

Abstract
Autoimmunity may contribute to the pathogenesis of chronic obstructive pulmonary disease (COPD). Studies have identified disease-specific autoantibodies (DSAAbs) in COPD patients, but natural autoantibodies (NAAbs) may also play a role. Previous studies have concentrated on circulating autoantibodies, but lung-associated autoantibodies may be most important. Our aim was to investigate NAAbs and DSAAbs in the circulation and lungs of COPD smoking (CS) patients compared to smokers (S) without airway obstruction and subjects who have never smoked (NS). Immunoglobulin (Ig)G antibodies that bind to lung tissue components were significantly lower in the circulation of CS patients than NS (with intermediate levels in S), as detected by enzyme-linked immunosorbent assay (ELISA). The levels of antibodies to collagen-1 (the major lung collagen) detected by ELISA were also reduced significantly in CS patients’ sera compared to NS. The detection of these antibodies in NS subjects indicates that they are NAAbs. The occurrence of DSAAbs in some CS patients and S subjects was indicated by high levels of serum IgG antibodies to cytokeratin-18 and collagen-5; furthermore, antibodies to collagen-5 eluted from homogenized lung tissue exposed to low pH (0·1 M glycine, pH 2·8) were raised significantly in CS compared to S and NS. Thus, this study supports a role in COPD for both NAAbs and DSAAbs.
Keywords: autoantibodies, autoimmunity, lung
Introduction
Chronic obstructive pulmonary disease (COPD), involving chronic bronchitis and emphysema, is characterized by progressive airflow limitation that is not fully reversible; it is a major and increasing cause of morbidity and mortality worldwide 1,2. Although tobacco smoke is the major aetiological factor, only 15–20% of smokers develop clinically significant COPD but, once disease is established, it is progressive, regardless of smoking cessation.
Immune mechanisms are central to the pathogenesis of COPD, with contributions of both innate and adaptive immunity 3–14, and the possible involvement of autoimmunity in COPD has been increasingly recognized 15. An association of lung pathology with a variety of well-characterized autoimmune diseases is evident 16,17, and cigarette smoking is a risk factor for several recognized autoimmune diseases 18. Antibodies specific to a variety of autoantigens have been described in pulmonary fibrosis 19–21. Animal experiments demonstrate that COPD-like pathology can be generated by autoimmune mechanisms 22.
More direct evidence for autoimmunity in COPD patients is provided by reports of autoantibodies (AAb) and T cells specific for elastin 23,24, although other studies were unable to confirm this 25–28. Antibodies to cytokeratin-18 have also been reported in COPD 29 and in non-allergic asthma 30. It has also been proposed that a broad range of tissue components may serve as autoantigens in COPD, thereby giving rise to a variety of AAb whose production differs between patients 25. This concept is supported by the demonstration, in differing proportions of COPD patients, of AAb to pulmonary and other tissues 31,32 including endothelium 33, carbonyl-modified proteins 34 and a broad range of tissue-specific and systemic autoantigens 35,36.
The studies cited above focus on the occurrence of disease-specific autoantibodies (DSAAb) in COPD, i.e. AAb that are detectable in patients but rarely, if at all, in healthy individuals. A further consideration is the potential involvement of natural autoantibodies (NAAb) that are present in everyone, but which may take on particular relevance in disease situations. NAAb are thought to have a variety of physiological roles, including first line of defence against infection, clearance of ageing cells and their constituents, antigen presentation, anti-tumour and anti-inflammatory activities and immune homeostasis 37. They show high ‘connectivity’ through idiotype/anti-idiotype interactions that may account for the fact that, unlike DSAAb, the levels of detectable NAAb are significantly higher in IgG purified from serum than they are in whole serum 38,39.
Previous studies have also concentrated on the detection of circulating AAb in COPD patients, but it could be argued that AAb sequestered to the lung are most relevant to the disease process. Thus, the aim of the present study was to investigate the occurrence of both NAAb and DSAAb in the circulation and in lung tissue of COPD patients compared to smokers and non-smokers without COPD.
