

Abstract
G protein–coupled receptors (GPCRs) are a prominent class of plasma membrane proteins that regulate physiologic responses to a wide variety of stimuli and therapeutic agents. Although GPCR oligomerization has been studied extensively in recombinant cells, it remains uncertain whether native receptors expressed in their natural cellular environment are monomers, dimers, or oligomers. The goal of this study was to determine the monomer/oligomer status of a native GPCR endogenously expressed in its natural cellular environment. Native 5-HT2C receptors in choroid plexus epithelial cells were evaluated using fluorescence correlation spectroscopy (FCS) with photon counting histogram (PCH). An anti–5-HT2C fragment antigen binding protein was used to label native 5-HT2C receptors. A known monomeric receptor (CD-86) served as a control for decoding the oligomer status of native 5-HT2C receptors by molecular brightness analysis. FCS with PCH revealed molecular brightness values for native 5-HT2C receptors equivalent to the molecular brightness of a homodimer. 5-HT2C receptors displayed a diffusion coefficient of 5 × 10−9 cm2/s and were expressed at 32 receptors/μm2 on the apical surface of choroid plexus epithelial cells. The functional significance and signaling capabilities of the homodimer were investigated in human embryonic kidney 293 cells using agonists that bind in a wash-resistant manner to one or both protomers of the homodimer. Whereas agonist binding to one protomer resulted in G protein activation, maximal stimulation required occupancy of both protomers. This study is the first to demonstrate the homodimeric structure of 5-HT2C receptors endogenously expressed in their native cellular environment, and identifies the homodimer as a functional signaling unit.
Introduction
G protein–coupled receptors (GPCRs) represent one of the largest families of cell membrane signaling proteins. They are present on the surface of most cells and regulate the physiologic functions of all major organ systems in the human body. GPCRs modulate physiologic responses to light, odorants, hormones, neurotransmitters, and therapeutic agents. Although physiologic processes regulated by GPCR activation and blockade have been studied for decades, it is still uncertain whether the functional signaling unit is a monomer, dimer, or higher-order oligomer.
Currently, there are hundreds of reports in the published literature describing the dimeric or oligomeric nature of GPCRs expressed in recombinant cell systems. Dimer/oligomer formation has been reported to regulate all aspects of GPCR function including synthesis, ligand binding, G protein coupling, and trafficking (reviewed in Herrick-Davis, 2013; Milligan, 2013). Even so, the presence and functional relevance of GPCR dimerization in vivo is widely debated (reviewed in Ferré et al., 2014). Concerns have been raised about the functional relevance of GPCR dimerization, as monomeric receptors have been reported to activate G proteins in reconstituted systems (Bayburt et al., 2007; Whorton et al., 2007), and the crystal structure of the agonist-occupied β2-adrenergic receptor revealed a monomeric receptor in complex with a single G protein (Rasmussen et al., 2011). On the other hand, structural studies of native rhodopsin receptors have revealed their dimeric organization in rod outer segments (Liang et al., 2003) and their association with G transducin (Jastrzebska et al., 2013b). Additionally, leukotriene, dopamine, and serotonin receptors have been solubilized as homodimers in complex with a single G protein (Banères and Parello, 2003; Han et al., 2009; Pellissier et al., 2011).
The presence of GPCR dimers or oligomers in intact native tissues and in vivo is inferred from studies using indirect biophysical methods, immunofluorescence in fixed tissue sections, and functional studies in transgenic animals. For example, ligand-based fluorescence resonance energy transfer studies identified oxytocin homodimers in rat mammary gland tissue (Albizu et al., 2010), and heterodimers of D1 and D2 dopamine receptors in striatal neurons are inferred from antibody-based fluorescence resonance energy transfer in fixed brain slices (Hasbi et al., 2011). Supporting evidence is provided by studies using heterodimer-selective antibodies (Berg et al., 2012) and transgenic mice expressing mutant receptors that restore (Rivero-Müller et al., 2010) or inhibit (González et al., 2012) normal receptor function. Pharmacological approaches have used heterodimer-selective agonists (Waldhoer et al., 2005; Fujita et al., 2014) or antagonists that bind in a pseudo-irreversible manner to one protomer within the homodimer (Smith et al., 2011). However, studies using direct biophysical methods capable of monitoring protein interactions in living tissues are needed to substantiate these findings.
Although recent studies have used direct methods with near single-molecule sensitivity, such as total internal reflection fluorescence (Calebiro et al., 2013; Kasai and Kusumi, 2014; Teichmann et al., 2014) and fluorescence correlation spectroscopy (FCS) (Briddon et al., 2008; Malengo et al., 2008; Ganguly and Chattopadhyay, 2010; Herrick-Davis et al., 2012, 2013; Corriden et al., 2014), these studies were performed using recombinant cell lines. The purpose of the present study was to determine the monomer/oligomer status of a native GPCR endogenously expressed in its natural cellular environment. The serotonin 5-HT2C receptor was selected because it is highly expressed in choroid plexus epithelial cells, an established primary cell culture for studying native 5-HT2C receptors (Esterle and Sanders-Bush, 1992), and monoclonal antibodies recognizing the native conformation of the 5-HT2C receptor have already been developed (Mancia et al., 2007). Previously, we demonstrated the application of FCS with photon counting histogram (PCH), a sensitive method for the direct quantification of photons emitted from individual fluorescent molecules, for determining the oligomeric status of GPCR expressed in recombinant cell lines (Herrick-Davis et al., 2012, 2013). In FCS, fluorescence-tagged proteins that associate with one another or are part of a larger protein complex will codiffuse with one another. Autocorrelation analysis reveals the diffusion characteristics of fluorescence-tagged proteins in the sample and provides an estimate of the number of fluorescent proteins codiffusing together as a complex within the plasma membrane.
In the present study, a monoclonal anti–5-HT2C fragment antigen binding protein was used as a probe for labeling native 5-HT2C receptors in choroid plexus epithelial cells. FCS and PCH were used to monitor the diffusion characteristics and oligomer status of 5-HT2C receptors expressed in their native cellular environment. The functional significance was investigated in human embryonic kidney 293 (HEK293) cells using ergots that bind in a wash-resistant manner to one protomer (cabergoline) or both protomers (ergotamine) of the homodimer. These studies provide the first pharmacological and functional evidence for the presence of 5-HT2C receptor homodimers and highlight the signaling capabilities of the individual protomers.
Materials and Methods
Cell Culture and Transfection.
HEK293 cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s minimal essential medium (DMEM; Cellgro, Manassas, VA) with 10% fetal bovine serum (FBS; HyClone, Logan, UT) in a humidified incubator at 37°C, 5% CO2. For the confocal microscopy and FCS experiments, HEK293 cells were plated in six-well plates fitted with 25-mm poly-d-lysine–coated glass coverslips (Fisher Scientific, Pittsburgh, PA) at a density of 4 × 105 cells per coverslip and transfected with 200 ng of the indicated plasmid DNA using lipofectamine reagent (Invitrogen, Carlsbad, CA) for 5 hours. Following transfection, cells were cultured in minimal essential medium (without phenol red) with 10% charcoal-stripped serum (Gibco/Life Technologies, Grand Island, NY) for 20 hours at 37°C, 5% CO2. Prior to FCS analysis, coverslips were washed twice in HEPES-buffered Krebs-Ringer solution.
Plasmid.
cDNAs encoding the 5-HT2A receptor and the VSV isoform of the 5-HT2C receptor were cloned into pEGFP-N1, pECFP-N1, and pmCherry-N1 vectors (Takara Clontech, Mountain View, CA) to create chimeric receptors with fluorescent tags on the C-terminus of each receptor. CD-86/GFP and CD-86/GFP-GFP were generously provided by G. Milligan (University of Glasgow, Glasgow, UK). Site-directed mutagenesis (Stratagene, La Jolla, CA) was used to create an A206K mutation in all green fluorescent protein (GFP) constructs to eliminate potential aggregation (Zacharias et al., 2002).
GFP-Tagged, Anti–5-HT2C Fragment Antigen Binding Protein (2C-Fab-GFP).
A monoclonal antibody recognizing an external domain of the 5-HT2C receptor was previously developed and shown to label 5-HT2C receptors in transfected HEK293 cells (Mancia et al., 2007). The cDNA sequences encoding the heavy and light chains of the monoclonal anti–5-HT2C fragment antigen binding protein (Fab) were subcloned into mammalian expression vectors (Assur et al., 2007) and plasmids containing clone 1C4 (Mancia et al., 2007) were used in the present study. The light-chain cDNA sequence was cloned into an expression vector upstream of an internal ribosomal entry site followed by red fluorescent protein as a selection marker (Assur et al., 2007). For the present study, the heavy-chain cDNA sequence was modified as follows: a His6 tag was added in place of the stop codon, and the cDNA was ligated into the pEGFP-N1 expression vector (Clontech) for direct labeling of the heavy-chain C-terminus with GFP. Site-directed mutagenesis (Stratagene) was used to create an A206K mutation in the GFP tag to eliminate potential aggregation (Zacharias et al., 2002). HEK293 cells were transfected by calcium phosphate precipitation with a mixture of the light- and heavy-chain plasmids. A stable cell line expressing both light and heavy chains was identified by fluorescence microscopy and Western blot. Stably transfected cells were plated at a density of 5 × 106 cells/100-mm dish and cultured (37°C, 5% CO2) for 1 week in DMEM with 10% low IgG FBS (HyClone). 2C-Fab-GFP was purified from the culture media on nickel resin columns (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. Following column purification, samples were dialyzed in HEPES-buffered Krebs-Ringer (pH 7.4), and concentrated to 0.1 μM in spin columns (30,000 nominal molecular weight cut-off; EMD Millipore, Billerica, MA).
Western Blot.
