

Abstract
Cleidocranial dysplasia (CCD) is a rare (1:1,000,000) congenital condition secondary to spontaneous mutation (40%) or autosomal dominant inheritance (60%) affecting skeletal and dental systems. Hypomineralization of the hypoplastic clavicles and/or cranium is the major feature observed by prenatal ultrasound. Radiologically clavicles are hypoplastic or absent in chest X-ray. Delayed closure of the fontanelle and the skull sutures in pediatric and adolescent population and increased mobility of shoulders in all age groups (exhibited by the ability to bring shoulders close to each other) are prominent clinical diagnostic features of CCD. The diagnosis of CCD is often missed or significantly delayed. The management of CCD involves a multidisciplinary approach and its early diagnosis is essential to select an optimum plan and therapeutic benefit. We present here a case of CCD in a 17-month-old girl referred to us for investigation of below average weight and height gain; we stress on the usefulness of early diagnosis in the management of CCD and discuss current management concepts.
Keywords: Absent or hypoplastic clavicles, cleidocranial dysplasia, short stature, supernumerary teeth, wide fontanelle
INTRODUCTION
Cleidocranial dysplasia (CCD, mutational dysostosis, Marie-Sainton disease, Scheuthauer-Marie-Sainton syndrome, cleidocranial dysostosis and dento-osseous dysplasia) is a rare (1:1,000,000) congenital condition affecting skeletal and dental systems.[1,2] Its radiographic and clinical features are illustrated in Table 1.[1,2,3,4,5,6] A wide range of manifestations is reported in CCD cases, ranging from mild to severe involving only dental system or syringomyelia or complete absence of ossification of parietal bones in addition to other skeletal abnormalities.[1,2,3,4,5,6] Interestingly in families with CCD, subsequent generations with inherited CCD have relatively milder forms of the disease. Here, we illustrate a case of 17-month-old girl presenting with failure to thrive and ultimately diagnosed with CCD. We stress on the usefulness of early diagnosis in the successful management of CCD and present important clinical, radiological and sonographic findings to aid in its early diagnosis.
Table 1.
Radiographic and clinical features of CCD
CASE REPORT
The present case report is about a 17-month-old girl child who was referred to us for investigation of below average weight and height gain. She was born at gestational age of 40 weeks by normal vaginal delivery in a hospital, after precipitous labor, to a healthy non-consanguineous couple and at birth weighed 3100 g. She started sitting without support at 11 months and walking without support at 15 months of age.
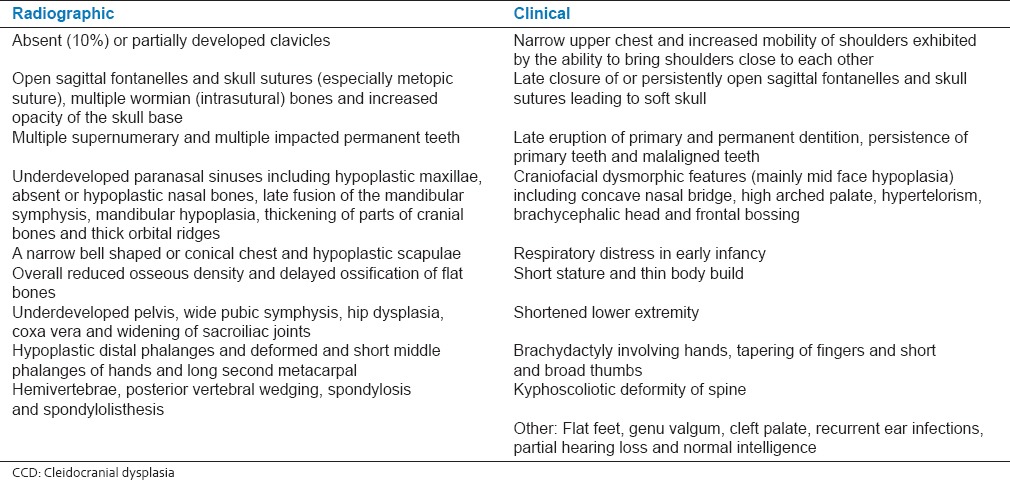
