

Abstract
Human natural killer (NK) cells play an important role in anti-viral immunity. However, studying their activation kinetics during infection is highly problematic. A clinical trial of a therapeutic virus provided an opportunity to study human NK cell activation in vivo in a controlled manner. Ten colorectal cancer patients with liver metastases received between one and five doses of oncolytic reovirus prior to surgical resection of their tumour. NK cell surface expression of the interferon-inducible molecules CD69 and tetherin peaked 24–48 h post-infection, coincident with a peak of interferon-induced gene expression. The interferon response and NK cell activation were transient, declining by 96 h post-infection. Furthermore, neither NK cell activation nor the interferon response were sustained in patients undergoing multiple rounds of virus treatment. These results show that reovirus modulates human NK cell activity in vivo and suggest that this may contribute to any therapeutic effect of this oncolytic virus. Detection of a single, transient peak of activation, despite multiple treatment rounds, has implications for the design of reovirus-based therapy. Furthermore, our results suggest the existence of a post-infection refractory period when the interferon response and NK cell activation are blunted. This refractory period has been observed previously in animal models and may underlie the enhanced susceptibility to secondary infections that is seen following viral infection.
Keywords: human infection, innate immunity, interferon response, natural killer cells, viral infection
Introduction
Infection induces the rapid activation of innate immunity. Innate immune activation serves two purposes: it limits pathogen replication while the clonal selection of B and T cells occurs, and it favours the development of the appropriate adaptive response 1. Current information on the kinetics of innate immune activation stems largely from animal models, yet there is a need to define these processes in humans; such knowledge promises to enhance the efficacy of vaccines and other immunotherapeutic strategies. However, studying the early stages of infection in humans presents both logistical and ethical problems.
Natural killer (NK) cells are important in the innate immune response to infected cells and to tumours 2,3. Early animal studies revealed that NK cell activation occurred within 2–3 days of viral infection 4,5 and NK cells are known to be critical in anti-viral immunity 3,6–9. Activated NK cells destroy infected cells directly and produce cytokines, such as interferon (IFN)-γ, that favour the development of a cytotoxic T cell response 2,10. Rare human NK cell deficiencies are associated with increased susceptibility to viral infection, revealing the importance of human NK cells in anti-viral immunity 11,12. However, analysing the time-line of human NK cell activation in response to viral infection in vivo remains difficult. Virus-infected patients show evidence of NK cell activation compared to uninfected controls, but while vaccination allows controlled studies to be performed, the analysis of pre-infection status and very early post-infection events remains challenging 3,13–18. Hence, our view of the early stages of NK cell activation is based largely on studies performed using model species.
Reovirus, a non-enveloped dsRNA virus, is pathogenic in mice and induces a type I IFN (IFN-I) response 19. While it is not a significant human pathogen, reovirus has the interesting property of preferentially killing tumour cells, leading to its evaluation as a therapeutic agent 20. The anti-cancer effects of reovirus and other oncolytic viruses appear to be linked to a twofold mode of action, namely the direct killing of tumour cells and the induction of innate and adaptive anti-tumour immunity 21–24. Intravenous delivery of reovirus into patients is associated with its rapid loss from the circulation; in eight out of ten treated patients the virus was undetectable in the bloodstream after 1 h post-infection 25. Despite the presence of neutralizing antibodies, reovirus reached the tumour and was associated with tumour cell apoptosis 25. This same trial allowed us to study infection-induced human NK cell activation under controlled conditions. Our results define the kinetics of human NK cell activation in response to viral infection in vivo.
Materials and methods
Ethical approval and the clinical trial
This study was undertaken following institutional and national ethical and regulatory approval. Patients were enrolled into the trial and provided blood samples following informed consent. The patient group and the trial are described elsewhere 25.
Antibodies
The following antibodies were used in this study; CD69 (clone FN50); CD56 (clone B159), CD16 (clone 3G8), CD3 (clone SK7), CD107a (clone H4A3), NKG2D (clone 1D11), DNAM-1 (clone DX11), NKp44 (clone p44-8·1), NKp46 (clone 9E2), CD158a (HP-3E4) and CD158b (CH-L), all from BD Biosciences (Oxford, UK); CD158e (clone DX9) from Miltenyi Biotec (Bisley, UK) and tetherin/CD317 (clone 26F8) from eBiosciences (Hatfield, UK). For the IFN-I blocking experiments, we used a cocktail of anti-IFN-α (clone MMHA-2), anti-IFN-β (clone 76703·111) and rabbit anti-IFN-α anti-serum, all from PBL Assay Science (Piscataway, NJ, USA).