Intact choroid plexus tissue isolated from the third and lateral ventricles of an adult female Sprague-Dawley rat was placed in a microcentrifuge tube containing 50 μl of cold RIPA buffer with protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The tube was placed on ice for 15 minutes, then sonicated in an ice-cold water bath for 15 minutes, and placed on ice again for 15 minutes prior to centrifugation at 30,000g for 30 minutes at 4°C. Ten microliters of the supernatant was diluted 1:1 in nonreducing Laemmli sample buffer. Purified 2C-Fab-GFP and prestained protein standards (Bio-Rad, Hercules, CA) were also diluted in nonreducing Laemmli sample buffer. The samples were heated at 70°C for 15 minutes and run on a 10% Tris-HCl Bio-Rad Ready Gel at 95 V for 70 minutes. Gel proteins were transferred to nitrocellulose (Hybond ECL; Amersham/GE Healthcare, Pittsburgh, PA) and probed with 2C-Fab-GFP overnight at 4°C in 1% milk/1% bovine serum albumin blocking solution. Following incubation with GFP(B-2)–horseradish peroxidase (1:3000; Santa Cruz, Dallas, TX), proteins were visualized using enhanced chemiluminescence (Amersham).
Choroid Plexus Epithelial Cell Culture and Immunostaining.
Primary choroid plexus epithelial cells were prepared as described by Esterle and Sanders-Bush (1992). In brief, choroid plexuses were dissected from the third and lateral ventricles of fetuses of timed-pregnant Sprague-Dawley rats at 19 days of gestation. The choroids were placed in digest buffer containing phosphate-buffered saline, pH 7.4, with 0.33 mg/ml pronase (Sigma-Aldrich) and 0.25 mg/ml DNase 1 (Sigma-Aldrich) for 25 minutes at 37°C, washed twice, then dissociated by trituration in DMEM/F12 (1:1) (Cellgro) with 0.13 mg/ml DNase 1. The supernatant containing dissociated epithelial cells was centrifuged at 1100 rpm for 3 minutes. Epithelial cells were resuspended in culture media containing DMEM/F12 (1:1) with 10% charcoal-stripped FBS (Gibco), 1% N2 supplement (Invitrogen), 10 ng/ml epidermal growth factor (Invitrogen), and 1% PenStrep (Invitrogen), then transferred to a 60-mm dish and incubated (37°C, 5% CO2) for 2 hours to allow fibroblasts to adhere to the dish. The culture medium containing unattached epithelial cells was removed from the dish, and the epithelial cells were plated on 25-mm glass coverslips (Fisher Scientific) coated with laminin (Gibco). After 3 weeks in culture, primary choroid epithelial cells were labeled with the monoclonal anti–5-HT2C–Fab-GFP antibody (2C-Fab-GFP, diluted 1:3 in HEPES-buffered Krebs-Ringer) for 40 minutes at 23°C immediately prior to FCS recording. For costaining with antitransthyretin (Bioss, Woburn, MA), 3-week-old cultures of choroid epithelial cells were fixed in phosphate-buffered 3.7% paraformaldehyde (10 minutes at room temperature), permeabilized with 0.1% triton, blocked with 4% donkey serum, and stained overnight at 4°C with 2C-Fab-GFP (diluted 1:10) and rabbit antitransthyretin (diluted 1:100) in HEPES-buffered Krebs-Ringer, pH 7.4, with 1% donkey serum. The rabbit antitransthyretin was visualized with an Alexa 488–conjugated donkey anti-rabbit IgG (diluted 1:1000; Invitrogen).
Inositol Phosphate Assay.
HEK293 cells were seeded at 2 × 105 cells/well, in 24-well plates, in DMEM with 10% FBS. Cells were transfected with 100 ng of plasmid containing cDNA encoding the VSV isoform of the 5-HT2C receptor using lipofectamine reagent for 5 hours at 37°C. Following transfection, cells were cultured in DMEM with 10% FBS for 24 hours, then washed and labeled overnight in inositol-free, serum-free DMEM with 0.5 μCi of [3H]myoinositol. At the start of the experiment, cells were washed twice with phosphate-buffered saline (PBS) and were pretreated with serum-free DMEM in the absence or presence of drug for 60 minutes (or as indicated), followed by drug washout. The washout period consisted of four washes with PBS over a 30-minute period: one wash immediately following the end of drug pretreatment, followed by three subsequent media changes at 10-minute intervals wherein the cells were returned to the incubator. Following drug washout, the inositol phosphate (IP) assay was initiated by adding lithium chloride to the assay media, with or without drugs as indicated, for an additional 60 minutes. Total [3H]IP production was measured by anion exchange chromatography as previously described (Berridge et al., 1983). Data were analyzed using GraphPad Prism software (GraphPad Software, La Jolla, CA).
Whole-Cell Radioligand Binding Assay.
HEK293 cells were seeded at 4 × 105 cells/well, in 12-well plates, in DMEM with 10% FBS. Cells were transfected with 200 ng plasmid containing cDNA encoding the VSV isoform of the 5-HT2C receptor in serum-free media using lipofectamine reagent for 5 hours at 37°C. Following transfection, cells were cultured in DMEM with 10% FBS for 24 hours, then washed and incubated overnight in serum-free DMEM. Cells were pretreated with the indicated drugs for 60 minutes followed by four washes with PBS over a 30-minute period. Washed cells were labeled with [3H]mesulergine (2.5 nM) in 0.5 ml of serum-free DMEM, in the absence and presence of 1 μM 5-HT to define specific binding to cell surface 5-HT2C receptors, for 20 minutes at 37°C. Cells were washed with PBS to remove unbound radioligand, and 0.5 ml of ice-cold 3% trichloroacetic acid was added to each well. The plate was incubated on ice for 20 minutes to release surface-bound ligand from the cells without cell lysis. Following incubation, 450 µl of the trichloroacetic acid solution was removed from each well, mixed with 5 ml of Ecoscint cocktail, and counted in a Beckman liquid scintillation counter (Beckman Coulter, Pasadena, CA).
Fluorescence Correlation Spectroscopy.
For FCS measurements, cells were washed twice with HEPES-buffered Krebs-Ringer (without glucose), and the coverslip was placed in an Attofluor Cell Chamber (Molecular Probes/Life Technologies, Grand Island, NY) with 1 ml of HEPES-buffered minimal essential medium (without phenol red). FCS measurements were made using a Zeiss LSM-780 confocal microscope (Zen 2010 software; Carl Zeiss, Jena, Germany) equipped with a 32 element gallium arsenide phosphide linear array detector, as previously described (Herrick-Davis and Mazurkiewicz, 2013). One-photon excitation with a continuous-wave argon ion laser was performed using a 40× (numerical aperture 1.2) C-apochromat water immersion objective to create an observation volume on the order of 10−15 liters. FCS measurements were made on the upper plasma membrane of transfected HEK293 cells and the apical surface of primary choroid plexus epithelial cells labeled with the 2C-Fab-GFP antibody. Positioning of the plasma membrane in the center of the observation volume was achieved by monitoring the photon counts per molecule in real time while simultaneously focusing upward through the cell cytosol and then plasma membrane to identify the focal plane corresponding to the maximal photon counts per molecule. FCS measurements were recorded at 23°C for 100 seconds, as 10 consecutive 10-second intervals. GFP was excited by the 488-nm laser set at 0.1% to minimize photobleaching. The time-dependent fluctuations in fluorescence intensity were recorded on the gallium arsenide phosphide detector for fluorescence emission in the range of 520–625 nm. A pinhole of one airy unit was used. FCS recordings were analyzed by a digital temporal correlator (using nonlinear least-squares minimization, Zeiss Aim 4.2 software) to calculate the autocorrelation function G(τ), which represents the time-dependent decay in fluorescence fluctuation intensity as in eq. 1:
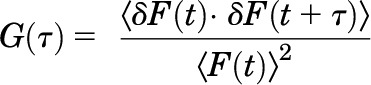 |
(1) |
where G(τ) is the <time average> of the change in fluorescence fluctuation intensity (δF) at some time point (t) and at a time interval later (t + τ), divided by the square of the average fluorescence intensity. Autocorrelation analyses were performed using the Zeiss Aim 4.2 software package with an autocorrelation bin time of 0.2 microseconds. FCS data were fit to a two-dimensional (2D) model (for lateral diffusion within the plasma membrane) with two components as in eq. 2:
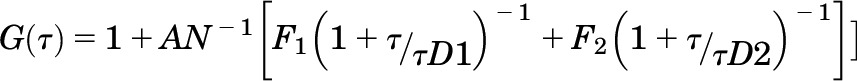 |
(2) |
where N is the number of molecules in the observation volume. F1, F2, and τD1, τD2 represent the respective fractions and diffusion times of the two components. A pre-exponential term is included to account for photophysical properties (e.g., blinking) of the fluorescent probe,
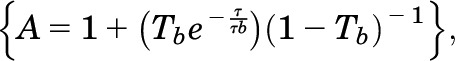 |
where Tb and τb represent the blinking fraction and relaxation time, respectively. It should be noted that individual GFP molecules are not always fluorescent. They can exhibit blinking, exist in a prolonged dark state, or be immature and nonfluorescent (Ulbrich and Isacoff, 2007).
The autocorrelation curve depicts the fluorescence intensity fluctuations as a function of particle number and diffusion time. The average dwell time of the fluorescent species within the observation volume (τD) is calculated from the midpoint of the autocorrelation curve. The diffusion coefficient (D) for lateral diffusion of fluorescence-tagged receptors within the plasma membrane is calculated as in eq. 3, where ωo is the radius of the observation volume in the horizontal dimension:
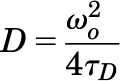 |
(3) |
The radius of the observation volume (ωo) was determined experimentally by measuring the width at the half-maximum height of a Gaussian distribution fitted to the image of subresolution fluorescent beads (0.1 μm FluoSpheres; Invitrogen), as previously described (Cole et al., 2011). In our experimental setup, ωo was determined to be 0.30 μm. Thus, the surface area of the plasma membrane in the observation volume is approximated by πωo2 to be 0.28 µm2.
The amplitude of the autocorrelation function G(0) (equal to the y-intercept) is inversely related to the number of molecules in the observation volume (NPSF) as in eq. 4:
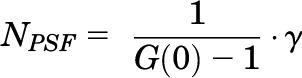 |
(4) |
where γ is the point spread function (PSF) which describes the shape of the observation volume. The numerical value of γ differs depending on the model selected for analysis and is 0.5 for 2D FCS analysis and 0.35 for a three-dimensional (3D) Gaussian model used in PCH analysis.
The average fluorescence intensity or average photon count rate (k) recorded for a given sample is determined by the number of fluorescent molecules (NPSF) and their molecular brightness (ε), as described in eq. 5:
| (5) |
Thus, dividing the count rate (k) by the number of molecules (NPSF) provides an estimate of the molecular brightness (ε) of the sample.
Photon Counting Histogram.
Fluorescence fluctuation data recorded during a FCS experiment can be used to generate photon counting histograms, which provide quantitative information about the number of fluorescent molecules and the number of photon counts per molecule (Chen et al., 1999). Ten measurements were made on the upper plasma membrane of each cell by monitoring the photon count rate for 100 seconds, as 10 consecutive 10-second observation periods. Although the laser intensity was set to 0.1% to minimize photo-bleaching, photo-bleaching was apparent during the first 10-second observation period. Therefore, the average molecular brightness from the second through 10th observation periods was calculated and reported as the molecular brightness for that cell. Segments of the fluorescence intensity trace that showed large spikes or drifts in fluorescence intensity (due to cell movement) were excluded from the analysis. To generate a histogram, each 10-second observation period was broken down into one million intervals or bins (PCH bin time = 10 microseconds ). Histograms were constructed using the PCH module in the Zeiss Aim 4.2 software in which the number of 10-microsecond bins was plotted on the y-axis and photon counts on the x-axis. The resulting histogram depicts the number of bins that registered 1, 2, 3 photon counts etc. during one 10-second observation period. Since a constant intensity light source produces a photon count distribution that follows Poisson statistics, as fluorescent molecules enter and diffuse through the nonhomogenously illuminated observation volume, the fluctuations in fluorescence intensity result in a broadening of the Poisson distribution. This super-Poisson characteristic is observed in the tail of the PCH curve. PCH data were fit to a one-component model in which concentration and molecular brightness were allowed to be free (and the first-order correction was fixed at zero) to determine the average molecular brightness of the sample. Residuals of the curve fit and reduced χ2 analyses were used to determine the goodness of fit (Müller et al., 2000).
Controls for Molecular Brightness Analysis.
Both cytosolic and plasma membrane controls were used to decode the molecular brightness of GFP-tagged 5-HT2C receptors. A known monomeric plasma membrane receptor with a single C-terminal GFP tag (CD-86/GFP) and with a tandem GFP tag (CD-86/GFP-GFP) was used to determine the molecular brightness of a single GFP and two GFP tags. All GFP constructs contained an A206K mutation to eliminate potential self-aggregation of GFP (Zacharias et al., 2002). To determine the contribution of background autofluorescence from cytoplasmic proteins, a dilute solution of purified monomeric GFP was evaluated. The molecular brightness of 2C-Fab-GFP in solution [8795 counts per second per molecule (CPSM)] was similar to GFP from pEGFP plasmid expressed in the cytosol of HEK293 cells (9075 CPSM), indicating that background autofluorescence from cytoplasmic proteins was minimal (approximately 3%) in our experimental setup.
Results
Creation of a Monoclonal, Monovalent Anti–5-HT2C Fragment Antigen Binding Protein with GFP Tag (2C-Fab-GFP).
Anti–5-HT2C monoclonal antibodies (mAbs) were generated by immunizing mice with a detergent-solubilized and purified form of the rat 5-HT2C receptor (Mancia et al., 2007). The mAb clone 1C4, recognizing an extracellular epitope and demonstrating selectivity for rat over human 5-HT2C receptors (Mancia et al., 2007), was selected for the present study. The rat and human 5-HT2C receptor protein sequences differ by only one or two amino acids in the extracellular loop regions, whereas the N-terminal domains differ by 11 amino acids (Saltzman et al., 1991). Native 5-HT2C receptors expressed in mammalian cells are glycosylated at two sites: N39 in the N-terminus and N204/205 in the second extracellular loop (Backstrom et al., 1995). Since the mAb recognizes the native conformation of the receptor, and binding of the mAb does not interfere with deglycosylation (Mancia et al., 2007), these observations implicate the N-terminus as the likely epitope for binding of the mAb to the 5-HT2C receptor.
Figure 1A illustrates the architecture of the anti–5-HT2C Fab. The heavy- and light-chain variable regions of the anti–5-HT2C Fab are linked to the constant regions of antilysozyme D1.3 from pASK84 (Skerra, 1994). The antilysozyme D1.3 allows proper assembly of the Fab via disulfide linkage of the constant regions following expression in mammalian cells. The light and heavy chains of the anti–5-HT2C Fab were subcloned into mammalian expression vectors as illustrated in Fig. 1B. The light-chain cDNA sequence is followed by an internal ribosomal entry site and red fluorescent protein as a selection marker (Assur et al., 2007). The heavy-chain cDNA sequence was subcloned into pEGFP, upstream and in frame with the GFP sequence for direct labeling of the heavy chain with a GFP tag. Following coexpression of the light- and heavy-chain plasmids in a HEK293 stable cell line, the anti–5-HT2C Fab with GFP tag (2C-Fab-GFP) was isolated from the culture media (structure shown in Fig. 1C). GFP was used instead of chemical labeling to ensure an exact 1:1 stoichiometry of GFP tag per Fab, essential for molecular brightness analysis. Although it is noted that individual GFP molecules are not always fluorescent, as GFP exhibits blinking and dark states (Ulbrich and Isacoff, 2007), the use of appropriately matched monomeric and dimeric GFP control samples in the present study allows accurate determination of GPCR oligomer status by molecular brightness analysis.
Fig. 1.

Design of the GFP-tagged anti–5-HT2C fragment antigen binding protein (2C-Fab-GFP). (A) The Fab is composed of the heavy- and light-chain variable regions (VH and VL) of a monoclonal antibody raised against the 5-HT2C receptor and the constant regions (CH1 and CL) of antilysozyme D1.3 from pASK84. (B) The light-chain cDNA sequence was cloned into an expression vector with an internal ribosomal entry site (IRES) followed by an red fluorescent protein (RFP) selection marker. The heavy-chain cDNA sequence was cloned into an expression vector upstream and in frame with the cDNA sequence for GFP, for direct labeling of the heavy chain with a GFP tag. (C) 2C-Fab-GFP was purified from the culture media of a stable HEK293 cell line coexpressing the heavy- and light-chain plasmids. CMV, cytomegalovirus.
Characterization of 2C-Fab-GFP.
Analysis of purified 2C-Fab-GFP by Western blot revealed a single band migrating near the 100 kDa standard molecular mass marker (Fig. 2A). These results are consistent with the predicted migration pattern of the light chain/heavy chain/GFP complex, in which each component contributes approximately 30 kDa to the overall size of the complex. To test the specificity of the 2C-Fab-GFP probe for labeling 5-HT2C receptors, a total protein extract was prepared from freshly isolated, intact choroid plexus tissue and analyzed by Western blot (Fig. 2B). Western blot analysis of the 5-HT2C receptor is predicted to show bands migrating between 45 and 65 kDa, representing immature and mature forms of the receptor (Backstrom et al., 1995). The 5-HT2C receptor has been shown to form detergent-sensitive homodimers (Herrick-Davis et al., 2004); thus, under mild denaturing conditions, fainter bands representing homodimers should appear between 90 and 130 kDa. Since several GPCRs have been solubilized as homodimers in a pentameric assembly with their cognate G protein (Banères and Parello, 2003; Han et al., 2009; Pellissier et al., 2011; Jastrzebska et al., 2013b), an additional band representing the 5-HT2C receptor homodimer in complex with a heterotrimeric G protein would be expected at approximately 200 kDa. The predominant bands identified with the 2C-Fab-GFP probe on the Western blot shown in Fig. 2B fall within these predicted size ranges.
Fig. 2.

Western blots of 2C-Fab-GFP. (A) Lane 1: molecular mass markers (kilodaltons); lane 2: 2C-Fab-GFP purified from the culture media of a stable HEK293 cell line coexpressing the heavy- and light-chain plasmids. The light chain, heavy chain, and GFP are approximately 30 kDa each. (B) Lane 1: molecular mass markers (kilodaltons); lane 2: total protein extract from choroid plexus tissue labeled with 2C-Fab-GFP. Bands within the predicted ranges for immature to mature 5-HT2C receptors (45–60 kDa), homodimers (90–120 kDa), and homodimers in complex with G protein (170–200 kDa) are present.
Next, the specificity of the 2C-Fab-GFP probe for labeling the native conformation of the 5-HT2C receptor expressed on the plasma membrane of living cells was examined. The 5-HT2A receptor is the protein most closely related to the 5-HT2C receptor in the genome. Therefore, the 5-HT2A receptor was used as a control to demonstrate the specificity of the 2C-Fab-GFP probe. Separate pools of HEK293 cells were independently transfected with plasmids containing cDNA encoding either the rat 5-HT2C receptor with a C-terminal mCherry tag or the 5-HT2A receptor with a C-terminal cyan fluorescent protein tag. Twenty-four hours after transfection, the separate pools of HEK293 cells were mixed together and plated on the same coverslip. The live cells were immunostained with purified 2C-Fab-GFP and examined by confocal microscopy (Fig. 3A). The 2C-Fab-GFP immunostaining colocalized with plasma membrane mCherry-tagged 5-HT2C receptors but was absent in untransfected cells and cells expressing cyan fluorescent protein–tagged 5-HT2A receptors.
Fig. 3.

2C-Fab-GFP specificity. (A) HEK293 cells were divided into two groups and were separately transfected with plasmid containing mCherry-tagged 5-HT2C (5-HT2C/mCherry, red) or cyan fluorescent protein (CFP)–tagged 5-HT2A (5-HT2A/CFP, blue). Following transfection, the cells were mixed and plated on a single coverslip and stained live with 2C-Fab-GFP (green) for 30 minutes at room temperature. White scale bar = 10 µm. The merged image (right panel) shows 2C-Fab-GFP plasma membrane labeling of cells expressing 5-HT2C/mCherry (shown as yellow) and no labeling of cells expressing the most closely related protein in the genome, the 5-HT2A receptor (no overlap of green and blue). (B) Native 5-HT2C receptors endogenously expressed in choroid plexus epithelial cells labeled with 2C-Fab-GFP (green) are shown in the left panel. The same field of cells costained with an antibody to transthyretin (red) is shown in the middle panel. The merged image is shown in the right panel. White scale bar = 50 µm. (C) Optical sections (0.5 µm thick) from a Z-stack (6.0 µm) showing the apical surface, lateral plasma membrane, and basolateral regions of cells costained with 2C-Fab-GFP and transthyretin. The relative position within the Z-stack is noted in the lower-left corner of each image. (D) Choroid plexus epithelial cells labeled with the 2C-Fab-GFP probe in the presence of 1 μM 5-HT. The left panel shows an entire field of cells (white scale bar = 50 µm). The middle panel shows a single cell at higher magnification (white scale bar = 50 µm). The right panel shows the differential interference contrast (DIC) overlay of the cell in the middle panel. The white arrows point to the lateral plasma membrane where there is little fluorescence, in contrast to cells cultured in the absence of 5-HT (Fig. 3C, middle panel).
A blast search of the rat genome was conducted to identify potential off-target labeling sites for the 2C-Fab-GFP probe in choroid epithelial cells. When the results of the search were limited to proteins expressed on the surface of epithelial cells, the protein with the highest homology to the N-terminal domain of the 5-HT2C receptor was aminopeptidase M. This protein displayed 43% sequence homology to residues 31–46 of the 5-HT2C N-terminal domain. However, the corresponding residues of aminopeptidase M (658–673) are located within an intracellular region of the protein and thus would not be accessible to interact with 2C-Fab-GFP when used to label native 5-HT2C receptors on the surface of intact epithelial cells. It is important to note that the anti–5-HT2C monoclonal Fab demonstrates species selectivity for rat over human 5-HT2C receptors (Mancia et al., 2007), even though the rat and human protein sequences share 78% homology within the N-terminal domain of the 5-HT2C receptor (Saltzman et al., 1991). These results indicate that the 2C-Fab-GFP probe is highly specific for labeling native 5-HT2C receptors on the surface of choroid plexus epithelial cells, with minimal to no off-target labeling.
Characterization of Primary Choroid Plexus Epithelial Cell Cultures.
Primary epithelial cell cultures expressing native 5-HT2C receptors were prepared from rat choroid plexus tissue. Of the 14 different 5-HT receptor subtypes identified to date, the 5-HT2C receptor is the only one that is expressed in the choroid plexus. Choroid plexus epithelial cells are the only cells in the brain that express transthyretin, a thyroxin and retinol carrier protein that is also found in the blood and liver (Dickson et al., 1985). Thus, transthyretin is commonly used as an intracellular biomarker to identify epithelial cells isolated from intact choroid plexus tissue.
The primary epithelial cell cultures displayed the characteristic cobblestone pattern and stained positive for 5-HT2C receptors and transthyretin (Fig. 3B). As the cultures mature over a period of 2 weeks, the epithelial cells become polarized (Villalobos et al., 1997). Radioligand binding studies have reported that 5-HT2C receptors are expressed on the apical surface of the epithelial cells (Hartig et al., 1990). This observation was confirmed in our cultures using confocal microscopy. The 2C-Fab-GFP immunostaining was present on the apical surface and lateral plasma membrane but not on the basolateral surface of the cultured epithelial cells (Fig. 3C). The functionality of the 5-HT2C receptors and the specificity of the 2C-Fab-GFP probe are clearly demonstrated by the pronounced redistribution of fluorescence from the plasma membrane to intracellular vesicles following exposure to 5-HT, as shown in Fig. 3D.
The epithelial cells were large and polygonal. The apical surface area was determined using the polygonal measuring tool in the Zeiss Aim 4.2 software. The mean and standard error of the mean from 10 different fields of cells (52 cells in total) was determined to be 3263 ± 387 µm2. This value was used to calculate the level of native 5-HT2C receptors endogenously expressed on the apical surface of the epithelial cells, as described later.
FCS and PCH Analysis of the 2C-Fab-GFP Probe.
FCS is performed using a high numerical aperture objective to focus a laser beam into a small diffraction-limited spot, creating a detection or observation volume on the order of 0.5 × 10−15 liters (reviewed in Elson, 2011; Herrick-Davis and Mazurkiewicz, 2013). As fluorescent molecules pass through the observation volume, they are excited by a laser and give off bursts of photons, which are recorded in real time by a sensitive photon counting detector. FCS recordings were made using a purified solution of 2C-Fab-GFP and on the upper plasma membrane of HEK293 cells expressing fluorescence-tagged 5-HT2C receptors. The resulting fluctuations in fluorescence, produced by the fluorescent molecules entering and leaving the observation volume, are shown in the fluorescence intensity traces in Fig. 4A. Autocorrelation analysis of the fluorescence intensity trace is performed using a nonlinear least-squares fitting routine which graphically represents the autocorrelation function G(τ) on the ordinate and diffusion time on the abscissa. The average dwell time of the fluorescent molecules within the observation volume is calculated from the midpoint of the autocorrelation decay curve.
Fig. 4.

FCS recordings from a solution of 2C-Fab-GFP, the plasma membrane of transfected HEK293 cells expressing the 5-HT2C receptor labeled with 2C-Fab-GFP, and the plasma membrane of transfected HEK293 cells expressing a chimeric 5-HT2C receptor with a C-terminal GFP tag (5-HT2C/GFP). (A) Fluorescence intensity traces for one 10-second observation period. (B) Autocorrelation analysis of the fluorescence intensity traces. The red line represents the autocorrelation of the observed fluorescence signal, and the green line represents the fit to a two-component model: a fast component (on the order of 300 microseconds) related to the photo-physical properties of GFP, and a slower component (on the order of 40 milliseconds) representing the translational diffusion of fluorescence-tagged receptors in the plasma membrane. Dividing the average photon count rate (kilohertz) determined from the fluorescence intensity trace (A) by the number of fluorescent molecules determined from the amplitude of the autocorrelation curve (B) predicts the average molecular brightness of the sample. (C) Photon counting histograms of the corresponding FCS recordings. To generate the histograms, each 10-second fluorescence intensity trace (A) was broken down into one million 10-microsecond intervals or bins (PCH bin time = 10 microseconds). The number of bins is plotted on the y-axis and photon counts on the x-axis. (D) Residuals of the PCH curve fit to a one-component model. The residuals of the curve fit are less than 2 standard deviations and are randomly distributed about zero, indicating a good fit to the selected model.
Autocorrelation curves for the 2C-Fab-GFP in solution and the labeled 5-HT2C receptors are shown in Fig. 4B. Autocorrelation analysis of the 2C-Fab-GFP solution was best fit by a 3D model for Brownian diffusion. The biphasic autocorrelation curves for the fluorescence-tagged 5-HT2C receptors were best fit by a 2D model for the lateral diffusion of proteins within the plasma membrane, with a very fast component related to the photo-physical properties of GFP (on the order of 300 microseconds) and a slower component representing the translational diffusion of receptors within the plasma membrane (on the order of 40–50 milliseconds). Autocorrelation analysis revealed diffusion coefficients on the order of 0.5–0.6 μm2/s for the fluorescence-tagged 5-HT2C receptors in HEK293 cells (Table 1).
TABLE 1.
Diffusion (FCS) and molecular brightness (PCH) of fluorescent tags in solution or in the cytoplasm, and fluorescence-tagged receptors on the plasma membrane
HEK293 cells expressing a known monomeric receptor (CD-86) were used as a control to determine the molecular brightness of a monomer (CD-86/GFP). The molecular brightness of a dimer was determined using a tandem GFP tag (CD-86/GFP-GFP). Diffusion reported in milliseconds represents the average dwell time of the receptor in the observation volume. Diffusion coefficients (micrometers squared per second) were calculated using a 2D model for the lateral diffusion of receptors within the plasma membrane. PCH molecular brightness values are reported as CPSM. Reduced χ2 values are reported for the PCH data fit to a one-component model for a single, homogeneous population of monomers or homodimers. Data represent the mean ± S.E.M. for the number of cells examined (N).
| Fluorescent Tags and Labeled Receptors |
FCS Diffusion |
PCH Brightness |
Reduced |
N |
|
|---|---|---|---|---|---|
| ms | μm2/s | CPSM | χ2 | ||
| 2C-Fab-GFP (S) | —a | — | 8795 ± 145 | 1.14 ± 0.03 | 35 |
| mGFP in HEK293 cells (C) | — | — | 9075 ± 287 | 1.07 ± 0.02 | 25 |
| CD-86/GFP in HEK293 cells (PM) | 37.0 ± 2.0 | 0.61 ± 0.03 | 10,333 ± 364 | 1.09 ± 0.07 | 12 |
| CD-86/GFP-GFP in HEK293 cells (PM) | 37.4 ± 2.2 | 0.60 ± 0.04 | 19,691 ± 530 | 1.33 ± 0.10 | 10 |
| 5-HT2C/GFP in HEK293 cells (PM) | 40.4 ± 1.9 | 0.56 ± 0.03 | 18,330 ± 674 | 1.19 ± 0.07 | 15 |
| 2C-Fab-GFP–labeled 5-HT2C receptors in HEK293 cells (PM) | 48.7 ± 2.2 | 0.46 ± 0.02 | 18,783 ± 384 | 1.51 ± 0.12 | 20 |
| 2C-Fab-GFP–labeled 5-HT2C receptors in choroid plexus epithelial cells (PM) | 42.6 ± 1.8 | 0.53 ± 0.02 | 18,466 ± 361 | 1.55 ± 0.22 | 10 |
Cytoplasm, C; plasma membrane, PM; solution, S.
Not calculated.
The amplitude (y-intercept) of the autocorrelation curve is inversely related to the number of fluorescent molecules present in the observation volume. For illustration purposes, autocorrelation curves from samples with similar numbers of fluorescent molecules or fluorescent complexes are shown in Fig. 4B. Note that, although the amplitude of the autocorrelation curve is the same for all three samples, the corresponding fluorescence intensity traces (Fig. 4A) show average photon count rates of approximately 65 kHz for 2C-Fab-GFP and 135 kHz for 2C-Fab-GFP–labeled 5-HT2C receptors and 5-HT2C receptors with a C-terminal GFP tag (5-HT2C/GFP). These results indicate that the fluorescence-tagged 5-HT2C receptors produce approximately twice as many photon counts and thus are approximately twice as bright as the 2C-Fab-GFP in solution. The amplitude of the autocorrelation curve for the 2C-Fab-GFP–labeled receptors expressed in HEK293 cells (Fig. 4B) was similar to that of native 5-HT2C receptors in choroid epithelial cells (Fig. 5B), indicating that the transfected HEK293 cells were expressing receptors within the native physiologic range.
Fig. 5.

FCS recordings from the plasma membrane of choroid plexus epithelial cells labeled with 2C-Fab-GFP, and the plasma membrane of transfected HEK293 cells expressing monomeric CD-86 with one GFP tag (CD-86–GFP) or two GFP tags (CD-86–GFP-GFP). (A) Fluorescence intensity traces for one 10-second observation period. (B) Autocorrelation analysis of the fluorescence intensity traces. The red line represents the autocorrelation of the observed fluorescence signal, and the green line represents the fit to a two-component model: a fast component (on the order of 300 microseconds) related to the photo-physical properties of GFP, and a slower component (on the order of 40 milliseconds) representing the translational diffusion of fluorescence-tagged receptors in the plasma membrane. Dividing the average photon count rate (kilohertz) determined from the fluorescence intensity trace (A) by the number of fluorescent molecules determined from the amplitude of the autocorrelation curve (B) predicts the average molecular brightness of the sample. (C) Photon counting histograms of the corresponding FCS recordings. To generate the histograms, each 10-second fluorescence intensity trace (A) was broken down into one million 10-microsecond intervals or bins (PCH bin time = 10 microseconds). The number of bins is plotted on the y-axis and photon counts on the x-axis. (D) Residuals of the PCH curve fit to a one-component model. The residuals of the curve fit are less than 2 standard deviations and are randomly distributed about zero, indicating a good fit to the selected model.
Fluorescence fluctuation data recorded during an FCS experiment can be used to generate a PCH, which provides quantitative information about the number of fluorescent molecules and the number of photon counts produced by individual fluorescent molecules (Chen et al., 1999). The number of photon counts per molecule provides an estimate of the molecular brightness. Since the molecular brightness is proportional to the number of fluorescent molecules traveling together within a protein complex, the oligomer size of a fluorescence-tagged protein can be determined by comparing molecular brightness values with established monomeric controls containing a single fluorescent tag or with two fluorescent tags (Chen et al., 1999). Thus, a GPCR homodimer with two fluorescent tags would be twice as bright as a monomer, a tetramer with four fluorescent tags would be 4 times as bright, and so forth.
Photon counting histograms generated from the FCS data are presented in Fig. 4C. To generate a histogram, each 10-second fluorescence intensity trace (shown in Fig. 4A) was broken down into one million 10-microsecond intervals or bins (PCH bin time = 10 microseconds). Histograms were constructed in which the number of 10-microsecond bins was plotted on the y-axis and photon counts on the x-axis. The shape of the histogram is a function of the number of fluorescent molecules and their molecular brightness. For the 2C-Fab-GFP–labeled receptors and 5-HT2C/GFP, the histograms show an average of 1.1 photon counts per 10-microsecond bin time, which is equivalent to 110,000 counts per second. Dividing by the average number of molecules, calculated from the amplitude of the autocorrelation curves in Fig. 4B (using eq. 4 with a 3D PCH model where N = 6), yields an average molecular brightness of 18,333 CPSM for the fluorescence-tagged receptors. The PCH for the 2C-Fab-GFP solution registered half the number of photon counts as the PCH for the fluorescence-tagged receptors, indicating that the solution was half as bright. Molecular brightness values for the 2C-Fab-GFP solution and for monomeric GFP expressed in the cytosol of HEK293 cells were on the order of 9000 CPSM (Table 1), roughly half the brightness of the fluorescence-tagged 5-HT2C receptors.
The PCHs shown in Fig. 4C were generated by fitting the data to a one-component model for a single population of fluorescent species, monomers for the 2C-Fab-GFP solution, and homodimers for the fluorescence-tagged receptors. The residuals of the PCH curve fit (Fig. 4D) are an indicator of how well the data fit the selected model. When the residuals are systematic with large variations in amplitude (greater than 2 standard deviations) and reduced χ2 values greater than 3.0, the selected model is a poor fit (Müller et al., 2000). However, when the residuals of the curve fit are randomly distributed about zero with small variations in amplitude (as in Fig. 4D) and reduced χ2 values close to unity (as in Table 1), the data are a good fit for the selected model (Müller et al., 2000). The residuals of the curve fit indicate that the PCH data are adequately described by a one-component model for homodimers, without monomers or tetramers, indicating that the 2C-Fab-GFP–labeled 5-HT2C receptors expressed in HEK293 cells are homodimers.
FCS and PCH Analysis of Native 5-HT2C Receptors in Choroid Plexus Epithelial Cells.
Native 5-HT2C receptors expressed in primary choroid plexus epithelial cells were labeled with 2C-Fab-GFP. The apical plasma membrane was positioned within the laser-illuminated observation volume by focusing upward from the middle of the cell to the top, while simultaneously monitoring the photon counts per molecule. Optimal positioning of the plasma membrane within the center of the observation volume is critical for accurate molecular brightness analysis because the observation volume is not illuminated homogeneously and the detected photon counts decrease as fluorescent molecules travel away from the center of the observation volume. As the freely diffusing, fluorescence-tagged receptors passed through the observation volume, they were excited by the laser, and the fluctuations in fluorescence intensity were recorded (Fig. 5A). Similar to 5-HT2C receptors transiently expressed in HEK293 cells, the autocorrelation curve for native 5-HT2C receptors in primary epithelial cells was best fit by a 2D model with two components, a fast component related to the photo-physical properties of the GFP tag and a slower component representing the translational diffusion of receptors within the plasma membrane. Autocorrelation analysis revealed diffusion coefficients on the order of 0.5 μm2/s for native 5-HT2C receptors in choroid plexus epithelial cells.
The amplitude of the FCS autocorrelation curve for native 5-HT2C receptors in primary epithelial cells presented in Fig. 5B indicates an average of nine fluorescent molecules within the observation volume (calculated using eq. 4 with a 2D model). A plasma membrane observation area of 0.28 µm2 (calculated from the radius of the observation volume as described in Materials and Methods) yields an approximate expression level of 32 receptors/µm2 of plasma membrane. An average apical surface area of 3263 µm2 predicts an expression level on the order of 105 5-HT2C receptors per choroid epithelial cell.
PCH analysis of native 5-HT2C receptors labeled with 2C-Fab-GFP (Fig. 5C) produced histograms with similar numbers of photon counts and molecular brightness values as observed for 5-HT2C receptors expressed in HEK293 cells (Table 1). A known monomeric receptor, CD-86, was used as a control for decoding the monomer/oligomer status of native 5-HT2C receptors in choroid epithelial cells. FCS and PCH analyses for CD-86 with a C-terminal GFP tag (CD-86–GFP) and tandem GFP tag (CD-86–GFP-GFP) are shown in Fig. 5. The fluorescence intensity trace for CD-86–GFP displayed a photon count rate that was approximately half that of CD-86–GFP-GFP (Fig. 5A), even though the amplitude of the autocorrelation curve was similar for both samples (Fig. 5B). These results predict that the CD-86 receptor with a single GFP tag is half as bright as CD-86 with two GFP tags.
The fluorescence intensity traces for native 5-HT2C receptors labeled with 2C-Fab-GFP and CD-86–GFP-GFP displayed similar photon count rates (Fig. 5A), and the corresponding histograms (Fig. 5C) predict similar molecular brightness values. PCH analysis produced a molecular brightness value for monomeric CD-86–GFP (10,333 CPSM) that was similar to the monomeric 2C-Fab-GFP in solution (8795 CPSM), but was approximately half the brightness of CD-86–GFP-GFP (19,691 CPSM) and native 5-HT2C receptors labeled with 2C-Fab-GFP (18,466 CPSM), as reported in Table 1. When fit to a one-component model for homodimers, without monomers or tetramers, the residuals of the PCH (fit deviation shown in Fig. 5D) were less than 2 standard deviations, were randomly distributed around zero, and yielded reduced χ2 values close to unity (Table 1), indicating that the PCH data are adequately described by the selected model. These results demonstrate the homodimeric nature of native 5-HT2C receptors in choroid plexus epithelial cells.
Signaling Properties of 5-HT2C Receptor Homodimers.
Teitler and colleagues used a pharmacological approach for identifying and investigating the signaling properties of GPCR homodimers (Smith et al., 2011). In their study, antagonists that bind in a wash-resistant, pseudo-irreversible manner to one or both protomers within the homodimer were identified and used to provide the first pharmacological evidence for the presence of 5-HT7 homodimers (Smith et al., 2011). We rationalized that if agonists with similar binding characteristics were available, it would be possible to investigate the signaling properties of individual protomers within the homodimer. Such studies would have the distinct advantage of allowing the investigation of the signaling properties of wild-type GPCR instead of requiring the use of mutant receptors with altered binding properties or signaling defective mutant receptors, as used in previous studies of this nature.
Ergot derivatives are known agonists with moderate to high affinity for 5-HT2A,2B,2C receptors (Newman-Tancredi et al., 2002; Knight et al., 2004). Recently, the crystal structures of the 5-HT1B and 5-HT2B receptors were solved in the presence of ergotamine (Wacker et al., 2013), indicating ergotamine forms a relatively stable complex with the receptor. These observations led us to investigate and subsequently identify ergot derivatives that bind in a wash-resistant manner to one or both protomers of the 5-HT2C receptor homodimer. The ergots were used to investigate the signaling properties of the homodimer, as described later.
Dose-response curves for the stimulation of IP production in HEK293 cells expressing wild-type 5-HT2C receptors are shown in Fig. 6. Ergotamine (Erg), 5-HT, and cabergoline (Cab) stimulated IP production in a dose-dependent manner, with EC50 values of 1.3, 2.3, and 527 nM, respectively, similar to previously published results (Newman-Tancredi et al., 2002; Knight et al., 2004). The data are expressed as the percentage of maximal IP production achieved with that drug. The efficacies of Erg and Cab with respect to 5-HT were 0.90 and 0.85, respectively.
Fig. 6.

Representative dose-response curves for stimulation of IP measured in HEK293 cells expressing 5-HT2C receptors. IP production was measured in the presence of increasing concentrations of each drug as indicated. Basal IP levels determined in cells treated with serum-free culture media were subtracted out. Data are expressed as the percentage of maximal IP production achievable with each drug. In comparison with 5-HT, the intrinsic efficacies of Erg and Cab were 0.90 and 0.85, respectively.
Next, the signaling and binding properties before and after drug washout were investigated (Fig. 7). For these experiments, each drug was tested at a single concentration 20-fold over the EC50 value of that drug. In the initial experiment shown in Fig. 7A, the drugs were added with the lithium chloride at the beginning of the IP assay, and IP production was measured. As expected, similar maximal levels of IP production were achieved with each drug. In a second set of experiments shown in Fig. 7, B and C, the cells were pretreated with drugs for 60 minutes to ensure equilibration, then washed four times over a 30-minute period (drug washout) to remove drug that binds in a competitive, reversible manner. At the start of the IP assay, the cells were only treated with serum-free culture media containing lithium chloride (to prevent IP breakdown). No drugs were added during the IP assay. As expected, pretreatment with 5-HT followed by drug washout resulted in minimal stimulation of IP production (Fig. 7B), indicating that 5-HT binds in a competitive, reversible manner to 5-HT2C receptors. In contrast, approximately half-maximal IP production remained following a 60-minute pretreatment with Cab and subsequent washout, and near-maximal IP production was observed following Erg pretreatment and drug washout (Fig. 7B).
Fig. 7.

IP production and radioligand binding in HEK293 cells expressing 5-HT2C receptors. (A) IP production was measured for 60 minutes in the presence of serum-free culture media (basal), 50 nM 5-HT, 10 μM Cab, or 25 nM Erg. The concentration of each drug was 20-fold over its EC50 value. Basal levels of IP production were typically 14% of maximal stimulation with 5-HT. Data represent the mean ± S.E.M. from six experiments. (B) Cells were pretreated in the presence of serum-free culture media (basal), 50 nM 5-HT, 10 μM Cab, or 25 nM Erg for 60 minutes. Following drug washout, IP production was measured in serum-free culture media without drugs for 60 minutes. Data are expressed as the percentage of maximal IP production achieved with each drug, with basal IP levels subtracted out. Data represent the mean ± S.E.M. from six experiments. *P < 0.01 versus 5-HT. (C) Whole-cell radioligand binding assay. Cells were pretreated for 60 minutes with 50 nM 5-HT, 10 μM Cab, or 25 nM Erg. Following drug washout, cells were labeled with 2.5 nM [3H]mesulergine in the absence and presence of 1 μM 5-HT to define specific binding to cell surface 5-HT2C receptors. Data are expressed as the percent inhibition of specific binding. Data represent the mean ± S.E.M. from four experiments. *P < 0.01 versus 5-HT.
Whole-cell radioligand binding studies were performed in parallel with the IP signaling experiments to monitor drug interactions with cell surface 5-HT2C receptors. Intact cells expressing 5-HT2C receptors were incubated with a saturating concentration of [3H]mesulergine in the absence and presence of 1 μM 5-HT to define specific binding to cell surface receptors (Fig. 7C). 5-HT pretreatment followed by drug washout resulted in minimal inhibition of [3H]mesulergine binding, demonstrating that 5-HT binds in a competitive, reversible manner to cell surface 5-HT2C receptors. However, pretreatment and washout of Cab and Erg inhibited specific radioligand binding by 45 and 81%, respectively (Fig. 7C). The inhibition of radioligand binding following drug washout indicates that Cab and Erg bind in a wash-resistant manner with slow dissociation kinetics or in a pseudo-irreversible manner to cell surface 5-HT2C receptors. The magnitude of the observed effect was identical to the half-maximal and near-maximal residual signaling properties displayed by Cab and Erg, respectively, in the IP assay.
The time course for establishing the wash-resistant signaling of Erg and Cab was investigated (Fig. 8). Cells were pretreated with drug for various times, ranging from 5 to 60 minutes, followed by drug washout and subsequent IP assay in the absence of drug. Establishment of the Cab wash-resistant IP signaling occurred gradually over the 60-minute test period, reaching maximal levels within 30 minutes of drug treatment. In contrast, the Erg wash-resistant IP signaling was rapidly established and reached near-maximal levels within 5 minutes of drug treatment. A gradual desensitization of the maximal IP response (20%) was observed over the 60-minute pretreatment period with Erg. Attempts to investigate the duration of the wash-resistant IP signaling were hampered by a desensitization of the IP response, due to prolonged receptor stimulation by the continual presence of the wash-resistant drug. Pretreatment with drug for 60 minutes followed by drug washout and a subsequent 90-minute delay prior to the IP assay resulted in a decrease in the wash-resistant Cab signaling from 45 to 16% and a decrease in Erg signaling from 80 to 30% of maximal levels. However, the magnitude of the residual Erg signaling was still twice that of Cab.
Fig. 8.

Time course for the onset of wash-resistant signaling. HEK293 cells expressing 5-HT2C receptors were treated with 25 nM Erg or 10 μM Cab for the indicated times, followed by drug washout. IP production was measured in serum-free culture media without drugs for 60 minutes. Data are expressed as the percentage of maximal IP production achievable with that drug, with basal IP levels subtracted out. Data represent the mean ± S.E.M. from three experiments.
In a subsequent set of experiments, the ability to modulate IP signaling following drug washout was investigated (Fig. 9). In these experiments, cells were pretreated with drug for 60 minutes followed by drug washout, and then the cells were challenged during the IP assay with 5-HT or a 5-HT2C receptor antagonist (mianserin). Pretreatment with 5-HT followed by washout resulted in minimal stimulation of IP production. However, subsequent challenge with 5-HT robustly stimulated IP production to near-maximal levels (80%). An average 20% desensitization of the maximal response was routinely observed following a 1-hour pretreatment with 5-HT (as noted for Erg in Fig. 8). Pretreatment with Cab, followed by drug washout, resulted in half-maximal IP stimulation. Subsequent exposure of the Cab-pretreated cells to 5-HT restored the IP signaling to maximal levels (Fig. 9).
Fig. 9.

Cells were pretreated with 50 nM 5-HT, 10 μM Cab, or 25 nM Erg for 60 minutes. Following drug washout, IP production was measured in the absence (−) or presence of 50 nM 5-HT or 1 μM mianserin (Mian) for 60 minutes. Data are expressed as the percentage of maximal IP production achievable with each drug, with basal IP levels subtracted out. Data represent the mean ± S.E.M. from four to six experiments. *P < 0.01 versus no drug (−).
Mianserin, a known competitive antagonist of the orthosteric binding site, was used to determine the physical arrangement of the Cab-occupied protomers responsible for generating the half-maximal IP signaling following Cab washout (Fig. 9). For mianserin to have an effect on the wash-resistant, Cab-stimulated IP production, mianserin must bind to an unoccupied receptor that is capable of communicating with a receptor that is occupied by Cab and stimulates an IP response. For example, if the washout of Cab was random, it would create a situation in which 50% of the homodimers would have one protomer occupied by Cab, 25% of the homodimers would be fully occupied by Cab, and 25% would be unoccupied. Mianserin could only influence the IP signal generated by the 50% that have one protomer occupied by Cab and the other protomer unoccupied. Mianserin would have no effect on the signal generated by the 25% that had both protomers occupied by Cab. In this case, mianserin would produce a partial inhibition of the IP response. For mianserin to produce a complete blockade of the IP response, as observed in Fig. 9, the 5-HT2C receptors participating in generating the IP signal must be present as homodimers (consistent with the results of the FCS studies) in which one protomer is occupied by Cab and the other protomer is unoccupied and available for mianserin to bind and shut off IP signaling. The observed results are consistent with a model in which Cab binds in a wash-resistant manner to one protomer of the homodimer, and that occupancy of one protomer by Cab produces a half-maximal response.
In contrast to Cab, the wash-resistant IP signaling established by pretreatment with Erg was not enhanced by exposure to 5-HT, but was similar to maximal IP levels following 5-HT stimulation of the 5-HT– and Cab-pretreated cells (Fig. 9). If the maximal IP signal produced by Erg resulted from occupancy of a single protomer within the 5-HT2C homodimer, then mianserin would be expected to block the response. However, mianserin had no effect on the wash-resistant IP signal produced by Erg. These results suggest that Erg binds in a wash-resistant manner to both protomers within the homodimer to produce a maximal response.
Discussion
Although many studies report the identification of homodimers and higher-order oligomers of GPCR expressed in recombinant cell systems (reviewed in Herrick-Davis, 2013; Milligan, 2013; Ferré et al., 2014), few studies have examined the properties of native GPCRs. This is due to the technical challenges associated with developing selective probes for labeling native receptors along with sensitive methods capable of directly monitoring native protein-protein interactions in living cells. In the present study, the monomer/oligomer status of a native GPCR endogenously expressed in its natural cellular environment was determined using a GFP-tagged Fab derived from a monoclonal antibody. The 2C-Fab-GFP probe selectively recognized the native conformation of the 5-HT2C receptor and demonstrated the polarized distribution of native 5-HT2C receptors on the apical surface of choroid plexus epithelial cells.
FCS with PCH was used to monitor native 5-HT2C receptor interactions. FCS is more sensitive than standard proximity-based resonance energy transfer techniques and diffraction-limited imaging techniques. In FCS, the combined use of confocal microscopy with avalanche photodiodes or gallium arsenide photon counting detectors provides a sensitive method for determining the number of photon counts arising from individual fluorescent molecules as a measure of their molecular brightness (reviewed in Elson, 2011). FCS recordings revealed that the 2C-Fab-GFP probe displayed similar spectral properties and molecular brightness values as established monomeric GFP control samples. In addition, FCS with PCH revealed similar molecular brightness values for 2C-Fab-GFP–labeled 5-HT2C receptors as for chimeric 5-HT2C receptors with a C-terminal GFP tag, previously demonstrated to form homodimers in HEK293 cells (Herrick-Davis et al., 2012). These results demonstrate that only one 2C-Fab-GFP probe binds per 5-HT2C receptor, and that the 2C-Fab-GFP probe is capable of binding to both protomers within a homodimeric receptor complex. Molecular brightness values for 2C-Fab-GFP–labeled native 5-HT2C receptors on the apical surface of choroid epithelial cells were equivalent to the predicted molecular brightness of a homodimer. Residuals of the PCH curve fit and reduced χ2 analysis indicate that the data fit a one-component model for homodimers.
Several studies using total internal reflection fluorescence (TIRF) have concluded that GPCRs exist in a monomer↔dimer equilibrium, with receptors associating and dissociating with one another on a time scale of seconds (Calebiro et al., 2013; Kasai and Kusumi, 2014; Teichmann et al., 2014). TIRF is a diffraction-limited technique, with a limit of resolution on the order of 200 nm. To achieve single-molecule sensitivity, TIRF requires extremely low receptor expression levels (0.15–0.45 receptors/µm2). The reported expression level of 20 receptors/µm2 for native β2-adrenergic receptors in alveolar epithelial cells (Hegener et al., 2004) is similar to the 32 receptors/µm2 observed for native 5-HT2C receptors in choroid epithelial cells in the present study. In TIRF studies of β1-adrenergic and β2-adrenergic receptors, the observed monomer↔dimer equilibrium was most pronounced at expression levels between 0.15 and 0.35 receptors/µm2, with a decline in the monomer fraction and shift toward a more predominant homodimer fraction at 0.45 receptors/µm2 (Calebiro et al., 2013). These results predict a predominantly homodimer fraction for β-adrenergic receptors at higher, native, physiologic expression levels. This is consistent with the results of the present study and the results of our previous FCS studies demonstrating that biogenic amine receptors are predominantly homodimers when expressed within their normal physiologic range (Herrick-Davis et al., 2013).
The functional significance of GPCR dimer/oligomer formation in vivo is largely unknown and widely debated. Speculations about the functional significance range from a requirement for trafficking from the endoplasmic reticulum (ER) to the plasma membrane (Salahpour et al., 2004; Herrick-Davis et al., 2006; Milligan, 2013) and homodimers representing the minimal functional signaling unit (Jastrzebska et al., 2013a), to heterodimerization generating novel signaling complexes (Ferré et al., 2014; Fujita et al., 2014). Studies with CCR5 receptors indicate that chaperone protein binding is involved in the preassembly of homodimers along with G proteins in the ER (Kuang et al., 2012). More recently, it was proposed that homodimer formation within the ER and Golgi may play an essential role in the apical sorting of proteins in polarized cells. FCS and PCH analysis of the neurotropin p75 receptor demonstrated the requirement of dimerization within the trans-Golgi network for targeting to the apical surface of polarized epithelial cells (Youker et al., 2013). It remains to be determined if dimerization plays a role in the apical sorting of native 5-HT2C receptors in choroid plexus epithelial cells.
Whether agonist binding to one or both protomers in a dimeric complex is necessary or sufficient for G protein activation is another area of uncertainty. The majority of studies performed to date have used recombinant cell lines expressing mutant receptors with altered binding and/or signaling properties. These studies concluded that agonist binding to one protomer within a dimer can elicit a response, and in general, the second protomer is not silent but plays a role in the signaling process in an asymmetric manner (Han et al., 2009; Pin et al., 2009; Maurice et al., 2011; Pellissier et al., 2011; Jastrzebska et al., 2013a). Additional studies using methods capable of monitoring the signaling properties of wild-type and native GPCR homodimers are necessary to substantiate these findings.
Teitler and colleagues have developed a simple and elegant pharmacological approach for investigating the properties of wild-type and native GPCR homodimers using antagonists that bind in a wash-resistant manner (Smith et al., 2011; Teitler and Klein, 2012). In the present study, we used a similar method using agonists that bind in a wash-resistant manner to investigate the signaling properties of wild-type 5-HT2C receptor homodimers. Ergot derivatives were found to display wash-resistant binding to one protomer (Cab) or both protomers (Erg) within the homodimer. The results obtained with Cab and Erg provide the first pharmacological evidence for the presence and functional significance of 5-HT2C homodimers. If 5-HT2C receptors exist as monomers, then mianserin could not abolish the wash-resistant, Cab-stimulated IP production. The results with Cab and Erg are consistent with the hypothesis that 5-HT2C receptors function as homodimers.
5-HT, Cab, and Erg displayed little to no cooperativity in their stimulation of IP production as evidenced by the monophasic concentration-response curves with Hill coefficients close to unity. Also, the half-maximal stimulation of IP production following Cab washout and the maximal stimulation produced by Erg following occupancy of both protomers are suggestive of a balanced signaling between protomers, with each protomer contributing equally to the overall response. However, several studies have reported allosteric communication between protomers of the homodimer, with one protomer contributing a greater response than the other (reviewed in Ferré et al., 2014). The degree of positive or negative cooperativity observed following ligand binding may be influenced by the resting state of the receptor. Changes in receptor conformation upon agonist binding would be predicted to be greater for receptors that are in an inactive conformation at rest than for receptors capable of reaching an active conformation prior to agonist binding. Thus, the conformational change across the dimer interface following agonist binding to one protomer of a constitutively active homodimer would be less. This hypothesis is supported by a recent study demonstrating a direct link between cooperativity and basal activity for glycoprotein hormone receptors (Zoenen et al., 2012). The receptors displaying the highest levels of constitutive activity lost nearly all of their cooperative allosteric regulation. The 5-HT2C receptor is known to display constitutive activity (Herrick-Davis et al., 1999). This may contribute to the more balanced signaling between protomers of 5-HT2C receptor homodimers observed in the present study.
In conclusion, the results of the present study provide direct biophysical evidence for the homodimeric structure of a native GPCR endogenously expressed in its natural cellular environment. FCS with PCH revealed homodimers of native 5-HT2C receptors on the apical surface of choroid plexus epithelial cells. The pharmacological studies performed in HEK293 cells highlight the signaling capabilities of the individual protomers and provide supporting evidence that 5-HT2C receptors function as homodimers. Whereas binding to one protomer of the 5-HT2C receptor homodimer stimulates G protein activation and IP production, binding to both protomers is required for maximal activation. Using a direct biophysical technique, such as FCS, combined with a pharmacological approach provides a powerful combination for evaluating the stoichiometry and signaling properties of native GPCRs. Studies using pharmacological approaches capable of predicting the oligomer number, as described in Teitler and Klein (2012), will be essential for determining the functional relevance of GPCR dimerization in vivo.
Abbreviations
- Cab
cabergoline
- CPSM
counts per second per molecule
- 2D
two-dimensional
- 3D
three-dimensional
- DMEM
Dulbecco’s minimal essential medium
- ER
endoplasmic reticulum
- Erg
ergotamine
- FBS
fetal bovine serum
- Fab
fragment antigen binding
- FCS
fluorescence correlation spectroscopy
- GFP
green fluorescent protein
- GPCR
G protein–coupled receptor
- HEK293
human embryonic kidney 293
- IP
inositol phosphate
- mAb
monoclonal antibody
- PBS
phosphate-buffered saline
- PCH
photon counting histogram
- TIRF
total internal reflection fluorescence
Authorship Contributions
Participated in research design: Herrick-Davis, Cowan, Grinde, Lindsley, Mancia, Mazurkiewicz, Teitler.
Conducted experiments: Herrick-Davis, Grinde, Lindsley, Mazurkiewicz.
Contributed new reagents or analytic tools: Cowan, Mancia.
Performed data analysis: Herrick-Davis, Grinde, Mazurkiewicz, Teitler.
Wrote or contributed to the writing of the manuscript: Herrick-Davis, Cowan, Lindsley, Mancia, Teitler.
Footnotes
Funding for this work was provided by a grant from the National Institutes of Health National Institute of Mental Health [R21-MH086796] awarded to K.H.-D.
References
- Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, et al. (2010) Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol 6:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assur Z, Schieren I, Hendrickson WA, Mancia F. (2007) Two-color selection for amplified co-production of proteins in mammalian cells. Protein Expr Purif 55:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backstrom JR, Westphal RS, Canton H, Sanders-Bush E. (1995) Identification of rat serotonin 5-HT2C receptors as glycoproteins containing N-linked oligosaccharides. Brain Res Mol Brain Res 33:311–318. [DOI] [PubMed] [Google Scholar]
- Banères JL, Parello J. (2003) Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J Mol Biol 329:815–829. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. (2007) Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J Biol Chem 282:14875–14881. [DOI] [PubMed] [Google Scholar]
- Berg KA, Rowan MP, Gupta A, Sanchez TA, Silva M, Gomes I, McGuire BA, Portoghese PS, Hargreaves KM, Devi LA, et al. (2012) Allosteric interactions between δ and κ opioid receptors in peripheral sensory neurons. Mol Pharmacol 81:264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Dawson RM, Downes CP, Heslop JP, Irvine RF. (1983) Changes in the levels of inositol phosphates after agonist-dependent hydrolysis of membrane phosphoinositides. Biochem J 212:473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briddon SJ, Gandía J, Amaral OB, Ferré S, Lluís C, Franco R, Hill SJ, Ciruela F. (2008) Plasma membrane diffusion of G protein-coupled receptor oligomers. Biochim Biophys Acta 1783:2262–2268. [DOI] [PubMed] [Google Scholar]
- Calebiro D, Rieken F, Wagner J, Sungkaworn T, Zabel U, Borzi A, Cocucci E, Zürn A, Lohse MJ. (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc Natl Acad Sci USA 110:743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Müller JD, So PT, Gratton E. (1999) The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys J 77:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RW, Jinadasa T, Brown CM. (2011) Measuring and interpreting point spread functions to determine confocal microscope resolution and ensure quality control. Nat Protoc 6:1929–1941. [DOI] [PubMed] [Google Scholar]
- Corriden R, Kilpatrick LE, Kellam B, Briddon SJ, Hill SJ. (2014) Kinetic analysis of antagonist-occupied adenosine-A3 receptors within membrane microdomains of individual cells provides evidence of receptor dimerization and allosterism. FASEB J 28:4211–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson PW, Howlett GJ, Schreiber G. (1985) Rat transthyretin (prealbumin). Molecular cloning, nucleotide sequence, and gene expression in liver and brain. J Biol Chem 260:8214–8219. [PubMed] [Google Scholar]
- Elson EL. (2011) Fluorescence correlation spectroscopy: past, present, future. Biophys J 101:2855–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterle TM, Sanders-Bush E. (1992) Serotonin agonists increase transferrin levels via activation of 5-HT1C receptors in choroid plexus epithelium. J Neurosci 12:4775–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. (2014) G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 66:413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita W, Gomes I, Devi LA. (2015) Heteromers of μ-δ opioid receptors: new pharmacology and novel therapeutic possibilities. Br J Pharmacol 172:375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly S, Chattopadhyay A. (2010) Cholesterol depletion mimics the effect of cytoskeletal destabilization on membrane dynamics of the serotonin1A receptor: A zFCS study. Biophys J 99:1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González S, Rangel-Barajas C, Peper M, Lorenzo R, Moreno E, Ciruela F, Borycz J, Ortiz J, Lluís C, Franco R, et al. (2012) Dopamine D4 receptor, but not the ADHD-associated D4.7 variant, forms functional heteromers with the dopamine D2S receptor in the brain. Mol Psychiatry 17:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. (2009) Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol 5:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartig PR, Hoffman BJ, Kaufman MJ, Hirata F. (1990) The 5-HT1C receptor. Ann N Y Acad Sci 600:149–166, discussion 166–167. [DOI] [PubMed] [Google Scholar]
- Hasbi A, O’Dowd BF, George SR. (2011) Dopamine D1-D2 receptor heteromer signaling pathway in the brain: emerging physiological relevance. Mol Brain 4:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegener O, Prenner L, Runkel F, Baader SL, Kappler J, Häberlein H. (2004) Dynamics of beta2-adrenergic receptor-ligand complexes on living cells. Biochemistry 43:6190–6199. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K. (2013) Functional significance of serotonin receptor dimerization. Exp Brain Res 230:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Cowan A, Mazurkiewicz JE. (2013) Fluorescence correlation spectroscopy analysis of serotonin, adrenergic, muscarinic, and dopamine receptor dimerization: the oligomer number puzzle. Mol Pharmacol 84:630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Lindsley T, Cowan A, Mazurkiewicz JE. (2012) Oligomer size of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor revealed by fluorescence correlation spectroscopy with photon counting histogram analysis: evidence for homodimers without monomers or tetramers. J Biol Chem 287:23604–23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Mazurkiewicz JE. (2004) Biochemical and biophysical characterization of serotonin 5-HT2C receptor homodimers on the plasma membrane of living cells. Biochemistry 43:13963–13971. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Niswender CM. (1999) Serotonin 5-HT2C receptor RNA editing alters receptor basal activity: implications for serotonergic signal transduction. J Neurochem 73:1711–1717. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Mazurkiewicz JE. (2013) Fluorescence correlation spectroscopy and photon-counting histogram analysis of receptor-receptor interactions. Methods Cell Biol 117:181–196. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Weaver BA, Grinde E, Mazurkiewicz JE. (2006) Serotonin 5-HT2C receptor homodimer biogenesis in the endoplasmic reticulum: real-time visualization with confocal fluorescence resonance energy transfer. J Biol Chem 281:27109–27116. [DOI] [PubMed] [Google Scholar]
- Jastrzebska B, Orban T, Golczak M, Engel A, Palczewski K. (2013a) Asymmetry of the rhodopsin dimer in complex with transducin. FASEB J 27:1572–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastrzebska B, Ringler P, Palczewski K, Engel A. (2013b) The rhodopsin-transducin complex houses two distinct rhodopsin molecules. J Struct Biol 182:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai RS, Kusumi A. (2014) Single-molecule imaging revealed dynamic GPCR dimerization. Curr Opin Cell Biol 27:78–86. [DOI] [PubMed] [Google Scholar]
- Knight AR, Misra A, Quirk K, Benwell K, Revell D, Kennett G, Bickerdike M. (2004) Pharmacological characterisation of the agonist radioligand binding site of 5-HT(2A), 5-HT(2B) and 5-HT(2C) receptors. Naunyn Schmiedebergs Arch Pharmacol 370:114–123. [DOI] [PubMed] [Google Scholar]
- Kuang YQ, Charette N, Frazer J, Holland PJ, Attwood KM, Dellaire G, Dupré DJ. (2012) Dopamine receptor-interacting protein 78 acts as a molecular chaperone for CCR5 chemokine receptor signaling complex organization. PLoS ONE 7:e40522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. (2003) Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem 278:21655–21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malengo G, Andolfo A, Sidenius N, Gratton E, Zamai M, Caiolfa VR. (2008) Fluorescence correlation spectroscopy and photon counting histogram on membrane proteins: functional dynamics of the glycosylphosphatidylinositol-anchored urokinase plasminogen activator receptor. J Biomed Opt 13:031215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia F, Brenner-Morton S, Siegel R, Assur Z, Sun Y, Schieren I, Mendelsohn M, Axel R, Hendrickson WA. (2007) Production and characterization of monoclonal antibodies sensitive to conformation in the 5HT2c serotonin receptor. Proc Natl Acad Sci USA 104:4303–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice P, Kamal M, Jockers R. (2011) Asymmetry of GPCR oligomers supports their functional relevance. Trends Pharmacol Sci 32:514–520. [DOI] [PubMed] [Google Scholar]
- Milligan G. (2013) The prevalence, maintenance, and relevance of G protein-coupled receptor oligomerization. Mol Pharmacol 84:158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller JD, Chen Y, Gratton E. (2000) Resolving heterogeneity on the single molecular level with the photon-counting histogram. Biophys J 78:474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman-Tancredi A, Cussac D, Quentric Y, Touzard M, Verrièle L, Carpentier N, Millan MJ. (2002) Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. III. Agonist and antagonist properties at serotonin, 5-HT(1) and 5-HT(2), receptor subtypes. J Pharmacol Exp Ther 303:815–822. [DOI] [PubMed] [Google Scholar]
- Pellissier LP, Barthet G, Gaven F, Cassier E, Trinquet E, Pin JP, Marin P, Dumuis A, Bockaert J, Banères JL, et al. (2011) G protein activation by serotonin type 4 receptor dimers: evidence that turning on two protomers is more efficient. J Biol Chem 286:9985–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Comps-Agrar L, Maurel D, Monnier C, Rives ML, Trinquet E, Kniazeff J, Rondard P, Prézeau L. (2009) G-protein-coupled receptor oligomers: two or more for what? Lessons from mGlu and GABAB receptors. J Physiol 587:5337–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero-Müller A, Chou YY, Ji I, Lajic S, Hanyaloglu AC, Jonas K, Rahman N, Ji TH, Huhtaniemi I. (2010) Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc Natl Acad Sci USA 107:2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, Angers S, Mercier JF, Lagacé M, Marullo S, Bouvier M. (2004) Homodimerization of the beta2-adrenergic receptor as a prerequisite for cell surface targeting. J Biol Chem 279:33390–33397. [DOI] [PubMed] [Google Scholar]
- Saltzman AG, Morse B, Whitman MM, Ivanshchenko Y, Jaye M, Felder S. (1991) Cloning of the human serotonin 5-HT2 and 5-HT1C receptor subtypes. Biochem Biophys Res Commun 181:1469–1478. [DOI] [PubMed] [Google Scholar]
- Skerra A. (1994) A general vector, pASK84, for cloning, bacterial production, and single-step purification of antibody Fab fragments. Gene 141:79–84. [DOI] [PubMed] [Google Scholar]
- Smith C, Toohey N, Knight JA, Klein MT, Teitler M. (2011) Risperidone-induced inactivation and clozapine-induced reactivation of rat cortical astrocyte 5-hydroxytryptamine₇ receptors: evidence for in situ G protein-coupled receptor homodimer protomer cross-talk. Mol Pharmacol 79:318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann A, Gibert A, Lampe A, Grzesik P, Rutz C, Furkert J, Schmoranzer J, Krause G, Wiesner B, Schülein R. (2014) The specific monomer/dimer equilibrium of the corticotropin-releasing factor receptor type 1 is established in the endoplasmic reticulum. J Biol Chem 289:24250–24262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitler M, Klein MT. (2012) A new approach for studying GPCR dimers: drug-induced inactivation and reactivation to reveal GPCR dimer function in vitro, in primary culture, and in vivo. Pharmacol Ther 133:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. (2007) Subunit counting in membrane-bound proteins. Nat Methods 4:319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos AR, Parmelee JT, Pritchard JB. (1997) Functional characterization of choroid plexus epithelial cells in primary culture. J Pharmacol Exp Ther 282:1109–1116. [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. (2013) Structural features for functional selectivity at serotonin receptors. Science 340:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, Portoghese PS, Whistler JL. (2005) A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc Natl Acad Sci USA 102:9050–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA 104:7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youker RT, Bruns JR, Costa SA, Rbaibi Y, Lanni F, Kashlan OB, Teng H, Weisz OA. (2013) Multiple motifs regulate apical sorting of p75 via a mechanism that involves dimerization and higher-order oligomerization. Mol Biol Cell 24:1996–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916. [DOI] [PubMed] [Google Scholar]
- Zoenen M, Urizar E, Swillens S, Vassart G, Costagliola S. (2012) Evidence for activity-regulated hormone-binding cooperativity across glycoprotein hormone receptor homomers. Nat Commun 3:1007–1017. [DOI] [PubMed] [Google Scholar]