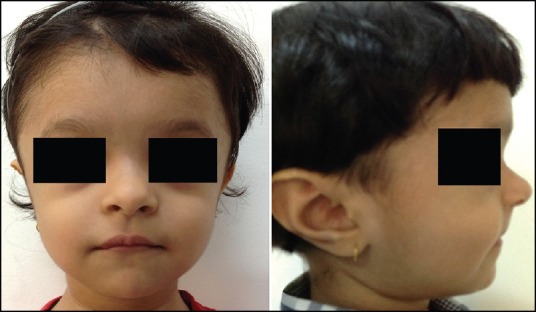
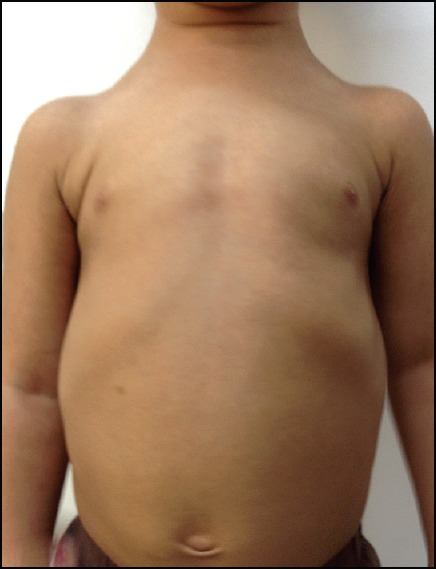
Clinical examination revealed a height of 73 cm (less than 3rd percentile) and weight of 7.8 kg (less than 3rd percentile). The head circumference was of 50 cm (50th percentile). Open anterior fontanelle was observed, with anteroposterior and transverse diameter of 6 and 4 cm, respectively. A high-arched palate, concave nasal bridge, hypertelorism, delayed appearance of primary dentition, mandibular hypoplasia, brachycephalic head and frontal bossing were also noted [Figure 1]. The chest was conical in shape, upper chest was narrow and the shoulders were hypermobile [Figures 2 and 3]. Clinical examination of other systems was normal. The family history was non-contributory. Thyroid function tests and serum vitamin D, serum calcium, serum phosphorus, serum alkaline phosphatase and serum parathyroid hormone levels were within the normal limits.
Figure 1.
A 17-month-old girl presented with below average height and weight gain, subsequently diagnosed as having cleidocranial dysplasia. Frontal (a) and lateral (b) views of face showing brachycephaly, frontal bossing, hypertelorism, depressed nasal bridge and hypoplastic mandible and maxilla
Figure 2.
A 17-month-old girl presented with below average height and weight gain, subsequently diagnosed as having cleidocranial dysplasia. Frontal view of the chest showing conical or bell shaped thorax and narrow upper chest
Figure 3.
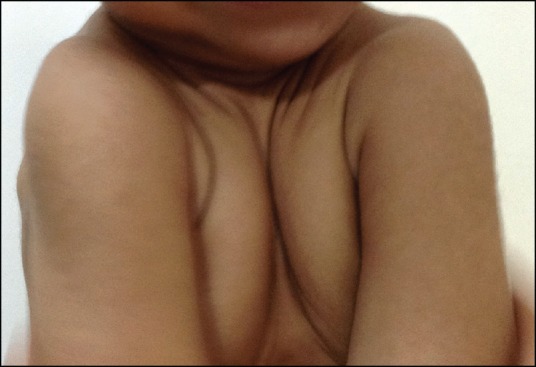
A 17-month-old girl presented with below average height and weight gain, subsequently diagnosed as having cleidocranial dysplasia. Frontal view of the chest showing hypermobile shoulders which can be brought close to each other anteriorly
Radiographic skeletal survey revealed a large anterior fontanelle, multiple wormian bones, increased opacity of the skull base, multiple unerupted primary teeth, absent right clavicle, underdeveloped left clavicle, a narrow bell shaped or conical chest, overall reduced bone density and broad pubic symphysis [Figures 4–6]. Retrospectively her antenatal ultrasound studies were reviewed which revealed hypomineralization of the cranium and wide-open fontanelles and skull sutures. CCD secondary to spontaneous mutation was diagnosed based on the above findings.
Figure 4.
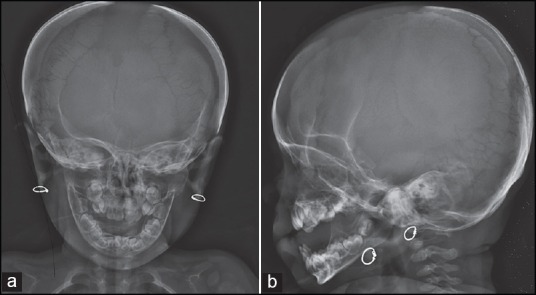
A 17-month-old girl presented with below average height and weight gain, subsequently diagnosed as having cleidocranial dysplasia. Frontal (a) and lateral (b) skull radiographs showing wide open sagittal fontanelles and sutures, multiple wormian bones, brachycephaly, frontal bossing, hypoplastic mandible and maxilla and multiple unerupted primary teeth
Figure 6.