Flow cytometry and gene expression analysis
Cell surface expression of CD69 and tetherin was determined using flow cytometry on either purified NK cells (P1-P4) or by gating on CD56+CD3– NK cells in peripheral blood mononuclear cells (PBMC; P5-P10). NK cells were purified by indirect magnetic immunoselection (Miltenyi Biotech). Flow cytometry was performed using a Becton Dickinson (BD) LSRII or BD FACSCalibur flow cytometer using BD FACSDiva software and BD CellQuest Pro software, respectively. For gene expression studies, mRNA was converted to cDNA (using random hexamer priming) and expression of interferon-induced protein with tetratricopeptide repeats 1 (IFIT1), interferon-induced protein 44-like (IFI44L), 18S RNA or ABL proto-oncogene 1 (ABL1) was analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR) using Taqman reagents from Applied Biosystems. Data were normalized to either 18S RNA or ABL1 mRNA (as indicated) and the fold-change induced during infection calculated using the ΔΔCt method.
In vitro studies
PBMCs from healthy donors were co-incubated with reovirus (Reolysin®; Oncolytics Biotech Inc., Calgary, AB, Canada) at a multiplicity of infection (MOI) of 0·2–1 in the presence of either the anti-human IFN-I antibody cocktail or matched serum/immunoglobulin (Ig)G controls. Degranulation assays were performed 48 h post-infection using the K562 target cell line in the presence of GolgiStop (BD Biosciences) and the anti-CD107a antibody 26. For analysis of isolated NK cells and fractionation of PBMC, the NK cells were purified using indirect magnetic immunoselection reagents (Miltenyi Biotec) and the NK cell-depleted PBMC (PBMCΔNK) were eluted from the column.
Results
Ten patients (P1–10; aged 50–74 years) with colorectal cancer liver metastases were enrolled into a clinical end-point trial to assess the delivery of reovirus to the metastatic tumour 25. Each patient received between one and five intravenous infusions of 1010 units of reovirus prior to planned surgical resection of their tumour. Seven of the 10 patients received reovirus daily for 5 days, P7 received four doses, P8 a single dose and P1 received three doses with an altered timing (Fig. 1a). Six of the 10 patients experienced fever and several experienced flu-like symptoms during treatment, consistent with viral infection 25.
Figure 1.

Human natural killer (NK) cell activation by reovirus in vivo. (a) Schedule of patient infection and sampling during the reovirus clinical trial. Four different schedules were employed. Patients (P) 2, 3, 4, 5, 6, 9 and 10 were treated according to the schedule shown at the top. The variations for P7, P8 and P1 are shown below. The time of infection is shown above the horizontal line (grey vertical arrow) and the time of blood sampling below (black vertical arrow). Times of blood sampling (time post-infection) are shown in hours (0, 1, 24, 48, 72, 96), immediately prior to surgery (Sgy) and in months (1 Mo, 3 Mo). The indicated variations were made according to clinical parameters which, together with patient details and timing of the surgery, have been reported previously 25. Each infusion consisted of 1010 units of reovirus, with one unit defined as the dose of virus required to kill 50% of cultured cells in vitro (the tissue culture infective dose 50%, or TCID50). (b) NK cell surface expression of CD69 in selected patients representing key variations to the schedule and in a healthy control (HC); P3 represents the patients with repeated doses, P8 had a single dose and P1 underwent treatment with altered timing to the other patients. The time post-infection is shown with 0 h immediately prior to the first infusion. The values in grey indicate the percentage of CD69 expressing NK cells compared to isotype control stains that were performed for all analyses (not shown). (c) Summary of CD69 expression in patients P2–10 and in healthy control (HC) donors (n = 7). The P-values were calculated using the Mann–Whitney U-test; ***P < 0·001. P1 data are omitted from this analysis because of the altered timing of therapy.
Blood samples taken before and during treatment were used to analyse the NK cell phenotype. Infection induced rapid expression of the lymphocyte activation marker CD69 on the NK cells, peaking 48-h post-infection (Fig. 1b,c). A single dose of reovirus was sufficient to induce this activation, as shown in P8, who received just one dose, and in P1, in whom more than 60% of peripheral blood NK cells were CD69+ before the second dose was administered (Fig. 1b). With the exception of P1 and P8, all patients received two doses of virus before the 48-h sample (when NK cell activation peaked) and a further two doses between the 48- and 96-h samples (Fig. 1a). However, NK cell activation declined after 48 h in all patients, suggesting that NK cells were refractory to further stimulation within this period.