Materials and methods
Study population
The Nottingham Local Research Ethics Committee (REC) as well as Nottingham University Hospitals Research and Development (R&D) approved the study protocol. Written informed consent was obtained from the participants. Serum samples were obtained from smokers with moderate-to-severe COPD (CS) (GOLD stages II–III, BODE index 2–9), smokers without airway obstruction (S) and never smokers (NS). COPD diagnosis was defined according to the American Thoracic Society guidelines, including spirometry criteria of a forced expiratory volume in 1 s (FEV1) below 80% of predicted with a FEV1/FVC (forced vital capacity) ratio of <70% and reversibility of inhaled beta-2-agonist of <10% or <200 ml absolute improvement; all were current smokers (Table 1). S and NS participants underwent pulmonary function tests, which revealed normal spirometry results (Table 1). Individuals who had α1-anti-trypsin deficiency, a history of physician-diagnosed asthma or had a positive skin prick test in response to the allergens grass pollen, house dust mite, cat dander or dog hair (ALK-Abello’, Reading, UK) were excluded from the study. CS patients were also excluded if they had experienced an exacerbation of disease within the previous 6 weeks.
Table 1.
Demographic characteristic of the study participants who provided serum samples.
| CS | S | NS | ||
|---|---|---|---|---|
| Number of patients | 6 | 6 | 6 | |
| Gender | Male/female | 3/3 | 1/5 | 1/5 |
| Age (years) | Mean | 63·67† | 51·83 | 50·83 |
| Median | 65 (54–72) | 49·5 (43–63) | 51·5 (45–57) | |
| FEV1 | Mean | 51·50*† | 98·50 | 115·2 |
| (% pred) | Median | 53 (17–71) | 100·5 (81–119) | 110 (103–140) |
| FVC | Mean | 83·50 | 106·8 | 122·2 |
| (%) | Median | 80 (44–135) | 108 (81–128) | 115·5 (112–144) |
| FEV1/ | Mean | 61·54*† | 92·61 | 94·60 |
| FVC (%) | Median | 65·46 (39·4–72·9) | 91 (84–101·7) | 5·73 (79·1–107·2) |
| Smoking | Mean | 47·86 | 29·52 | 0 |
| (pack/year) | Median | 57·50 (23·8–62·2) | 29·38 (15–42) | 0 |
CS = smokers with chronic obstructive pulmonary disease; S = smokers without airway obstruction; NS = never smokers; FEV1 = forced expiratory volume in 1 s; FVC = forced vital capacity; Pred = predicted value.
Indicates significant difference between CS and S (P < 0·001).
Indicates significant difference between CS and NS (P < 0·001).
Lung samples
Lung samples were obtained from CS patients, S and NS subjects (five of each). Samples were either donated generously by GlaxoSmithKline (GSK, Stevenage, UK) or obtained from Nottingham University Hospitals NHS Trust. Ethical approval was obtained through GSK and Nottingham University Hospitals R&D. Patients in Nottingham were informed and consented before entering the study. Lung samples were obtained from Nottingham patients with COPD who had undergone bullectomy or lung volume reduction surgery as a part of the management of severe emphysema. S and NS Nottingham participants were lung cancer patients who underwent lung biopsy or surgery in the course of the case management. Normal lung tissue distal to the tumour, as judged by the pathologist, was used from these patients’ samples. Lung samples were snap-frozen in liquid nitrogen and stored at −80°C. The lung samples provided by GSK were autopsy specimens from CS, S and NS individuals and were obtained within the normal framework of autopsy procedures.
Homogenization of human lung tissue samples and elution of lung-bound antibodies
Lung tissue samples – previously snap-frozen in liquid nitrogen from CS, S and NS subjects – were defrosted, adjusted to approximately 0·52 g of tissue and placed on ice. Samples were cut into small pieces (about 3–4 mm in diameter) and added to 10 ml of ice-cold 10 mM Tris-HCl buffer pH 7·5 (Sigma Aldrich, Poole, UK) containing one tablet of complete protease inhibitor cocktail (Roche Diagnostics, Lewes, UK). Cell disruption was performed using a 2-ml screw-cap vial filled to three-quarters with 1·0-mm-diameter glass beads. The remaining vial volume was filled with preprepared lung tissue pieces and the cap was secured. Three cycles of tissue disruption were performed using the Mini-Bead Beater Type BX-4 Cell Disrupter (Glen Mills Inc., Clifton, NJ, USA), for 30 s each, at 5000 rpm. Samples were cooled on ice between runs to avoid warming. Disrupted lung samples were microcentrifuged for 2–4 h, at 17 968 g at 4°C to separate soluble material of the lung homogenates from the insoluble pellets. Homogenates and pellets were stored at −20°C to be used in enzyme-linked immunosorbent assay (ELISA).
For each lung sample fraction, the protein concentration was determined using a NanoDrop® ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Antibodies were eluted from the pellets using elution buffer consisting of cold 0·1 M glycine, pH 2·8 (Sigma Aldrich) and 0·5 M NaCl. Pellet-supernatants were pipetted several times to mix well and incubated on ice for 1 h with intermittent mixing every 10 min. The pH of the supernatants was neutralized by the addition of 1 M Tris pH 8 (Sigma Aldrich) and centrifuged. The protein concentration was measured by Nanodrop and the protein concentrations were standardized to 1 mg/ml. Samples were diluted 1 : 4 and tested by ELISA for quantitative measurement of eluted antibodies.
ELISA
ELISA was used for the detection of serum antibodies to lung tissue or specific candidate antigens. Duplicate wells of Nunc Maxi-Sorb 96-well plates (Scientific Laboratory Supplies Ltd, Nottingham, UK) were coated with 100 μl of diluted antigen preparation, using whole lung homogenate soluble fractions (10 μg total protein/ml) or the individual candidate protein antigens shown in Table 2. The plates were incubated overnight at room temperature. In order to determine background binding, duplicate wells were coated overnight with 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) (both from Sigma Aldrich). The plates were washed three times with wash buffer containing 0·05% Tween-20 (Sigma Aldrich) in PBS and then blocked with 300 μl of reagent diluent (1% BSA in PBS) and incubated for 1 h at room temperature. The washing step was then repeated. Participants’ serum samples were diluted to 1 : 40 with reagent diluent. One hundred μl of diluted serum sample and controls were added to the wells in duplicate and the plate was incubated for 2 h at room temperature. The plate was washed three times as before. One hundred μl of the appropriate biotinylated secondary antibody : goat anti-human immunoglobulin (Ig)A, IgG, IgM or polyclonal Igs (Biosource International, Nivelles, Belgium) diluted to 1 : 5000 in reagent diluent was added to each well and incubated for 2 h at room temperature. The plate was washed as before. One hundred μl of streptavidin–alkaline phosphatase (Sigma Aldrich) (1 : 500 dilution in reagent diluent) was added to each well. The plate was incubated for 20 min at room temperature then washed three times. One hundred μl of the alkaline phosphatase yellow p-nitrophenyl phosphate (pNPP) liquid substrate (Sigma Aldrich) was added to each well and the plate was incubated for 20 min at room temperature. The optical density (OD) was measured after 30 min at 405 nm using a Vmax microplate reader (Molecular Devices, Wokingham, UK). The data are presented as ODs, as it was not possible to generate a standard curve for specific antibody levels.
Table 2.
Candidate antigens – the source, the concentration used and the manufacturing company.
| Antigen | Source | Concentration μg/ml | Company |
|---|---|---|---|
| Collagen-1 | Human lung | 10 | Sigma, Aldrich, Poole, UK |
| Collagen-2 | Human cartilage | 1 | Chemicon International, Watford, UK |
| Collagen-3 | Human placenta | 10 | Sigma, Aldrich, Poole, UK |
| Collagen-4 | Human placenta | 10 | Sigma, Aldrich, Poole, UK |
| Collagen-5 | Human placenta | 10 | Sigma, Aldrich, Poole, UK |
| Fibronectin | Human plasma | 10 | Sigma, Aldrich, Poole, UK |
| Laminin | Human plasma | 10 | Sigma, Aldrich, Poole, UK |
| Elastin peptides | Human lung elastin peptides | 25 | Elastin product company, Owensville, MO, USA. |
| Vitronectin | Human plasma | 1 | Sigma, Aldrich, Poole, UK |
| Vimentin | Human recombinant protein | 1 | Progen Biotechnik, Heidelberg, Germany |
| Cytokeratin-8 | Human recombinant protein | 1 | GenWay Biotech, San Diego, CA, USA |
| Cytokeratin-18 | Human recombinant protein | 1 | GenWay Biotech, San Diego, CA, USA |
| Thyroglobulin | Purified human | 10 | Sigma, Aldrich, Poole, UK |
| Bovine serum albumin (BSA) | Bovine serum | 10 000 | Sigma, Aldrich, Poole, UK |
Statistics
Differences between groups were assessed by Mann–Whitney U-test or Student's t-test using spss version 15·0 and Prism version 3·0 software. P-values ≤0·05 were considered to be significant.
Results
Serum autoantibodies reactive with human lung tissue antigens
Human lung samples were homogenized, as described in Materials and methods, and used in ELISA as the target antigen preparation for detecting IgG autoantibodies in sera from COPD patients who smoke (CS), smokers without airway obstruction (S) and never smokers (NS) (Fig. 1). The lung samples and the sera were obtained from different subjects. Six homogenates were used as antigen preparations: two each from the lung tissues of CS patients (Fig. 1a,b), S subjects (Fig. 1c,d) and NS subjects (Fig. 1e,f). The results in Fig. 1 show that, regardless of the source of the lung antigen preparations, the sera from CS patients showed significantly and reproducibly lower levels of IgG antibody binding to lung antigens than the sera from NS individuals. In all cases, the median levels of IgG anti-lung antibodies in the sera of S individuals were between the medians for the NS sera and the CS sera, but were not significantly different from either.
Figure 1.
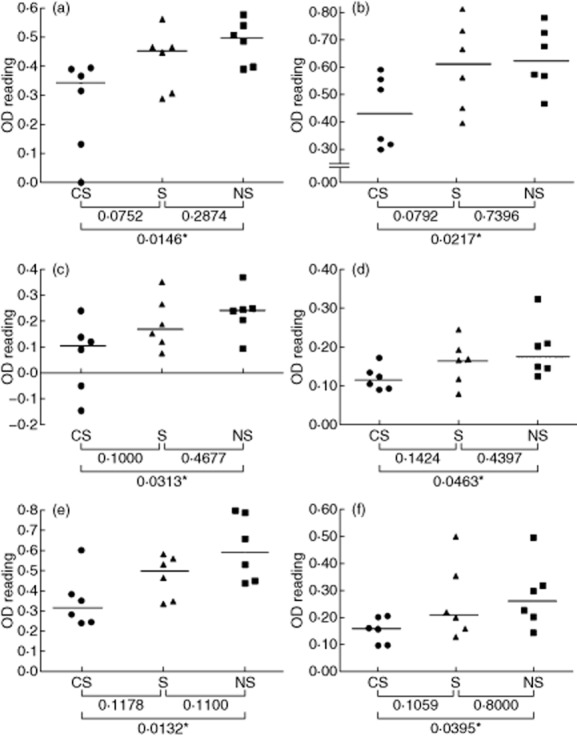
Detecting serum immunoglobulin (Ig)G autoantibodies that bind to constituents of human lung tissue. Sera from smokers with chronic obstructive pulmonary disease (CS), smokers (S) and non-smokers (NS) were analysed by enzyme-linked immunosorbent assay (ELISA) for IgG antibodies that bind to human lung tissue homogenates. The sources of the lung tissue were as follows: (a,b) CS patients; (c,d) S subjects; (e,f) NS subjects. Each point represents an individual serum sample, and the horizontal line is the median optical density (OD) value of the group; statistical comparisons between groups were performed using the Mann–Whitney U-test. The lung samples and the sera were obtained from different subjects.
The finding that the level of IgG anti-lung antibodies is highest in the NS sera suggests that these are ‘natural autoantibodies’ rather than disease-specific autoantibodies, and that their circulating levels are reduced significantly in CS patients. An alternative explanation could be that there is non-specific binding of IgG immunoglobulins to the lung homogenates, and that the lower levels of IgG binding observed with CS sera are due to a general reduction of circulating levels of IgG in CS patients. The total levels of serum IgG were therefore determined by nephelometry in the CS, S and NS sera; the results in Fig. 2 show that there were no significant differences between the three groups, indicating that non-specific IgG binding to the lung homogenates is not the explanation for the differences observed between groups.
Figure 2.
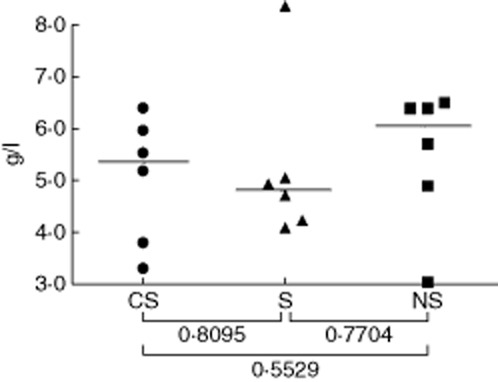
Total immunoglobulin (Ig)G immunoglobulin levels in sera. Sera from smokers with chronic obstructive pulmonary disease (CS) patients, smokers (S) and non-smokers (NS) were assayed for total IgG levels by nephelometry. Each point represents an individual serum sample, and the horizontal lines are the median IgG concentrations (g/l) of the groups; statistical comparisons between groups were performed using the Mann–Whitney U-test.
Serum autoantibody reactivity with specific candidate antigens
Having demonstrated a generalized decrease in IgG autoantibodies showing reactivity with lung tissue in sera from CS patients compared to S and NS controls, the reactivity of these sera with a series of potential candidate antigens relevant to lung was investigated. The purified proteins employed (Table 2) were chosen as candidate antigens on the basis of previous evidence of their immunogenicity in various lung diseases and/or their major representation within lung tissue. BSA was used as the blocking agent in the ELISA, and the ODs given for binding of serum IgG to BSA alone were subtracted from the ODs for all candidate antigens; there was no significant difference in binding to BSA by IgG in CS, S and NS sera. By contrast, the reactivity of serum IgG antibodies with collagen-1 (a major extracellular matrix component of lung tissue) mirrored the findings for antibody binding to lung homogenates described above. Thus, the CS sera showed significantly lower levels of IgG autoantibodies to collagen-1 than the NS sera, with the S sera showing intermediate levels (Fig. 3a). None of the other candidate antigens showed this pattern of reactivity with the sera, with the exception of elastin, to which overall levels of IgG binding were very low (data not shown).
Figure 3.
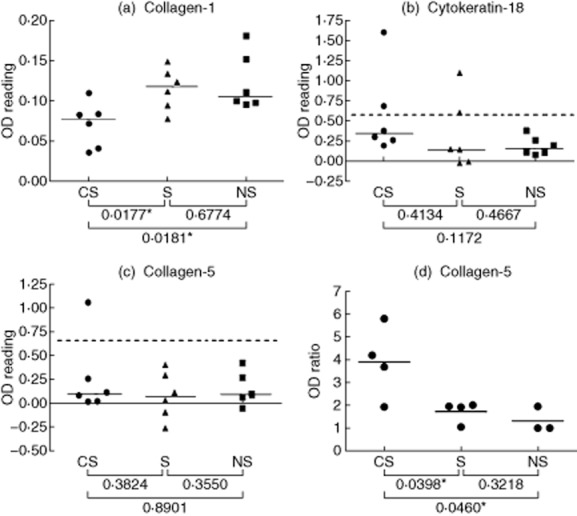
Detecting serum and lung-associated immunoglobulin (Ig)G autoantibodies that bind to candidate lung antigens. (a–c) Sera from smokers with chronic obstructive pulmonary disease (CS), smokers (S) and non-smokers (NS) subjects were analysed by enzyme-linked immunosorbent assay (ELISA) for IgG antibodies that bind to pure protein antigens [collagen-1, (a); cytokeratin-18, (b); collagen-5 (c)]. Each point represents an individual serum sample, and the horizontal lines are the median optical density (OD) values of the groups. Statistical comparisons between groups were performed using the Mann–Whitney U-test. Dashed lines represent 1·5 times the upper limit of the range of ODs given by the NS sera. (d) Eluates of lung tissue from CS, S and NS subjects were analysed by ELISA for IgG antibodies that bind to pure protein antigens. Each point represents an individual lung tissue eluate, and the standardized values shown are the ratio of the OD for eluted IgG binding to collagen-5 divided by the OD for the binding of the same eluted IgG to thyroglobulin (negative control antigen); the horizontal bars are the mean standardized values of the groups. Significant differences between groups were tested by Student's t-test.
Several of the candidate antigens showed high reactivity with IgG in individual CS and/or S sera; in these cases, positive binding was defined by sera giving an OD that was at least 1·5 times the upper limit of the range of ODs given by the NS sera (indicated by the dotted lines in Fig. 3b,c). Thus, individual CS and S sera showed strong IgG binding to cytokeratin-18 and/or collagen-5 (Fig. 3b,c); one S serum also showed IgG reactivity with collagen-4 (data not shown).
Autoantibodies sequestered to the lung in COPD
Lung tissue homogenates from CS, S and NS individuals were treated with glycine-HCl buffer to elute tissue-associated antibodies, as described in Materials and methods. These eluates were investigated in ELISA for IgG autoantibody reactivity with the purified candidate antigens used previously to detect serum IgG autoantibodies, as described above. In order to compare the levels of antibodies eluted from different lung specimens, the OD values generated for IgG binding to candidate lung antigens were standardized by expressing them as ratios to the OD values of the same eluates for binding to the negative control antigen, thyroglobulin. Figure 3d shows that the standardized levels of IgG antibodies specific for collagen-5 were higher in the eluates from CS lung tissue than from S or NS lung tissue. No significant differences were found for eluted antibodies specific for the other candidate antigens tested (collagen-1, collagen-3, cytokeratin-18, elastin, fibronectin, vimentin, vitronectin), although this may be at least partly a consequence of the small number of lung samples available for elution of antibodies, which is a clear limitation of the present study. Larger numbers of samples will be required to determine whether eluted antibodies specific for some of these other candidate antigens are, in fact, raised in CS lungs.
Discussion
In this study we show that detectable levels of IgG antibodies that bind to lung tissue components are lower in the circulation of CS patients than in NS subjects. The fact that such antibodies are detectable (and, indeed, highest) in the NS group indicates that these are NAAbs that occur naturally in everyone. The same phenomenon was seen for antibodies that bind to collagen-1 specifically; i.e. their detectable levels were reduced significantly in the sera of CS patients compared to NS subjects. Collagen-1 is the major type of collagen in lung tissue 40, and our findings suggest that collagen-1 is a significant target of the lung-reactive NAAbs in human sera. Interestingly, a similar observation was reported by Wood et al. with respect to antibodies to elastin; i.e. higher levels were detected in NS controls than in CS patients 28. Indeed, we also observed significantly lower levels of IgG antibodies to elastin peptides in CS patients’ sera than in NS subjects, but the levels of anti-elastin detected in our ELISA were generally very low (data not shown).
A possible explanation of our finding of reduced levels of lung-reactive NAAbs in CS patients’ sera is that, in COPD, NAAbs that can bind to a variety of lung constituents (including collagen-1) are sequestered to the lung from the circulation. Other explanations for the apparent reduction of lung-reactive NAAbs in CS patients’ sera are also possible. For example, binding of circulating antibodies to tissue constituents released into the bloodstream from inflamed, damaged lungs in COPD could reduce detection of these antibodies in ELISA due to their involvement in immune complex formation. Another possibility is that circulating NAAbs show higher connectivity in COPD than is normally the case; i.e. they are more connected into idiotype/anti-idiotype networks, thereby reducing their availability for binding to antigens 38,39.
If NAAbs are involved in COPD this leaves open the question of their role in the disease, which could be either protective or pathogenic. A physiological anti-inflammatory function of NAAbs is thought to involve binding to damaged and senescent cells, thereby promoting their clearance from tissues; an example is the proposed anti-atherogenic role of NAAbs by binding to oxidized tissue components 41. Conversely, NAAbs have been shown to promote intestinal ischaemia/reperfusion injury through binding to the damaged tissues and activating complement 42,43. Such effects may be tissue-specific 42, and further studies are required to determine whether NAAbs may inhibit or promote damage to the lungs in COPD. Therapeutic intravenous immunoglobulin (IVIG) preparations are known to contain NAAbs 44 that may account, at least in part, for the beneficial effects of treatment with IVIG in certain autoimmune and inflammatory diseases 45,46. Thus, if NAAbs are shown to be protective to the lung in COPD, the possibility of IVIG administration is raised as a potential therapy for this disease.
It is of note that the results in this study for smokers without COPD were intermediate between those for smokers with COPD and never smokers – i.e. serum antibodies of S individuals gave a level of binding to lung tissue intermediate between those of NS subjects and CS patients (which was not significantly different from either) (Fig. 1). This suggests that smokers without overt COPD may have subclinical features of the disease, and our findings are consistent with the increase in bronchus-associated lymphoid tissue in smokers compared to never smokers 47.
Our results also support the occurrence of DSAAbs in some CS and S subjects, although the number of subjects in this study was too low for statistical significance. Even so, the observation of individual CS and S sera with high levels of IgG antibodies to cytokeratin-18 and collagen-5 (and also to cytokeratin-8 and collagen-4), compared to NS sera, is consistent with others’ findings in COPD or other inflammatory lung diseases 19,29,40,48. For example, others have reported the occurrence of antibodies to cytokeratin-18 in both COPD 29 and in non-allergic asthma 30. It is also particularly interesting that we found antibodies to collagen-5 eluted from lung tissue to be raised significantly in CS patients compared to S and NS subjects. Immunity to collagen-5 is also associated with lung transplant dysfunction and bronchiolitis obliterans syndrome 40,49–51, lung cancer 52 and experimental scleroderma 53. Collagen-5 is a minor collagen in the lung, where it is sequestered within the major collagen-1 fibrils 40; thus, it may be its exposure during inflammation and damage to lung tissues that permits collagen-5 to be recognized as an autoantigen.
This raises the issue of whether autoimmunity in COPD is a primary or secondary event in the disease pathogenesis; it has been argued that autoimmunity in COPD may be secondary to the release of tissue components from damaged lung parenchyma rather than autoimmunity initiating this damage 54. Even if it were the case that the initial smoke-induced events involve antigen non-specific inflammation and tissue damage, this would not necessarily diminish the potential importance of autoimmunity in the propagation and transition to a self-perpetuating, smoke-independent process (which could explain why COPD persists after cessation of smoking). This would be consistent with a mechanism of induction of autoimmunity in COPD that involves smoke-induced tissue damage and exposure of sequestered lung autoantigens, as proposed above for collagen-5. This could be analogous to statin-induced autoimmune myopathy that involves autoimmunity to HMGCoA-reductase that is up-regulated in muscle tissue as a consequence of the inhibition of this enzyme by statins; but, once established, the autoimmune reaction continues, despite statin withdrawal 55.
A further consideration is whether autoantibodies that are associated with lung tissue itself (as demonstrated for anti-collagen-5 in this study) are produced in conventional lymphoid tissues and sequestered to the lung from the circulation, or whether they are produced by lymphoid structures that develop within the lung during COPD. This latter possibility seems highly likely, given the clearly documented development of lymphoid follicles within the lungs in COPD 13,14. These follicles have B cell cores that may be a source of lung-reactive autoantibodies 56. It would therefore be very interesting to investigate the specificity of antibodies produced by these lung follicular B cells.
A limitation of our study is the relatively small number of subjects studied, and the results reported here need to be confirmed in larger cohorts of participants. In addition, the panel of autoantigens investigated should be expanded in order to ensure that the profile of autoantibodies detected is as comprehensive as possible. We are currently aiming to fulfil these criteria using reverse-phase protein microarray technology. Direct identification of target antigens within lung homogenates by Western blotting would also provide further valuable information.
In summary, this study is consistent with a role for both NAAbs and DSAAbs (that bind to lung antigens) in the pathogenesis of COPD. Whether NAAbs ameliorate or exacerbate the inflammation and tissue damage remains to be determined.
Acknowledgments
This work was supported by King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia and by the Jones 1986 Charitable Trust.
Disclosure
The authors have no commercial or financial conflicts of interest to declare.
Author contributions
N. I. D. performed the experiments; I.T., L. C. F. and J. M. C. conceived and designed the study; all authors contributed to interpretation of data and to preparation and review of the manuscript.
References
- Lopez AD, Shibuya K, