

Abstract
Synaptotagmin 1 (Syt1), a major Ca2+ sensor in neuroexocytosis, utilizes SNARE- and membrane-binding to regulate vesicle fusion, a required process for neurotransmitter release at the synapse. However, the mechanism by which Syt1 orchestrates SNARE- and membrane- binding to control individual vesicle fusion steps is still unclear. In this study, we used a number of single vesicle assays that can differentiate intermediates of neuroexocytosis, to focus on Syt1 mutants that might impair Syt1-SNARE/PIP2 interaction, Ca2+-binding, or membrane penetration. Our results show that, although putative Syt1-SNARE/PIP2 coupling through the polybasic region of the C2B domain is critical for vesicle docking, its disruption does not affect content release. In contrast, Ca2+-binding and membrane-penetration mutants significantly reduce content release. Our results thus delineate multiple functions of Syt1 along the pathway of Ca2+-triggered exocytosis in unprecedented detail.
In the pre-synapse, Ca2+-triggered neurotransmitter release from synaptic vesicles is the key process for maintaining signal transduction in the neuronal system1. SNARE proteins, comprised of presynaptic t-SNAREs syntaxin 1A and SNAP-25 and vesicle v-SNARE VAMP2 (vesicle-associated membrane protein 2), function as the core fusion machinery in vesicle exocytosis2,3,4,5. For tight control of neurotransmitter release by Ca2+, another vesicle protein, synaptotagmin 1 (Syt1), has been identified as a major Ca2+ sensor6,7. It is believed that Syt1 plays a key role in synchronizing neurotransmitter release by sensing Ca2+ influx and subsequently triggering rapid fusion of the vesicle with the plasma membrane1,7,8,9,10.
At the molecular level, Syt1 contains an N-terminal transmembrane domain, a flexible linker region, and tandem Ca2+-binding C2 (C2A and C2B) domains11. Cytoplasmic C2A and C2B domains share a β-sandwich structure containing a bottom loop region that can bind two and three Ca2+ ions, respectively12,13,14. Syt1 can interact with t-SNARE and the ternary SNARE complex via the polybasic region on the C2B domain in both Ca2+-independent and -dependent manners15,16,17,18,19. Also, Syt1 could bind to the negatively charged lipid PIP2 (Phosphatidylinositol 4,5-bisphosphate) on the plasma membrane probably via the same polybasic region on the C2B domain in the absence of Ca2+, while the loop regions could penetrate into the acidic membrane upon Ca2+ binding20,21,22,23,24.
In functional studies, both SNARE and membrane binding by the C2 domains have proven essential in achieving vesicle fusion18,25,26,27,28,29,30. Mutations in the polybasic region resulted in the impairment of SNARE-mediated lipid mixing in vitro as well as Ca2+-triggered neurotransmitter release in vivo21,31,32. Mutations in the Ca2+ binding sites revealed that Syt1 is the Ca2+ sensor and that Ca2+ binding to the loop region is required for vesicle exocytosis6,29,33,34,35. Finally, the loss and the gain of function mutations on the loop region showed that its membrane penetration is a critical step for Ca2+-triggered membrane fusion26,27,33,36,37. It has recently been shown that membrane attachment through its transmembrane domain is important for Syt1 to function properly as a Ca2+-sensor29,38,39,40.
It is thought that vesicle fusion proceeds in at least three sequential steps: vesicle docking, hemifusion (or lipid mixing), and fusion-pore opening (or content release)9,41,42, and studies have shown that Syt1 might be involved in all three steps. Apparently, Syt1's interactions with the SNARE complex and with the membrane are responsible for these regulations. For example, the putative Syt1-t-SNARE/PIP2 interaction assists docking of vesicles to the plasma membrane18,30,43. Syt1's penetration into the membrane is likely to play roles in both lipid mixing and fusion-pore opening6,26,27,29,33,34,35,36,37. The Syt1-ternary SNARE interaction might regulate those later steps as well in response to the Ca2+ signal, although this has not been demonstrated experimentally. Because of experimental difficulties for previous ensemble fusion assays in resolving individual steps along the fusion pathway, many ambiguities still remain with respect to understanding the molecular origins of the Syt1's involvement in individual steps of Ca2+-triggered neuroexocytosis.
To address these issues, we generated polybasic region mutants, which are likely to impair the Syt1-SNARE/PIP2 interaction, Ca2+ binding site mutants to hamper its Ca2+ affinity, and loop region mutants to alter its membrane penetration capacity. With well-established single vesicle assays40,44,45,46 that, can resolve the vesicle fusion process into docking and content release reflecting fusion pore opening, we dissected the effect of these mutations on individual fusion steps. The results show that Ca2+-independent t-SNARE and PIP2 binding, mediated by the polybasic region of Syt1, is essential for vesicle docking. To our surprise, however, content release is regulated only by the Ca2+-dependent insertion of the loop region and Ca2+ binding sites of Syt1 into the membrane, and not at all by its SNARE/PIP2 interaction.