Reovirus dsRNA, and indeed other viral nucleic acids, induce IFN-I responses in animals via pathogen-associated molecular pattern receptor recognition. The cytoplasmic RNA sensor retinoic acid-inducible gene 1 (RIG-I) recognizes the 5′-diphosphate present on reovirus dsRNA and induces IFN gene expression and it is long established that IFN treatment activates human NK cells in vivo 27–30. CD69 is induced via IFN-I responses and we have shown previously that reovirus treatment of PBMC in vitro induces CD69 expression by NK cells in an IFN-I-dependent manner 23. Expression of the IFN-stimulated genes (ISGs) IFIT1 and IFI44L in the reovirus-treated patients showed similar kinetics to the induction of NK cell CD69 expression, peaking 48 h post-infection (Fig. 2a). Like CD69, expression of the ISGs was transient and declined after this initial post-infection peak. Collectively, these results are consistent with the virus-mediated induction of an IFN-I response in vivo and the IFN-I dependent activation of human NK cells within 24–48 h post-infection.
Figure 2.

Interferon (IFN) responses and changes in the natural killer (NK) cell surface phenotype in vivo. (a) Expression of the IFN-stimulated genes (ISGs) IFIT1 and IFI44L following reovirus treatment, as determined by quantitative reverse transcription–polymerase chain reaction (RT–PCR). The analyses were performed using NK cells purified from P5 or from whole peripheral blood mononuclear cells (PBMC) isolated from P7. The data show expression in the treated patient (black lines and squares) and in an uninfected control (grey lines and diamonds). Expression was calculated as the fold-change in expression compared to the pre-infection (0 h) time-point (assigned a value of 1 unit of expression). (b) Expression of NK cell receptors in patients at pre-infection (0 h) time-point (–) and 48 h post-infection (+). Data shows the change in mean fluorescence intensity (MFI) with the expression at the 0 h time-point assigned a value of 1. The number of patients in each group is indicated (n). Data (where n > 2) was analysed using the Student's t-test and statistically significant differences are shown; *P < 0·05. (c) Flow cytometric analysis of expression of patient NK cell surface molecules throughout the treatment course. The grey dotted line shows the approximate position of the median fluorescence intensity of the signal at the pre-infection time-point. The plots are from individual patients (shown) and are representative of data collected across the treatment group; only tetherin (and CD69, Fig. 1) show substantial alterations in expression. Tetherin expression was not determined at the 3-month (3Mo) time-point.
Tetherin is an IFN-I inducible anti-viral restriction factor, and its expression at the cell surface provides a convenient marker for IFN-I responses during viral infection 31–33. Tetherin was expressed constitutively at the NK cell surface and expression was enhanced significantly following reovirus treatment in vivo, exhibiting similar induction kinetics to CD69 and the ISGs (Fig. 2b,c). Human NK cells express several activating receptors that have been implicated in the detection of virus-infected cells, including NKG2D (CD314), DNAM-1 (CD226), NKp30 (CD337) and NKp44 (CD336) 2,3. Expression of these molecules was not altered significantly on patient NK cells at the peak of the IFN-I response and did not show further alterations in expression during the course of treatment (Fig. 2b,c).
We then performed experiments to analyse the response to reovirus in vitro. We treated PBMC with reovirus in the presence or absence of antibodies that block the IFN-I response. Treated PBMC were then co-cultured with tumour target cells and the tumour-mediated degranulation of the NK cells in the PBMC analysed using flow cytometry 26. This demonstrated that reovirus treatment of PBMC resulted in the IFN-I-dependent, functional activation of the NK cells (Fig. 3a), consistent with previously published data 23. We then treated PBMC with reovirus for 48 h, purified the NK cells (using immunomagnetic selection) and analysed the expression of IFIT1 mRNA in the NK cell population and in the PBMC depleted of NK cells (PBMCΔNK). Both the NK cells and the PBMCΔNK fraction demonstrated substantial induction of IFIT1 mRNA (Fig. 3b). Furthermore, flow cytometry of the reovirus-treated PBMC showed the induction of CD69 and tetherin expression on the NK cell surface (Fig. 3c), as we observed in the reovirus-treated patients (Figs 1 and 2). Similar to the situation observed in vivo, the in vitro reovirus treatment did not result in substantial changes in the cell surface expression of NKG2D, DNAM-1, NKp30, NKp44 or NKp46 on NK cells (Fig. 3c). We observed a significant increase in NKp46 expression in vitro, but this only represented an approximately 1·4-fold increase compared to an approximately 5-fold increase in tetherin expression (Fig. 3c). Cytokines such as IL-2 and IL-15 increase the cell surface expression of NKG2D and DNAM-1 in vitro 26 and a comparison of IL-15 and IFN-I stimulation of purified NK cells showed that IL-15 induced expression of NKG2D, DNAM-1, CD69 and tetherin, whereas IFN-I induced only CD69 and tetherin, similar to the effects of reovirus treatment we observed in vitro and in vivo (Fig. 3d). In conclusion, reovirus treatment, both in vivo and in vitro, was associated with the induction of CD69 and tetherin expression at the NK cell surface, but with little change in the expression of other NK cell activation receptors analysed. The induction of CD69 and tetherin in vivo coincided with the peak of IFN-I-induced gene expression and both CD69 and tetherin were IFN-I-inducible in NK cells in vitro.
