (Fig. 1). Thus, the observed increase in cellular levels of the uPA–PAI-1 complex 60 minutes after UCD38B treatment can be explained by drug-induced disruption of uPAS endosomal recycling and trafficking causing reduced clearance of transmembrane uPAR bound to uPA and PAI-1 in the plasmalemma. There is a basal, small colocalization of LRP-1 with PAI-1 that significantly increases following 60-minute treatment with UCD38B and is relocated to early and late endosomes along with PAI-1, uPA, and uPAR.
Figure 11 summarizes our proposed model for the cellular mechanisms by which intracellular UCD38B binds to the active site of intracellular uPA, causing uPA, PAI-1, uPAR, and LRP-1 to be rapidly endocytosed in early and late endosomes. Following UCD38B treatment, early and late endosomes (Steps 1 and 2, respectively) are relocated to mitochondrial regions and followed by mitochondrial swelling. This mitochondrial swelling corresponds with previously observed mitochondrial depolarization, leading to the release and nuclear translocation of AIF. In the nucleus, AIF causes nuclear condensation and caspase-independent glioma necrosis (Step 3). Under normal conditions, uPAR and LRP-1 are recycled back to the cell membrane via recycling endosomes that are disrupted by UCD38B (Step 4). We posit that disrupting this recycling step by UCD38B is responsible for the increase in uPA–PAI-1 protein complexes. Further analysis and refinement of this proposed drug mechanism should provide important insights into why increased uPA–PAI-1 and PAI-1 expression paradoxically has been shown to correspond with a worsened clinical prognosis in several cancer cell types (Andreasen, 2007).
Fig. 11.

Proposed model of altered endosomal trafficking following treatment with UCD38B. Cell permeant drug (UCD38B) targets the intracellular uPAS complex, relocating the early (EE) (1) and late endosomes (LE) (2) to be in close proximity with mitochondrial regions. AIF is released and translocated to the nucleus (3), leading to glioma cell death. We hypothesize that UCD38B disrupts endosomal recycling (4), thereby increasing the intracellular uPA–PAI-1 complex.
To our knowledge, this is the first report of a class of anticancer, cytotoxic small molecules that enter the cancer cell and initiate mis-trafficking of a subset of early and late endosomes containing uPA, PAI-1, and uPAR to mitochondrial domains and trigger irreversible programmed glioma necrotic cell death. Surprisingly, the effect of UCD38B and its 50-fold more potent congeners bypass the plasmalemmal uPAR complex and instead act intracellularly to disrupt uPAS endosomal transport. Earlier, we reported the downregulation of protein expression of uPA by small interfering RNA and demonstrated its effect on drug cytotoxicity (Pasupuleti et al., 2013). In another study, we have investigated the downregulation and overexpression of additional uPAS proteins in glioma cells and utilizing a congener of UCD38B, verified AIF-mediated cell death mechanism (unpublished data) within intracerebral glioma xenografts implanted in immunodeficient mice. Remarkably, these small molecules have demonstrated minimal toxicity in rodents when administered systemically or intracerebrally (N. Pasupuleti and F. Gorin, manuscript in preparation). In subsequent studies, the properties of UCD38B and its 50-fold more potent congeners are being extended to include patient-derived xenografts established from primary human glioblastoma tumors and maintained in immunodeficient mice.
This unusual drug mechanism of action explains why the antiglioma cytotoxicity of UCD38B was identified as being independent of the cell cycle in human glioma and breast cancer cell lines (Leon et al., 2013; Pasupuleti et al., 2013) and appears to be restricted to cancer cell types having altered uPAS expression, notably uPA and PAI-1.
Supplementary Material
Acknowledgments
The authors thank Dr. Pamela J. Lein for generous use of her laboratory and sharing her Olympus IX81 confocal microscope and ImageXpress Micro XL System.