Results
Syt1 mutants and their SNARE interactions
Syt1 mutants were designed on the basis of three reported functions of Syt1 in synaptic vesicle exocytosis: its interaction with SNARE and/or PIP2, Ca2+ binding, and membrane penetration. To alter these features of Syt1, we generated three groups of previously well-characterized Syt1 mutants. First, we intended to disrupt the SNARE/PIP2-binding polybasic region of the C2B domain by changing three basic amino acids Lys326, Lys327, and Lys331 to either acidic amino acid Glu (EEE), or neutral amino acid Gln (QQQ)21,31,32. We note however that there might be other binding modes between SNAREs and Syt1 (see Refs. 19,47). Another mutant, Y311N, was generated, because, although Y311 is a residue buried inside the polybasic region, this mutation has been shown to affect Syt1's binding to binary t-SNARE18,48, thereby hampering vesicle docking (blue in Fig. 1A). We also note though that these mutations might disrupt the Syt1 structure, thereby impacting functions. Such disruption is especially concerning for partially buried position 311(Ref. 48). The circular dichroism (CD) spectra however show that the overall folding was not affected significantly by the Tyr to Asn mutation at this position (Supplementary Fig. S1). Second, to block the Ca2+ binding capability, we mutated the amino acids Asp230 and Asp232 in the C2A domain to Ala (C2A*), the amino acids Asp363 and Asp365 in the C2B domain to Ala (C2B*), or generated all four mutations in the C2A and C2B domains (C2A*B*)6,14,29,33,34,35,49 (red in Fig. 1A). Third, we generated loop region mutants to reduce the membrane penetration ability of Syt1. We mutated amino acids Met173 and Phe234 in C2A to Ala (2A(A)), amino acids Val304, Ile367 in C2B to Ala (2A(B)), or generated all four mutations in C2AB (4A)27,33,50. Conversely, to enhance membrane penetration ability, we changed these amino acids to Trp instead of Ala (2W(A), 2W(B), 4W)26,27,33,36,37 (magenta in Fig. 1A).
Figure 1. SNARE binding of Syt1 mutants.

(A) Ribbon diagram of the cytoplasmic domain of Syt1 (C2AB). The model is derived from the crystal structural of the cytoplasmic domain of Syt1 (C2AB, PDB id: 2R83)69. The C2B domain was rotated about 180° in the horizontal plane relative to C2A and the linker between C2A and C2B to show the Ca2+ binding loops of both domains in the same plane. Two important Ca2+ binding residues (D230/D232) on the C2A domain and the other two Ca2+ binding residues (D363/D365) on the C2B domain are shown as red sticks. Four residues proposed to be important for membrane insertion (M173/F234 on C2A, V304/I367 on C2B) are shown in magenta. Four residues on the C2B domain (K326/K327/K331/Y311) that are proposed to bind the SNARE complex, are shown in blue. (B and C) GST pull-down assays of SNARE-Syt1 binding. Mutations in the polybasic region of the C2B domain (EEE, QQQ, and Y311N) hamper SNARE binding while the Ca2+ binding (C2A*, C2B*, and C2A*B*) and loop region mutations (2A(A), 2A(B), 4A, 2W(A), 2W(B), and 4W) mutations do not change the SNAREs binding capability of Syt1. The binary complex is formed between GST-tagged soluble syntaxin 1A (191–266) and His-tagged SNAP-25 and the ternary complex is formed among GST-tagged soluble VAMP2 (1–96), syntaxin 1A (191–266), and His-tagged SNAP-25.
Because it has been shown that Syt1 binding to the binary complex of t-SNAREs syntaxin 1A and SNAP-25 plays a role in vesicle docking prior to the rise of the Ca2+ level18, we first examined the Syt1 binding to the t-SNARE complex. The polybasic region mutants EEE, QQQ, and Y311N showed significant reduction of binding to the binary t-SNARE complex in the absence of Ca2+ (Fig. 1B). To further understand molecular mechanism of the t-SNARE-Syt1 interaction, we performed the GST pull-down assay in the present of 200 μM inositol 1,4,5-trisphosphate (IP3) to mimic PIP2 in solution. Our results showed that IP3 reduces the interaction between wild-type Syt1 and the binary t-SNARE complex somewhat (Supplementary Figs. S2B and S2C), indicating that Syt1 binding to PIP2 might compete with t-SNARE binding somewhat. Controls BSA and Ca2+ did not affect the interaction between Syt1 and the binary t-SNARE complex (Supplementary Figs. S2B and S2C), indicating that Syt1-binary t-SNARE interaction is specific and Ca2+-independent.
On the other hand, it has been postulated that Syt1 binding to the ternary SNARE complex regulates Ca2+-triggered opening of fusion-pores7. The polybasic region mutations reduced Syt1 binding to the ternary SNARE complex in the presence of Ca2+ (Fig. 1C). As controls, adding BSA or removing Ca2+ did not affect the interaction between Syt1 and the ternary SNARE complex (Supplementary Figs. S2D and S2E). IP3 (200 μM) did not alter the interaction between wild-type Syt1 and the ternary SNARE complex in the presence of 500 μM Ca2+ (Supplementary Figs. S2D and S2E). With these mutants we are now ready to test, with the single vesicle fusion assay whether or not there is direct correlation between Syt1-t-SNARE coupling and vesicle docking and, more importantly, between Syt1-ternary SNARE complex coupling and content release.