Figure 3.

Analysis of natural killer (NK) cell responses to reovirus treatment in vitro. (a) Reovirus and interferon (IFN)-I-mediated activation of NK cell granule exocytosis. The left-hand panel shows the display of cell surface CD107 (gated on CD56+CD3– NK cells within peripheral blood mononuclear cells (PBMC) in the presence or absence of K562 target cells. The experiment was performed using PBMC from healthy donors without further treatment (untreated), in the presence of 0·2 multiplicity of infection (MOI) reovirus (+virus) and in the presence of reovirus and an anti-IFN antibody or a control antibody (cAb). The percentage values indicate the proportion of CD107+ NK cells for each treatment. Statistical analysis was performed between the indicated pairs of treatments using Student's t-test; *P < 0·05. Limitations in the size of samples available from the clinical trial made cytotoxicity assays from the in vivo study difficult to perform. However, of three patients analysed, one showed increased cytotoxic activity 48 h post-infection (data not shown). (b) Expression of IFIT1 mRNA in NK cells and NK cell-depleted PBMC (PBMCΔNK) with (+) and without (–) reovirus treatment in vitro. Whole PBMC (from healthy donors) were treated with reovirus (at an MOI of 1), cultured for 48 h and fractionated into NK cells and NK-depleted PBMC (using magnetic indirect selection of NK cells). Reverse transcription–polymerase chain reaction (RT–PCR) for IFIT1 and ABL1 was performed using mRNA isolated from these fractions; IFIT1 expression was normalized to ABL1 mRNA and the fold-change induced during infection calculated via the ΔΔCt, with the untreated cells (–) assigned an expression value of 1 unit. The data shown are from two different donors. (c) The expression of NK cell surface markers ± reovirus treatment in vitro, analysed 48 h post-infection. The left-hand panel indicates the percentage of CD69 expressing NK cells in the PBMC population, the right-hand panel indicates the change in mean fluorescence intensity of the indicated markers. Statistical analysis was performed using Student's t-test; *P < 0·05; **P < 0·01; ***P < 0·001. (d) Expression of NK cell surface molecules following cytokine treatment in vitro. Purified NK cells (from healthy donors) were treated with 20 ng/ml of interleukin (IL)-15 or 100 IU interferon (IFN)-I for 48 h and expression of the indicated markers was analysed by flow cytometry. The dotted grey line shows the approximate position of the mean fluorescence intensity of the isotype control for CD69 and, for other markers, their expression in untreated cells.
These results, showing that reovirus treatment modulates NK cell activation in the early post-infection period, are consistent with a role for NK cells in controlling viral infection while adaptive immunity is developing. Recently, NK cells have been shown to have a more durable role in the immune response. The identification of so-called memory NK cells and the ability of activated NK cells to limit T cell responses have revealed that NK cell activity persists beyond this initial wave of activation 14,34–36. Interestingly, we found a significant increase in the absolute numbers of NK cells in the pre-surgery samples; in two patients (P9 and P7) we detected a 6-fold and a 13-fold increase, respectively (Fig. 4). Expression of killer-cell immunoglobulin-like receptor (KIR) molecules is clonal and maintained following cell division and KIRs thus provide markers for analysis of putative clonal expansions. Cell surface expression of CD158a, CD158b and CD158e antigens identified eight distinct populations constituting between approximately 2% to approximately 50% of total NK cells (Supporting information, Table S1). However, we did not detect clonal expansions that could account for the changes in absolute numbers seen between the 96 h and pre-surgery samples; this suggests that the increase in the absolute numbers of NK cells was due to polyclonal expansion.
Figure 4.
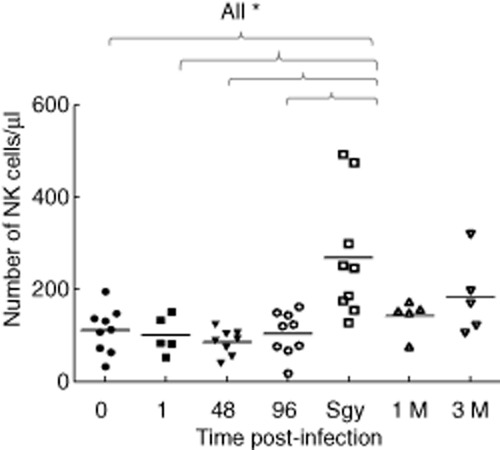
Absolute numbers of natural killer (NK) cells (cells per μl) in patients during the treatment course. The values at each time-point were compared to the pretreatment 0-h time-point using the Mann–Whitney U-test, only the statistically significant differences are shown; *P < 0·05. The normal range for the absolute number of NK cells in healthy donors is ∼90–600 cells per μl.