Abbreviations
- AIF
apoptosis-inducing factor
- BSA
bovine serum albumin
- DMSO
dimethylsulfoxide
- ELISA
enzyme-linked immunosorbant assay
- HGG
high-grade gliomas
- LDH
lactate dehydrogenase
- PBS
phosphate-buffered saline
- RAP
receptor associated protein
- UCD38B
5-benzylglycinyl-3-amino-6-chloro-N-(diaminomethylene) pyrazine-2-carboxamide
- UCD74A
5-glycinyl-3-amino-6-chloro-N-(diaminomethylene) pyrazine-2-carboxamide
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
- uPAS
urokinase plasminogen activator system
Authorship Contributions
Participated in research design: Pasupuleti, Gorin.
Conducted experiments: Pasupuleti.
Performed data analysis: Pasupuleti, Grodzki, Gorin.
Contributed to the writing of the manuscript: Pasupuleti, Gorin.
Footnotes
This research was funded by the National Institutes of Health, Neurologic Sciences [Grants R01-NS040489 and R01-NS060880], University of California Davis School of Medicine, University of California Davis Research Investments in Science and Engineering, and MIND Institute Intellectual and Developmental Disabilities Research Center [Grant U54-HD079125].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Andreasen PA. (2007) PAI-1 - a potential therapeutic target in cancer. Curr Drug Targets 8:1030–1041. [DOI] [PubMed] [Google Scholar]
- Andreasen PA, Kjoller L, Christensen L, Duffy MJ. (1997) The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer 72:1–22. [DOI] [PubMed] [Google Scholar]
- Bajou K, Maillard C, Jost M, Lijnen RH, Gils A, Declerck P, Carmeliet P, Foidart JM, Noel A. (2004) Host-derived plasminogen activator inhibitor-1 (PAI-1) concentration is critical for in vivo tumoral angiogenesis and growth. Oncogene 23:6986–6990. [DOI] [PubMed] [Google Scholar]
- Blasi F, Vassalli JD, Danø K. (1987) Urokinase-type plasminogen activator: proenzyme, receptor, and inhibitors. J Cell Biol 104:801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner JC, Brown PD, O’Neill BP, Meyer FB, Wetmore CJ, Uhm JH. (2007) Central nervous system tumors. Mayo Clin Proc 82:1271–1286. [DOI] [PubMed] [Google Scholar]
- Calore F, Genisset C, Casellato A, Rossato M, Codolo G, Esposti MD, Scorrano L, de Bernard M. (2010) Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ 17:1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conese M, Nykjaer A, Petersen CM, Cremona O, Pardi R, Andreasen PA, Gliemann J, Christensen EI, Blasi F. (1995) alpha-2 Macroglobulin receptor/Ldl receptor-related protein(Lrp)-dependent internalization of the urokinase receptor. J Cell Biol 131:1609–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese K, Sahores M, Madsen CD, Tacchetti C, Blasi F. (2008) Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS ONE 3:e3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Czekay RP, Aertgeerts K, Curriden SA, Loskutoff DJ. (2003) Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol 160:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn IF, Black PM. (2003) The neurosurgeon as local oncologist: cellular and molecular neurosurgery in malignant glioma therapy. Neurosurgery 52:1411–1422. [DOI] [PubMed] [Google Scholar]
- Ellis V, Scully MF, Kakkar VV. (1989) Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J Biol Chem 264:2185–2188. [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Panayotova-Dimitrova D, Leverkus M. (2012) Pick your poison: the Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 11:460–467. [DOI] [PubMed] [Google Scholar]
- Fulda S. (2013) The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther 14:999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Ruiz C, Colell A, Morales A, Calvo M, Enrich C, Fernández-Checa JC. (2002) Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J Biol Chem 277:36443–36448. [DOI] [PubMed] [Google Scholar]
- Grant BD, Donaldson JG. (2009) Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbeck N, Schmitt M, Meisner C, Friedel C, Untch M, Schmidt M, Sweep CG, Lisboa BW, Lux MP, Beck T, et al. Chemo-N 0 Study Group (2013) Ten-year analysis of the prospective multicentre Chemo-N0 trial validates American Society of Clinical Oncology (ASCO)-recommended biomarkers uPA and PAI-1 for therapy decision making in node-negative breast cancer patients. Eur J Cancer 49:1825–1835. [DOI] [PubMed] [Google Scholar]
- Harley W, Floyd C, Dunn T, Zhang XD, Chen TY, Hegde M, Palandoken H, Nantz MH, Leon L, Carraway KL, 3rd, et al. (2010) Dual inhibition of sodium-mediated proton and calcium efflux triggers non-apoptotic cell death in malignant gliomas. Brain Res 1363:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Clouthier DE, Hammer RE. (1992) LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 71:411–421. [DOI] [PubMed] [Google Scholar]
- Herz J, Hamann U, Rogne S, Myklebost O, Gausepohl H, Stanley KK. (1988) Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J 7:4119–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon LJ, Pasupuleti N, Gorin F, Carraway KL., 3rd (2013) A cell-permeant amiloride derivative induces caspase-independent, AIF-mediated programmed necrotic death of breast cancer cells. PLoS ONE 8:e63038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall KD, Baines CP. (2014) Necroptosis: is there a role for mitochondria? Front Physiol 5:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HL, Prévost MC, Xie Z, Matsuyama S, Reed JC, et al. (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281:2027–2031. [DOI] [PubMed] [Google Scholar]
- Massey AP, Harley WR, Pasupuleti N, Gorin FA, Nantz MH. (2012) 2-Amidino analogs of glycine-amiloride conjugates: inhibitors of urokinase-type plasminogen activator. Bioorg Med Chem Lett 22:2635–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD. (2000) Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci 23:222–229. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, McGraw TE. (2004) Endocytic recycling. Nat Rev Mol Cell Biol 5:121–132. [DOI] [PubMed] [Google Scholar]
- Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. (2006) AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 281:34870–34879. [DOI] [PubMed] [Google Scholar]
- Olson D, Pöllänen J, Høyer-Hansen G, Rønne E, Sakaguchi K, Wun TC, Appella E, Danø K, Blasi F. (1992) Internalization of the urokinase-plasminogen activator inhibitor type-1 complex is mediated by the urokinase receptor. J Biol Chem 267:9129–9133. [PubMed] [Google Scholar]
- Ouasti S, Matarrese P, Paddon R, Khosravi-Far R, Sorice M, Tinari A, Malorni W, Degli Esposti M. (2007) Death receptor ligation triggers membrane scrambling between Golgi and mitochondria. Cell Death Differ 14:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palandoken H, By K, Hegde M, Harley WR, Gorin FA, Nantz MH. (2005) Amiloride peptide conjugates: prodrugs for sodium-proton exchange inhibition. J Pharmacol Exp Ther 312:961–967. [DOI] [PubMed] [Google Scholar]
- Pasupuleti N, Leon L, Carraway KL, 3rd, Gorin F. (2013) 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase-independent necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp Ther 344:600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M, Harbeck N, Thomssen C, Wilhelm O, Magdolen V, Reuning U, Ulm K, Höfler H, Jänicke F, Graeff H. (1997) Clinical impact of the plasminogen activation system in tumor invasion and metastasis: prognostic relevance and target for therapy. Thromb Haemost 78:285–296. [PubMed] [Google Scholar]
- Setyono-Han B, Stürzebecher J, Schmalix WA, Muehlenweg B, Sieuwerts AM, Timmermans M, Magdolen V, Schmitt M, Klijn JG, Foekens JA. (2005) Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UK1. Thromb Haemost 93:779–786. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Hooper NM. (2007) The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem J 402:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulisse S, Baldini E, Sorrenti S, D’Armiento M. (2009) The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets 9:32–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.