In contrast to these polybasic region mutations, neither the mutations in the Ca2+-binding sites nor those in the membrane-penetrating loops affected the Syt1's ability to bind the binary t-SNARE complex in the absence of Ca2+ or the ternary SNARE complex in the presence of Ca2+ (Figs. 1B and 1C).
Polybasic region mutations in Syt1 reduce PIP2-binding and vesicle docking
To study the effect of the Syt1 mutants on individual fusion steps, we reconstituted wild-type Syt1 or its mutants and VAMP2 (molar ratio of 1:1) onto a population of vesicles (v-vesicles) (Supplementary Fig. S3). Premixed t-SNAREs (syntaxin 1A: SNAP-25 = 1:1.5) were reconstituted into another population of vesicles (t-vesicles) (Supplementary Fig. S3A). The wild-type Syt1 and its mutants had all similar reconstitution efficiencies when reconstituted together with VAMP2 to v-vesicles (Supplementary Fig. S3B).
Previously, we had shown that both binary t-SNARE and PIP2 play roles in Syt1-mediated vesicle docking30. Therefore, we performed the single vesicle membrane-binding assay to examine Syt1's binding to SNARE free vesicles (Supplementary Fig. S4A). In this assay, SNARE-free vesicles with 2 mol% PIP2 were immobilized (or tethered) on the surface by the avidin-biotin conjugation (see Materials and Methods section), Syt1- or its mutants-reconstituted vesicles doped with 2 mol% fluorescence acceptor lipid DiD (Syt1-vesicles) were loaded, and the number of Syt1-vesicles docked (or bound) to the SNARE-free vesicles in the imaging area (45 × 90 μm2) were counted. We found that only the polybasic region mutants showed impaired binding to the PIP2-containing vesicles by as much as 60% in the absence of Ca2+ (Fig. 2A and Supplementary Fig. S5), showing that Syt1 binding to the negatively charged PIP2 might contribute to vesicle docking, confirming our previous results30.
Figure 2. Interaction between the polybasic region of the C2B domain and PIP2 is important for vesicle docking.

(A–C) PIP2 binding abilities of wild-type Syt1 and its mutants with alterations at the polybasic region, Ca2+ binding sites, and the loop region. (D–F) Single vesicle docking probabilities of wild-type Syt1 and its mutants with alterations at the polybasic region, Ca2+ binding sites, and the loop region. The fraction is defined as the number of docked t-vesicles divided by the number of immobilized v-vesicles. Results are shown as the mean ± S.D. (*P < 0.05, n = 3, and n.s. means ‘not significant').
Then, we tested the effect of these mutations on vesicle docking by applying the single vesicle docking assay with t-vesicles containing DiD and v-vesicles containing fluorescence donor lipid DiI (2 mol% each) (Supplementary Figs. S4B and S4C)40,51,52. Our results showed that polybasic region-disrupted mutants Syt1 EEE and Y311N exhibited reduced vesicle docking by as much as ~70% when compared with wild-type Syt1. The QQQ mutant, however, had a mild inhibition by ~30% (Fig. 2D). Unlike the polybasic region mutants, the Ca2+ binding site and loop region mutants had negligible effects on vesicle docking (Figs. 2E and F). Similar to our previous work using solution single-vesicle assay, when PIP2 was removed from t-vesicles, vesicle docking is reduced significantly (Supplementary Fig. S6)30, suggesting that the Syt1-PIP2 interaction may well be the dominant force for vesicle docking. When the control IP3 or BSA was injected into the chamber together with t-vesicles, vesicle docking was not affected (Supplementary Fig. S6). Although our results may not be sufficient to pinpoint the detailed binding mechanism, they establish a direct correlation between Syt1-SNARE/PIP2 coupling and vesicle docking, and show that the polybasic region of Syt1 plays a role in vesicle docking.
Ca2+ binding and the penetration of the loop region of Syt1 into the membrane are essential for content release
Ca2+ triggered neurotransmitter release at the synapse requires a fusion-pore encompassing the vesicle membrane and the plasma membrane. To study the effect of various Syt1 mutations on the fusion-pore opening step, we first examined the Ca2+ triggered insertion of Syt1 into the membrane. This time PIP2 was removed from SNARE-free vesicles and Syt1-vesicles were injected into the chamber in the presence of 500 μM Ca2+. We found that both Ca2+ binding site mutants and alanine mutants on the loop region showed impaired vesicle docking (Supplementary Fig. S7), consistent with the previous report that Ca2+ bridges Syt1 and the membrane and both C2A and C2B domains of Syt1 bind to the membrane in the presence of Ca2+ (Ref. 43).
Then we applied the single vesicle content mixing assay and monitored the diffusion of sulforhodamine B from a v-vesicle to an empty t-vesicle, that results in a sudden increase of the fluorescence signal due to dilution-induced fluorescence dequenching40,46 (Supplementary Fig. S8A). We used v-vesicles reconstituted with Syt1 and VAMP2 in a 1:1 molar ratio for wild-type Syt1 and its mutants, although the vesicles containing Syt1 and VAMP2 at the 1:4 molar ratio also exhibited robust content mixing (Supplementary Fig. S9). The polybasic region mutants EEE, QQQ, and Y311N showed no difference in term of the fusion percentage among docked vesicles from the wild-type Syt1 in Ca2+-triggered content release (Fig. 3A and Supplementary Fig. S8B). We note that the disruption of polybasic region by these mutations could still affect neurotransmitter release in fast (submillisecond) time scale after the Ca2+ influx in vivo. However, the Ca2+ binding sites mutants Syt1-C2A*, Syt1-C2B*, and Syt1-C2A*B* exhibited a decrease in Ca2+-triggered content release by 30, 60, and 70%, respectively (Fig. 3B and Supplementary Fig. S8C). The results indicate that Ca2+ binding to the C2 domain is important for Ca2+-triggered content release. We notice that the mutation in C2B is more severely disruptive than those in C2A, consistent with previous findings25.
Figure 3. Content mixing of wild-type Syt1 and its mutants.

The bar graphs represent single vesicle content mixing events within 1 min. of the fusion reaction. Pink bars represent the cases without Ca2+ while red bars represent the cases with 500 μM Ca2+. Content mixing probabilities are for wild-type Syt1 and its mutants in the polybasic region (A), for the Ca2+ binding sites (B), and for the loop region (C). The fraction is defined as the number of content mixing events divided by the number of docked vesicles. Results shown represent the mean ± S.D. (*P < 0.05, n = 3 and n.s. means ‘not significant').
Next, we tested loop region mutants that either reduce or enhance the membrane penetration ability of the loops, depending on the side-chain size of the corresponding amino acids. We observed an anticipated inhibition of content release for alanine mutants (2A(A), 2A(B), 4A) by 40%, 20%, 70% (Fig. 3C and Supplementary Fig. S8D), respectively. The results thus indicate that membrane penetration of Syt1 is important for opening fusion pores. In contrast, we observed no enhancement for two tryptophan mutants (2W(A), 2W(B)), although the 4W mutant was able to increase content mixing by 20% (Fig. 3C and Supplementary Fig. S8D). As controls, in the absence of Ca2+ none of these mutants supported content mixing effectively (pink bars in Fig. 3 and dotted lines in Supplementary Fig. S8).
Our results thus show that the polybasic region that mediates Syt1-SNARE/PIP2 coupling plays a role in vesicle docking but not in the final fusion step. It appears, however, that Ca2+-binding and the loop penetration into the membrane are important elements in content release although they have minimal effect on vesicle docking.
Discussion
In this work, our single vesicle fusion assay revealed that mutations in the polybasic region in Syt1 cause reduced SNARE and PIP2 binding and result in an apparent decrease in vesicle docking, but produce little change in the content release. It has been long speculated that Syt1-SNARE interaction plays a critical role in Ca2+-triggered exocytosis25. Our results show that the coupling between the polybasic region of Syt1 and SNARE/PIP2 is limited to vesicle docking (Fig. 4) and does not extend its influence on to the final content release step.
Figure 4. A mechanistic model for multiple synaptotagmin 1 functions along the vesicle fusion pathway.
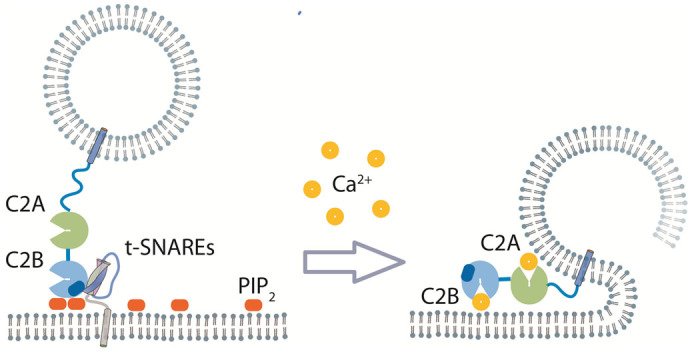
Before Ca2+, the polybasic region of the C2B domain interacts with t-SNAREs and PIP2 molecules to promote docking of synaptic vesicles. Upon calcium arrival, the membrane association and insertion of Ca2+-bound C2A and C2B domains become critical for content release.
However, the Syt1-ternary SNARE complex coupling might still play an important role in regulating Ca2+-triggered exocytosis. A current mechanistic model predicts that complexins, a family of small presynaptic proteins, bind to the SNARE complex and inhibit full zippering, thereby clamping membrane fusion53,54,55,56,57,58. It has been thought that Syt1 would unclamp the complexin clamp in the presence of Ca2+, requiring Syt1's binding to the SNARE complex53,54. It thus appears that Syt1-ternary SNARE coupling is required for replacing the complexin clamp. In a separate note, since polybasic region mutants cannot completely block the Syt1/ternary SNARE interaction (Fig. 1 and Supplementary Fig. S2) we cannot rule out the possibility that there might be another unidentified region mediating the Syt1/ternary SNARE interaction besides the polybasic region19,47.
Although the Syt1-SNARE/PIP2 interaction has little influence on fusion pores, our results show that Syt1's membrane insertion is indeed critical for Ca2+-triggered opening of the fusion (Fig. 4), consistent with the conclusion of many previous in vivo and in vitro studies7,25. For Syt1 it is unclear if SNARE- and membrane-binding is sequential. Our previous work raised the possibility that Syt1 simultaneously binds the SNARE complex and the PIP2-containing membrane30,39. Here, based on experiments involving Syt1 mutants, we show this concurrent SNARE/PIP2 interaction through the polybasic region on Syt1 in the absence of Ca2+, might be essential for synaptic vesicle docking.
How might the Syt1's insertion into the membrane promote the opening of fusion-pores for content release? McMahon and Chapman have independently shown that Syt1 has the capacity to induce a positive curvature of the bilayer33,50. They demonstrated that this newly discovered function is well correlated with the ability to stimulate lipid mixing (or hemifusion) with Ca2+. It is also possible that this curvature-inducing ability of Syt1 might provide some thrust to overcome the energy barrier for pore opening. Alternatively, it has long been thought that transmembrane domains of fusion proteins, including those in viral membrane fusion as well as in intracellular membrane fusion, play a critical role in driving the fusion-pore59,60. It has been shown that Syt1 binds the basic membrane proximal linker region of SNAREs where PIP2 molecules can cluster43. We speculate that this interaction might activate transmembrane domains to drive fusion pore opening, although more work is needed to support this idea. Very recently, Südhof and coworkers have disputed the critical involvement of SNARE transmembrane domains for Ca2+-triggered exocytosis, warranting further work in this area61.
In summary, using a series of single vesicle assays40,46,62,63,64 resolving the individual fusion steps in the single vesicle fusion event, we have shown that Syt1-SNARE/PIP2 coupling through the polybasic region has little to do with content release although it plays a significant role in vesicle docking. On the other hand, our results demonstrate that Ca2+-induced insertion of Syt1 into the membrane is essential for content release. Our results thus delineate multiple functions of Syt1 along the pathway of Ca2+-triggered exocytosis.
Methods
Plasmid constructs and site-directed mutagenesis
DNA sequences encoding rat syntaxin 1A (amino acids 1–288 with three native cysteines C145, C271, and C272 replaced by alanines), rat VAMP2 (amino acids 1–116 with C103 replaced by alanine), rat SNAP-25 (amino acids 1–206 with four native cysteines C85, C88, C90, and C92 replaced by alanines), soluble syntaxin 1A (amino acid 191–266), and soluble VAMP2 (amino acids 1–96) were inserted into the pGEX-KG vector as N-terminal glutathione S-transferase (GST) fusion proteins. SNAP-25 was inserted into a pET-28b vector also as an N-terminal 6xHistidine (His)-tag fusion protein. Recombinant synaptotagmin 1 (Syt1, amino acids 50–421 with four native cysteines C74, C75, C77 and C79 replaced by alanines and another C82 replaced by serine, (we denoted this recombinant form as wild-type Syt1 in this work and used it as the template for generating the point mutants) was inserted into a pET-28b vector as a C-terminal His-tagged protein. It has been previously shown that the deleted first 49 N-terminal residues does not affect the Syt1 function in exocytosis33,37,38. The Quick Change site-directed mutagenesis kit (Stratagene) was used to generate all Syt1 mutants, including Syt1 EEE (L326/327/331E), Syt1 QQQ (L326/327/331Q), Syt1 Y311N, Syt1 2A(A) (M173A/F234A), Syt1 2A(B) (V304A/I367A), Syt1 4A (M173A/F234A/V304A/I367A), Syt1 2W(A) (M173W/F234W), Syt1 2W(B) (V304W/I367W), Syt1 4W (M173W/F234W/V304W/I367W), Syt1-C2A* (D230A/D232A), Syt1-C2B* (D363A/D365A), and Syt1-C2A*B* (D230A/D232A/D363A/D365A). DNA sequences were confirmed by the Iowa State University DNA Sequencing Facility.
Protein expression and purification
The GST-tagged proteins were expressed in E. coli Rosetta (DE3) pLysS (Novagene). Details can be found in our previous work39,65. The His-tagged proteins were expressed in E. coli BL21 (DE3) (Novagen) and purified using previously-described protocol39,66.
Membrane reconstitution
The lipid molecules used in this study were 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), phosphatidylinositol-4,5-bisphosphate (PIP2, from porcine brain), cholesterol, and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamin-N-(biotinyl) (biotin-DPPE). All lipids were obtained from Avanti Polar Lipids. 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate (DiI), 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindodicarbocyanine Perchlorate (DiD), and sulforhodamine B were obtained from Invitrogen.
For the single vesicle docking assay, the molar ratios of lipids were 15:61:20:2:2 (DOPS:POPC:cholesterol:PIP2:DiD) for the t-SNARE-reconstituted (t-)vesicles, and 5:73:20:2 (DOPS:POPC:cholesterol:DiI) for the v-SNARE-reconstituted (v-)vesicles, respectively. To fix v-vesicles on the TIR imaging surface coated with neutravidin, 0.1 mol% biotin-DPPE was added to the v-vesicle lipid mixture prior to the rehydration-freeze thaw-extrusion steps (see below). The lipid mixture was first completely dried and then rehydrated with dialysis buffer (25 mM HEPES, pH 7.4, 100 mM KCl). After five freeze–thaw cycles, protein-free large unilamellar vesicles (~100 nm in diameter) were obtained by extrusion through a 100 nm polycarbonate filter (Whatman). For membrane reconstitution, SNARE proteins and Syt1 were mixed with protein-free vesicles at a protein-to-lipid molar ratio of 1:200 for each protein component (this ratio was kept the same for all experiments, including the single vesicle content mixing assay) with ~0.8% OG in the dialysis buffer at 4°C for 15 min. The mixture was diluted by a factor of two with the dialysis buffer and this diluted mixture was then dialyzed overnight in 2 L dialysis buffer at 4°C. Details for the reconstitution process were discussed in our previous work39,64.
For the single vesicle content mixing assay using a small sulforhodamine B content indicator, lipid mixtures were prepared as described for the single vesicle docking assay except that the fluorescent lipid dyes (DiI and DiD) were replaced by equal amounts of POPC. The t-vesicles were prepared as described earlier, except that 50 mM sulforhodamine B in the dialysis buffer was kept throughout all the sample preparation steps for the v-vesicles. Free sulforhodamine B was removed through dialysis and a further PD-10 column desalting step (GE healthcare).
Membrane reconstitution efficiency was confirmed using liposome co-sedimentation assay followed by a SDS-page analysis. Briefly, the aggregated protein was first removed after dialysis by centrifugation at 10,000 g for 10 min at 4°C. Then the membrane-bound protein was pelleted through high-speed liposome sedimentation using an Airfuge Air-Driven Ultracentrifuge (Beckman) at 150,000 g for 30 min at 4°C. Pelleted vesicles were re-suspended in the dialysis buffer and re-subjected to the centrifuge for twice more. Finally, the pellets were re-suspended in 60 μl (about 1/3 of the initial volume) of dialysis buffer and analyzed by SDS-page.
Single vesicle binding assay
Slide preparation was the same as that in the vesicle docking assay. SNARE-free vesicles (with or without 2 mol% PIP2) were immobilized on the PEG-coated surface. After two rounds of 200 μl dialysis buffer washing, Syt1 or its mutants reconstituted vesicles (Syt1-vesicles, DiD-labled without PIP2, 100 ~ 200 nM) were injected into the flow chamber for 30 min docking at room temperature. After washing out free Syt1-vesicles, the docked Syt1-vesicles number in the imaging area (45 × 90 μm2) was counted (Supplementary Figs. S10A-C).
Single vesicle docking and content mixing assays
After coating the quartz surface with a solution of methoxy-polyethylene glycol (mPEG) and biotin-PEG molecules to eliminate non-specific binding of vesicles, the quartz slide was assembled into a flow chamber and coated with neutravidin (0.2 mg ml−1). Following 30 minutes of incubation at room temperature (~25 °C), the v-vesicles were immobilized on the PEG-coated surface. After two rounds of 200 μl dialysis buffer washing, t-vesicles (100 ~ 200 nM in lipid concentrations) were injected at room temperature into the flow chamber for 30 min of docking. After washing out free t-vesicles, we acquired images (45 × 90 μm2) from 5 ~ 40 random locations within the flow chamber using 635 nm laser excitation for docked t-vesicles and 532 nm laser excitation for immobilized v-vesicles, respectively. All spots appeared in the green channel were considered as docked t-vesicles. The spots in the image were identified by the smCamera program (kindly provided by Dr. Taekjip Ha's group) based on the criteria: peak radius, 3 pixel; peak threshold/data scaler, 1%. The docking probability was calculated as the ratio of average docked t-vesicles to average anchored v-vesicles (Supplementary Figs. S10D-F). Details of the single vesicle docking assay were reported in our previous work67,68.
For real-time imaging of small sulforhodamine B content release, sulforhodamine B containing v-vesicles was immobilized on the PEG-coated surface. After two rounds of washing using 1 ml dialysis buffer, t-vesicles were injected into the channel to make them bind to v-vesicles. After 30 min of incubation at room temperature (~25 °C), dialysis buffer, with or without 500 μM Ca2+, was injected into the flow chamber by a motorized syringe pump at a speed of 33 μl sec−1. The detail of the single vesicle content mixing assay was reported in previous work45,46,62.
GST pull-down assay
To form a binary complex, His-tagged SNAP-25 and GST-tagged soluble Syntaxin 1A (191–266) cell lysates were mixed and loaded onto Ni-NTA beads. The binary complex was purified following the procedure described for His-tagged SNAP-25, and the purified binary complex was then loaded onto Glutathione-agarose beads. After washing thoroughly with a cleavage buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.8% OG) to remove unbound His-tagged SNAP-25, the binary complex immobilized beads were separated into many equal parts in 1.7 ml Eppendorf tubes. Equal amounts of wild-type Syt1 or its mutants and cleavage buffer were added to the immobilized binary complex and the mixture was incubated at 4°C for 1 h. The beads were then thoroughly washed with the cleavage buffer. 5xSDS-loading buffer (313 mM Tris-HCl, pH 6.8, 10% SDS, 0.05% bromophenol blue, 50% glycerol, 0.5 M DTT) was added to the samples and the mixture was boiled for 10 min. Proteins were resolved on precast 12% SDS-page and visualized using Coomassie blue staining. To test the binding of wild-type Syt1 and its mutants to the ternary SNARE complex, purified soluble Syntaxin 1A (191–266) was mixed with His-tagged SNAP-25 and GST-tagged soluble VAMP2 (1–96) cell lysates before loading onto Ni-NTA beads. The ternary complex was purified following the same procedure described for His-tagged SNAP-25. The purified ternary complex was then loaded onto Glutathione-agarose beads. After washing thoroughly with cleavage buffer to remove unbound His-tagged SNAP-25 and the binary complex, binding of wild-type Syt1 or its mutants was performed following the procedure described above for the binary complex, except that 500 μM Ca2+ was added to the cleavage buffer.
CD spectroscopy
The CD spectra were measured with an AVIV stop-flow Circular Dichroism spectropolarimeter at 190 to 260 nm using a cell with the 1 mm path-length. The sample containing 10 μM of either wild-type Syt1 or the Y311N mutant was measured at 25°C. For the correction of the baseline error, the signal from a blank run with PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4) containing 0.8% OG was subtracted from all the experimental spectra.
Author Contributions
Y.Lai, X.L. and Y.-K.S. designed the experiments, Y.Lai, X.L. and J.D. carried out the experiments, and Y.Lai, X.L. and Y.-K.S. wrote the paper.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr. Taekjip Ha (University of Illinois) for providing the analysis program for the single vesicle assay. This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM051290 (to Y.-K.S.), the Korea Institute of Science and Technology (KIST Institutional Project #2E25000), and the Ministry of Science and Technology of China (973 Program Grands 2015CB856304).
References
- Südhof T. C. The synaptic vesicle cycle. Annu Rev Neurosci 27, 509–547 (2004). [DOI] [PubMed] [Google Scholar]
- Poirier M. A. et al. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol 5, 765–769 (1998). [DOI] [PubMed] [Google Scholar]
- Sutton R. B., Fasshauer D., Jahn R. & Brunger A. T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353 (1998). [DOI] [PubMed] [Google Scholar]
- Weber T. et al. SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 (1998). [DOI] [PubMed] [Google Scholar]
- Stein A., Weber G., Wahl M. C. & Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature 460, 525–528 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R. et al. Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49 (2001). [DOI] [PubMed] [Google Scholar]
- Chapman E. R. Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat Rev Mol Cell Biol 3, 498–508 (2002). [DOI] [PubMed] [Google Scholar]
- Rizo J. & Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol 15, 665–674 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R. & Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J. & Südhof T. C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices--guilty as charged? Annu Rev Cell Dev Biol 28, 279–308 (2012). [DOI] [PubMed] [Google Scholar]
- Perin M. S., Brose N., Jahn R. & Südhof T. C. Domain structure of synaptotagmin (p65). J Biol Chem 266, 623–629 (1991). [PubMed] [Google Scholar]
- Sutton R. B., Davletov B. A., Berghuis A. M., Südhof T. C. & Sprang S. R. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80, 929–938 (1995). [DOI] [PubMed] [Google Scholar]
- Shao X., Davletov B. A., Sutton R. B., Südhof T. C. & Rizo J. Bipartite Ca2+-binding motif in C2 domains of synaptotagmin and protein kinase C. Science 273, 248–251 (1996). [DOI] [PubMed] [Google Scholar]
- Ubach J., Zhang X., Shao X., Südhof T. C. & Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J 17, 3921–3930 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerona R. R., Larsen E. C., Kowalchyk J. A. & Martin T. F. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem 275, 6328–6336 (2000). [DOI] [PubMed] [Google Scholar]
- Earles C. A., Bai J., Wang P. & Chapman E. R. The tandem C2 domains of synaptotagmin contain redundant Ca2+ binding sites that cooperate to engage t-SNAREs and trigger exocytosis. J Cell Biol 154, 1117–1123 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Kim-Miller M. J., Fukuda M., Kowalchyk J. A. & Martin T. F. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron 34, 599–611 (2002). [DOI] [PubMed] [Google Scholar]
- de Wit H. et al. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell 138, 935–946 (2009). [DOI] [PubMed] [Google Scholar]
- Vrljic M. et al. Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat Struct Mol Biol 17, 325–331 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J., Tucker W. C. & Chapman E. R. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol 11, 36–44 (2004). [DOI] [PubMed] [Google Scholar]
- Li L. et al. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem 281, 15845–15852 (2006). [DOI] [PubMed] [Google Scholar]
- Kuo W., Herrick D. Z., Ellena J. F. & Cafiso D. S. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. J Mol Biol 387, 284–294 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A., Stein A., Jahn R. & Fasshauer D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J Biol Chem 284, 25749–25760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G., Meyenberg K., Diederichsen U. & Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem 287, 16447–16453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E. R. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem 77, 615–641 (2008). [DOI] [PubMed] [Google Scholar]
- Rhee J. S. et al. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc Natl Acad Sci U S A 102, 18664–18669 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch K. L. et al. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell 19, 5093–5103 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. L., Huang H., Herrick D. Z., Epp N. & Cafiso D. S. Synaptotagmin 1 and SNAREs form a complex that is structurally heterogeneous. J Mol Biol 405, 696–706 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennekate W. et al. Cis- and trans-membrane interactions of synaptotagmin-1. Proc Natl Acad Sci U S A 109, 11037–11042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Y. et al. Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J 31, 2144–2155 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen C. A., Lee S. M., Shin Y. K. & Reist N. E. C2B polylysine motif of synaptotagmin facilitates a Ca2+-independent stage of synaptic vesicle priming in vivo. Mol Biol Cell 17, 5211–5226 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffaney J. D., Dunning F. M., Wang Z., Hui E. & Chapman E. R. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem 283, 31763–31775 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E., Johnson C. P., Yao J., Dunning F. M. & Chapman E. R. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2+)-regulated fusion. Cell 138, 709–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E. et al. Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol 18, 813–821 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G. et al. Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat Struct Mol Biol 18, 805–812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin O. H., Rizo J. & Südhof T. C. Synaptotagmin function in dense core vesicle exocytosis studied in cracked PC12 cells. Nat Neurosci 5, 649–656 (2002). [DOI] [PubMed] [Google Scholar]