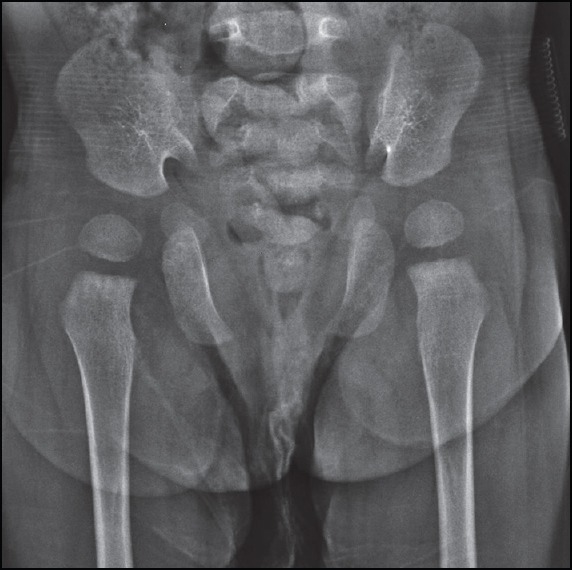
A 17-month-old girl presented with below average height and weight gain, subsequently diagnosed as having cleidocranial dysplasia. Frontal pelvic radiograph showing wide pubic symphysis
Figure 5.
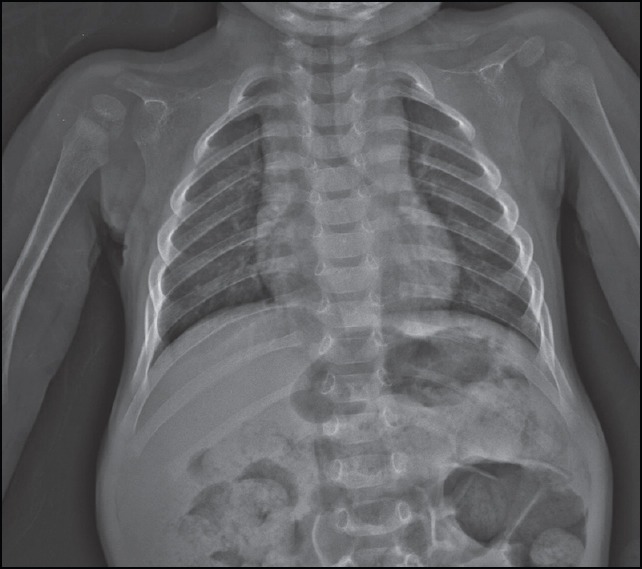
A 17-month-old girl presented with below average height and weight gain, subsequently diagnosed as having cleidocranial dysplasia. Frontal chest radiograph showing absent right and hypoplastic left clavicles, bell shaped thorax and narrow upper chest
This case offered many opportunities to clinicians for the diagnosis of CCD even before her birth (mother's antenatal ultrasound studies), however the diagnosis was missed. A second opportunity, when the child was admitted for three days post birth for transient tachypnea of newborn was also not successful in identifying CCD, although the chest x-rays indicated absent and hypoplastic clavicles. Wide open fontanelles were considered as normal variation as transcranial ultrasound at 1 day of age and thyroid function test were normal. A third opportunity presented at 3 months of age when she was admitted in a pediatric hospital for a week for treatment of high grade fever. High grade fever in association with boggy fontanelles was diagnosed as due to viral meningitis. Magnetic resonance imaging of the brain performed at that time was normal. Subsequent opportunities for CCD diagnosis were also missed when she received proper immunizations and sought medical care for minor ailments.
DISCUSSION
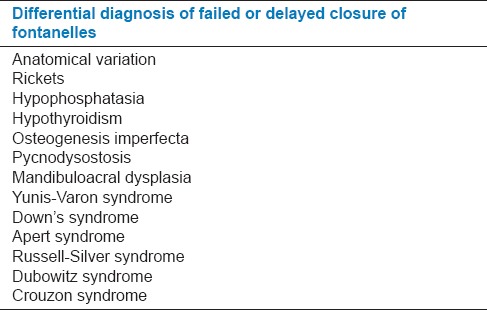
Varying degrees of clavicular hypoplasia is noted in CCD patients,[1,2,3,4,5,6] which needs differential diagnosis[3] from other conditions [Tables 2 and 3]. CCD can be clearly distinguished from these conditions, if, its other features are taken into consideration. Prominently the finding of osteosclerosis differentiates pycnodysostosis from CCD.[3]
Table 2.
Differential diagnosis of absent or hypoplastic clavicles

Table 3.
Differential diagnosis of failed or delayed closure of fontanelles
Heterozygous mutations in RUNX2 gene located on 6p21 chromosome are known to lead to CCD.[1,2,7,8,9] This gene encodes a protein or a transcription factor required for dentinogenesis, differentiation of osteoblasts and maturation of chondrocytes, thus affecting intramembranous as well as endochondral ossification.[1,7,8,9] CCD can be diagnosed prenatally[1] if specific genetic mutation leading to CCD in an affected family member is identified. Prenatal diagnosis is also possible by ultrasound [Table 4].[1] In addition, hypomineralization of the cranium on routine prenatal ultrasound should prompt one to look for hypomineralization of the hypoplastic clavicles or absent clavicles, to diagnose the spontaneous cases of CCD.
Table 4.
Antenatal ultrasound features of CCD

Initial evaluation of CCD should include a complete skeletal survey, audiologic and dental evaluation and consultation with a medical geneticist.[1]
Clinical management of CCD is also multidisciplinary, for instance growth retardation is managed by growth hormone treatment,[1] appropriate dental interventions to manage dental abnormalities, speech therapy and consultation with a psychiatrist or psychologist as required. Subnormal bone density (on dual energy X-ray absorptiometry) is treated with vitamin D and calcium supplements with frequent checks to identify osteoporosis.[1] Infections of the paranasal sinuses and the middle ear need aggressive treatment with antibiotics.[1] Cranioplasty may be considered for permanent closure of large cranial vault defects. Depressed forehead can be surgically corrected and the underdeveloped clavicles can be surgically lengthened for esthetic reasons.[1] Scoliosis, coxa vara, pseudarthrosis of the tibia and other skeletal abnormalities can be managed with the help of orthopedic treatments.
The diagnosis of CCD is often delayed, although, it is possible to diagnose it even during the prenatal stage. As the number of cases due to spontaneous mutation is relatively high, the diagnosis is challenging, especially in milder forms of the disease. Appropriate imaging and clinical examinations are necessary to identify the cases of CCD. Early diagnosis of CCD is important in achieving optimum treatment outcome. CCD should always be kept in mind when evaluating pediatric patients presenting with failure to thrive. Thus, we wish to draw attention to the important features of CCD to aid in its early diagnosis and discuss the current concepts in its clinical management.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Mendoza-Londono R, Lee B. Cleidocranial dysplasia. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong C, Smith RJ, et al., editors. GeneReviews™. Seattle (WA): University of Washington; 1993-2013. [2006 Jan 03; Last updated on 2013 Aug 29]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1513/ [Google Scholar]
- 2.Roberts T, Stephen L, Beighton P. Cleidocranial dysplasia: A review of the dental, historical, and practical implications with an overview of the South African experience. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:46–55. doi: 10.1016/j.oooo.2012.07.435. [DOI] [PubMed] [Google Scholar]
- 3.Karagüzel G, Aktürk FA, Okur E, Gümele HR, Gedik Y, Okten A. Cleidocranial dysplasia: A case report. J Clin Res Pediatr Endocrinol. 2010;2:134–6. doi: 10.4274/jcrpe.v2i3.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broeks I, Veenstra-Knol IE, Kamps AW. A rare presentation of cleidocranial dysplasia. BMJ Case Rep. 2012 doi: 10.1136/bcr-03-2012-6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chopra R, Marwaha M, Chaudhuri P, Bansal K, Chopra S. Hypodontia and delayed dentition as the primary manifestation of cleidocranial dysplasia presenting with a diagnostic dilemma. Case Rep Dent 2012. 2012 doi: 10.1155/2012/262043. 262043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martins RB, de Souza RS, Giovani EM. Cleidocranial dysplasia: Report of six clinical cases. Spec Care Dentist. 2014;34:144–50. doi: 10.1111/scd.12045. [DOI] [PubMed] [Google Scholar]
- 7.Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–9. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 8.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 9.Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–10. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]