Discussion
The use of a therapeutic virus within the context of a clinical trial has allowed us to analyse the kinetics of human NK cell activation in response to viral infection under controlled conditions. Our results demonstrate that human viral infection results in the rapid and transient activation of NK cells in the bloodstream and that this activation, which occurred within 24–48 h post-infection, was associated with an IFN-I response. Blood samples from healthy volunteers given a poly IC-like molecule (a mimic of dsRNA) exhibited similar kinetics of ISG induction and other gene expression responses that were consistent with the activation of innate immune responses, including those involving NK cells 37. This work, together with the results presented here, are consistent with early studies using mouse models in which viral infection resulted in IFN production and the induction of NK cell activation within 2–3 days of infection 4,5,38. In the absence of an IFN-I response, viral pathology is enhanced and this is associated with a reduction in infection-induced NK cell activity 38–40. However, the effect of IFN-I on NK cells is largely indirect, with IFN-I inducing IL-15 production and expression of the IL-15 receptor on NK cells; IL-15 then acts upon NK cells 40,41. Indeed, several viruses (including reovirus) induce IL-15 mRNA in PBMC and activate NK cells in an IL-15-dependent manner 42. The early in vivo activation of NK cells in response to reovirus treatment is highly suggestive of IFN-I- and IL-15-mediated events. However, the contribution of other NK cell-activating cytokines, such as IL-12 and IL-18 or indeed IL-2 (produced predominantly by activated T cells during adaptive immunity), cannot be discounted.
Expression of both CD69 and tetherin is IFN-I-inducible. Tetherin was identified originally as an IFN-I-inducible anti-viral restriction molecule with the ability to prevent release of human immunodeficiency virus (HIV) 31. This activity extends to a number of enveloped viruses, and IFN-I induction of tetherin allows it to act as a broad defence against viral spread. Tetherin provides a convenient cell surface marker of an IFN-I-induced anti-viral response 32,33. However, our in vitro data show that both tetherin and CD69 are inducible in NK cells by IFN-I and IL-15. Others have shown that several cytokines can induce tetherin and that its induction can precede IFN-I responses 43–45; it remains possible that other cytokines or signals induce CD69 and tetherin in response to reovirus infection. The actual role of CD69 in NK cell activity is poorly defined. Activated mouse NK cells traffic from the periphery to the lymph nodes, where NK cell-derived IFN-γ helps to promote cytotoxic T cell responses 10. In mouse B and T lymphocytes, IFN-I induction of CD69 decreases the activity of the sphingosine-1-phosphate receptor 1 (S1P1), thereby inhibiting egress from mouse secondary lymphoid tissue (SLT) 46. It is possible that human NK cell expression of CD69 causes similar effects, allowing NK cells that traffic from the blood to other tissues (such as the SLT) to remain there. However, while CD69 inhibits S1P1 responses in B and T lymphocytes, mouse and human NK cells express S1P5 preferentially, and this receptor is not inhibited by CD69 47,48. Thus, the role of NK cell CD69 remains unclear. Reovirus-activated NK cells may traffic to the liver (the site of the colorectal metastases in these patients), where they would be able to attack the tumour directly. Whether reovirus-activated NK cells participate directly in tumour lysis or whether these activated NK cells mediate other pathways of anti-tumour immunity via cytokine secretion, for example, remains unknown. A limitation of our study is the inability to analyse NK activation and trafficking beyond the peripheral blood. However, the trial established that reovirus reaches the tumour 25, suggesting that liver-resident NK cells might be activated. Indeed, we have shown previously that reovirus can activate liver-derived NK cells in vitro and enhance their response to colorectal tumour cell lines 23.