- Yao J., Kwon S. E., Gaffaney J. D., Dunning F. M. & Chapman E. R. Uncoupling the roles of synaptotagmin I during endo- and exocytosis of synaptic vesicles. Nat Neurosci 15, 243–249 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Liu H., Gu Y. & Chapman E. R. Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J Cell Biol 195, 1159–1170 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y. & Shin Y. K. The importance of an asymmetric distribution of acidic lipids for synaptotagmin 1 function as a Ca2+ sensor. Biochem J 443, 223–229 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y., Lou X., Jho Y., Yoon T. Y. & Shin Y. K. The synaptotagmin 1 linker may function as an electrostatic zipper that opens for docking but closes for fusion pore opening. Biochem J 456, 25–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Zhang F., Su Z., McNew J. A. & Shin Y. K. Hemifusion in SNARE-mediated membrane fusion. Nat Struct Mol Biol 12, 417–422 (2005). [DOI] [PubMed] [Google Scholar]
- Diao J. J., Zhao M. L., Zhang Y. X., Kyoung M. & Brunger A. T. Studying protein-reconstituted proteoliposome fusion with content indicators in vitro. Bioessays 35, 658–665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigmann A. et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol 20, 679–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon T. Y. et al. Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat Struct Mol Biol 15, 707–713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J. et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. eLife 1, e00109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y. et al. Fusion pore formation and expansion induced by Ca2+ and synaptotagmin 1. Proc. Natl. Acad. Sci. U S A 110, 1333–1338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi U. B. et al. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol 17, 318–324 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C. et al. Conserved prefusion protein assembly in regulated exocytosis. Mol Biol Cell 17, 283–294 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I. et al. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069 (2001). [DOI] [PubMed] [Google Scholar]
- Martens S., Kozlov M. M. & McMahon H. T. How synaptotagmin promotes membrane fusion. Science 316, 1205–1208 (2007). [DOI] [PubMed] [Google Scholar]
- Lee H. K. et al. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 328, 760–763 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J. J. et al. Complexin-1 Enhances the On-Rate of Vesicle Docking via Simultaneous SNARE and Membrane Interactions. Journal of the American Chemical Society 135, 15274–15277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126, 1175–1187 (2006). [DOI] [PubMed] [Google Scholar]
- Schaub J. R., Lu X., Doneske B., Shin Y. K. & McNew J. A. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat Struct Mol Biol 13, 748–750 (2006). [DOI] [PubMed] [Google Scholar]
- Giraudo C. G. et al. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science 323, 512–516 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar S. S. et al. A conformational switch in complexin is required for synaptotagmin to trigger synaptic fusion. Nat Struct Mol Biol 18, 934–940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Cao P. & Südhof T. C. Deconstructing complexin function in activating and clamping Ca2+-triggered exocytosis by comparing knockout and knockdown phenotypes. Proc Natl Acad Sci U S A 110, 20777–20782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar S. S. et al. Conformational dynamics of calcium-triggered activation of fusion by synaptotagmin. Biophys J 105, 2507–2516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J., Borbat P. P., Freed J. H. & Shin Y. K. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc Natl Acad Sci U S A 106, 5141–5146 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J. M. et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336, 1581–1584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Bacaj T., Yang X., Pang Z. P. & Südhof T. C. Lipid-anchored SNAREs lacking transmembrane regions fully support membrane fusion during neurotransmitter release. Neuron 80, 470–483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyoung M. et al. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A 108, E304–313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyoung M., Zhang Y., Diao J., Chu S. & Brunger A. T. Studying calcium-triggered vesicle fusion in a single vesicle-vesicle content and lipid-mixing system. Nat Protoc 8, 1–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J. et al. A single vesicle-vesicle fusion assay for in vitro studies of SNAREs and accessory proteins. Nat Protoc 7, 921–934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y., Lou X., Wang C., Xia T. & Tong J. Synaptotagmin 1 and Ca2+ drive trans SNARE zippering. Sci Rep 4, 4575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y. et al. Nonaggregated alpha-Synuclein Influences SNARE-Dependent Vesicle Docking via Membrane Binding. Biochemistry 53, 3889–3896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon T. Y. et al. Complexin and Ca(2+) stimulate SNARE-mediated membrane fusion. Nature Structural & Molecular Biology 15, 707–713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J., Yoon T. Y., Su Z. L., Shin Y. K. & Ha T. C2AB: A Molecular Glue for Lipid Vesicles with a Negatively Charged Surface. Langmuir 25, 7177–7180 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuson K. L., Montes M., Robert J. J. & Sutton R. B. Structure of human synaptotagmin 1 C2AB in the absence of Ca2+ reveals a novel domain association. Biochemistry 46, 13041–13048 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information