Nine of the 10 patients in the trial received multiple doses of reovirus, yet CD69, tetherin, IFI44L and IFIT1 all exhibited just a single peak of expression approximately 48 h after the first dose. For example, all patients except P1 and P8 received two further doses of virus between the 48- and 96-h time-points, yet we did not observe a second peak of activation (or IFN-I response) and, in all cases, responses declined in all patients after 48 h post-infection. Furthermore, P1 and P8 revealed that a single dose of virus gave a similar magnitude of response to those patients receiving multiple doses; P1 also showed that a strong response could be detected within 24 h of treatment. The results suggest that the initial IFN-I response (and NK cell activation) was followed by a refractory period during which the patients were unable to respond to further exposure to reovirus. Most adults have been exposed to reovirus and all patients in the trial had neutralizing antibodies that increased in titre around days 3–5 post-infection 25. Whether this boost in antibody titre blocks the IFN-I response to subsequent doses of reovirus seems unlikely, but nevertheless remains unclear. An intriguing alternative is that the refractory period is related to that observed in mouse viral infection models 49,50; the initial viral infection induces an IFN response which is followed by a refractory period in which further IFN responses to unrelated pathogens are blunted. This refractory period has been suggested to contribute to the enhanced susceptibility to unrelated, secondary infections that can follow viral infection. In mice, the mechanisms underlying this refractory period include a reduced capacity of plasmacytoid dendritic cells (pDC) to produce IFN 50 and the induction of OASL1, a negative regulator of IFN production 51. However, other homeostatic control mechanisms that halt responses, including molecules that target IFN production and downstream signalling pathways, may also influence responses 52–56. Interestingly, tetherin was proposed to act as a feedback inhibitor of IFN production by engaging the receptor ILT7 on pDC 57. However, while ILT7 ligation was confirmed to halt IFN production, a role for tetherin in this process was subsequently called into question 58. To the best of our knowledge, the data presented here are the first demonstration of this refractory period in humans. However, the constraints of working within a clinical trial make these conclusions speculative. Furthermore, applying these findings to the general population also warrants caution, because all the patients in the trial have metastatic cancer, associated presumably with alterations in immune status. Notwithstanding the limitations of our study, the clinical importance of opportunistic infections following acute viral infection (e.g. with influenza) or chronic infections (such as HIV) cannot be understated. From a cancer therapy perspective, our results indicate that the scheduling of oncolytic viruses will require optimization, if IFN-I and NK cell responses are to be maximized.
While profound effects were observed in the first 48 h post-infection, we also observed a later change in NK cells, namely a significant increase in the absolute numbers of NK cells. In HIV infection there is an expansion of particular KIR expressing cells 16. We did not detect particular clonal expansions using antibodies that detect CD158 family molecules that include KIR2DL1, KIR2DL3 and KIR3DL1 (as well as related short-form KIRs). This panel identified approximately 30–50% of total NK cells, consistent with more detailed KIR phenotyping where approximately 50% of NK cells lack KIR expression 59. However, we did not analyse NKG2C-expressing cells; this population expands in both cytomegalovirus and hantavirus infection and for the latter, expanded cells expressed at least one KIR molecule against a self-major histocompatibility complex (MHC) class I molecule, indicating that the expanded NK cells were functionally licensed 14,60. The KIR and MHC haplotype of the patients within our study was unavailable to us, and we do not know whether the expansions we detected were confined to licensed populations. The significance of this relatively late post-infection phenotype is unclear. There are emerging data that suggest a role for NK cells beyond the immediate post-infection stage 14. The significance of these more durable NK cell responses, for example whether they represent the formation of a memory-like NK cell population 34 or a role for NK cells in the cessation of T cell responses, currently remains unclear 35,36.
The ability of reovirus to induce NK cell activation is likely to contribute to its oncolytic activity in vivo. However, effective oncolytic virus treatment will depend upon achieving the correct balance of anti-viral and anti-tumour activity 20,61. For example, depletion of NK cells limits the efficacy of both vesicular stomatitis virus and reovirus virus treatment, consistent with stimulation of the anti-tumour effector function of this population 62,63. However, the anti-viral activity of NK cells has been shown to impede the action of oncolytic herpes simplex virus against glioblastoma 64. In the patients treated here, replication-competent reovirus was recovered from the colorectal liver metastases but not from surrounding healthy tissue, suggesting effective targeting of the virus to the tumour 25. Furthermore, ex vivo studies show that reovirus activates NK cells from the liver and enhances their cytotoxic activity towards colorectal cancer cell lines 23. However, viral infection in the liver can induce potent immunosuppressive activity [via IL-10 and transforming growth factor (TGF)-β] that limits NK cell production of IFN-γ; similar effects would be expected to blunt oncolytic virus-induced NK cell activation and anti-tumour immunity 65.
In summary, the use of a therapeutic virus in a clinical trial has enabled us to study the kinetics of NK cell activation in response to viral infection. The increasing use of therapeutic viruses promises to provide new opportunities to study the activation and resolution of the human immune response in vivo and provide key information that is currently inferred from studies performed in other species.
Acknowledgments
We are grateful to Josie Meade and Debbie Beirne for their help and encouragement with this work and to Stephen Waggoner for helpful discussion of the early literature on NK cell activation kinetics. Work in our laboratories is funded by Cancer Research UK, the Medical Research Foundation, Yorkshire Cancer Research and the Leeds Experimental Cancer Medicine Centre.
Author contributions
R. A. A., R. D., K. J. S., R. S. M. M., G. J. T. and A. A. M. designed and implemented the clinical trial and sample collection. M. C. provided clinical grade virus. T. D. H., A. A. M. and G. P. C. designed the experimental study. Y. M. E. S., T. D. H. and L. F. W. performed the bulk of the experimental work, with additional contributions from E. V. I. B., E. B. W., S. L. P. and G. B. S. G. P. C., L. F. W., E. B.W., Y. M. E. S. and T. D. H. analysed the data and G. P. C., L. F. W., Y. M. E. S., T. D. H. and E. B. W. wrote the paper.
Disclosure
M. C. is an employee of Oncolytics Biotech Inc. and an author on a patent for the clinical use of reovirus. A. A. M. has received commercial research grants from Oncolytics Biotech Inc. The other authors declare that they have no competing interests.
Supporting Information
Table S1. Clonal analysis of natural killer (NK) cell populations.
References
- Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50–53. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- Vidal SM, Khakoo SI, Biron CA. Natural killer cell responses during viral infections: flexibility and conditioning of innate immunity by experience. Curr Opin Virol. 2011;1:497–512. doi: 10.1016/j.coviro.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh RM., Jr Cytotoxic cells induced during lymphocytic choriomeningitis virus infection of mice. I. Characterization of natural killer cell induction. J Exp Med. 1978;148:163–181. doi: 10.1084/jem.148.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh RM, Jr, Zinkernagel RM. Heterospecific cytotoxic cell activity induced during the first three days of acute lymphocytic choriomeningitis virus infection in mice. Nature. 1977;268:646–648. doi: 10.1038/268646a0. [DOI] [PubMed] [Google Scholar]
- Bukowski JF, Woda BA, Habu S, Okumura K, Welsh RM. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J Immunol. 1983;131:1531–1538. [PubMed] [Google Scholar]
- Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- Gazit R, Gruda R, Elboim M, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7:517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]
- Brown MG, Dokun AO, Heusel JW, et al. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- Martin-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- Orange JS. Unraveling human natural killer cell deficiency. J Clin Invest. 2012;122:798–801. doi: 10.1172/JCI62620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. 2013;132:515–525. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadei B, Urbani S, Cazaly A, et al. Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology. 2010;138:1536–1545. doi: 10.1053/j.gastro.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkstrom NK, Lindgren T, Stoltz M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med. 2011;208:13–21. doi: 10.1084/jem.20100762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves PC, Matos DC, Marcovistz R, Galler R. TLR expression and NK cell activation after human yellow fever vaccination. Vaccine. 2009;27:5543–5549. doi: 10.1016/j.vaccine.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Alter G, Rihn S, Walter K, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol. 2009;83:6798–6805. doi: 10.1128/JVI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost S, Quillay H, Reardon J, et al. Changes in cytokine levels and NK cell activation associated with influenza. PLOS ONE. 2011;6:e25060. doi: 10.1371/journal.pone.0025060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis FA, Meager A, Beare AS, et al. Interferon induction and increased natural killer-cell activity in influenza infections in man. Lancet. 1981;2:891–893. doi: 10.1016/s0140-6736(81)91390-8. [DOI] [PubMed] [Google Scholar]
- Sherry B. Rotavirus and reovirus modulation of the interferon response. J Interferon Cytokine Res. 2009;29:559–567. doi: 10.1089/jir.2009.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Exp Rev Anticancer Ther. 2008;8:1581–1588. doi: 10.1586/14737140.8.10.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- Errington F, White CL, Twigger KR, et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008;15:1257–1270. doi: 10.1038/gt.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair RA, Scott KJ, Fraser S, et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int J Cancer. 2013;132:2327–2338. doi: 10.1002/ijc.27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Ilett EJ, Errington F, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15:4374–4381. doi: 10.1158/1078-0432.CCR-09-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4:138ra77. doi: 10.1126/scitranslmed.3003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade JL, Wilson EB, Holmes TD, et al. Proteolytic activation of the cytotoxic phenotype during human NK cell development. J Immunol. 2009;183:803–813. doi: 10.4049/jimmunol.0713829. [DOI] [PubMed] [Google Scholar]
- Goubau D, Schlee M, Deddouche S, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddlestone JR, Merigan TC, Jr, Oldstone MB. Induction and kinetics of natural killer cells in humans following interferon therapy. Nature. 1979;282:417–419. doi: 10.1038/282417a0. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- Homann S, Smith D, Little S, Richman D, Guatelli J. Upregulation of BST-2/tetherin by HIV infection in vivo. J Virol. 2011;85:10659–10668. doi: 10.1128/JVI.05524-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmberg AR, Neidermyer WJ, Jr, Breed MW, et al. Tetherin upregulation in simian immunodeficiency virus-infected macaques. J Virol. 2013;87:13917–13921. doi: 10.1128/JVI.01757-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Lopez-Verges S, Kim CC, DeRisi JL, Lanier LL. NK cells and immune ‘memory’. J Immunol. 2011;186:1891–1897. doi: 10.4049/jimmunol.1003035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang PA, Lang KS, Xu HC, et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl Acad Sci USA. 2012;109:1210–1215. doi: 10.1073/pnas.1118834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2012;481:394–398. doi: 10.1038/nature10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey M, Lefebvre F, Filali-Mouhim A, et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. 2011;208:2357–2366. doi: 10.1084/jem.20111171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS, Biron CA. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156:4746–4756. [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Salazar-Mather TP, Dalod MY, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol. 2002;169:4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- Baranek T, Manh TP, Alexandre Y, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe. 2012;2:571–584. doi: 10.1016/j.chom.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Fawaz LM, Sharif-Askari E, Menezes J. Up-regulation of NK cytotoxic activity via IL-15 induction by different viruses: a comparative study. J Immunol. 1999;163:4473–4480. [PubMed] [Google Scholar]
- Guzzo C, Jung M, Graveline A, Banfield BW, Gee K. IL-27 increases BST-2 expression in human monocytes and T cells independently of type I IFN. Sci Rep. 2012;2:974. doi: 10.1038/srep00974. doi: 10.1038/srep00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos Jimenez V, Booiman T, de Taeye SW, et al. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci Rep. 2012;2:763. doi: 10.1038/srep00763. doi: 10.1038/srep00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bego MG, Mercier J, Cohen EA. Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J Virol. 2012;86:3513–3527. doi: 10.1128/JVI.06971-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- Jenne CN, Enders A, Rivera R, et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206:2469–2481. doi: 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzer T, Chiossone L, Chaix J, et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol. 2007;8:1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- Alsharifi M, Regner M, Blanden R, et al. Exhaustion of type I interferon response following an acute viral infection. J Immunol. 2006;177:3235–3241. doi: 10.4049/jimmunol.177.5.3235. [DOI] [PubMed] [Google Scholar]
- Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe. 2008;4:374–386. doi: 10.1016/j.chom.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Park CH, Jeong YH, Kim YJ, Ha SJ. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8(+) T-cell exhaustion. PLOS Pathog. 2013;9:e1003478. doi: 10.1371/journal.ppat.1003478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KH, Macdonald A. Putting the brakes on the anti-viral response: negative regulators of type I interferon (IFN) production. Microbes Infect. 2011;13:291–302. doi: 10.1016/j.micinf.2010.12.007. [DOI] [PubMed] [Google Scholar]
- van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5:a009076. doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Cao W, Bover L, Cho M, et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J Exp Med. 2009;206:1603–1614. doi: 10.1084/jem.20090547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavano B, Galao RP, Graham DR, et al. Ig-like transcript 7, but not bone marrow stromal cell antigen 2 (also known as HM1·24, tetherin, or CD317), modulates plasmacytoid dendritic cell function in primary human blood leukocytes. J Immunol. 2013;190:2622–2630. doi: 10.4049/jimmunol.1202391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkstrom NK, Svensson A, Malmberg KJ, Eriksson K, Ljunggren HG. Characterization of natural killer cell phenotype and function during recurrent human HSV-2 infection. PLOS ONE. 2011;6:e27664. doi: 10.1371/journal.pone.0027664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Verges S, Milush JM, Schwartz BS, et al. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci USA. 2011;108:14725–14732. doi: 10.1073/pnas.1110900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Breckenridge CA, Yu J, Kaur B, Caligiuri MA, Chiocca EA. Deciphering the multifaceted relationship between oncolytic viruses and natural killer cells. Adv Virol. 2012;2012:702839. doi: 10.1155/2012/702839. doi: 10.1155/2012/702839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz RM, Galivo F, Kottke T, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- Prestwich RJ, Errington F, Diaz RM, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20:1119–1132. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Breckenridge CA, Yu J, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppa D, Micco L, Javaid A, et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLOS Pathog. 2010;6:e1001227. doi: 10.1371/journal.ppat.1001227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clonal analysis of natural killer (NK) cell populations.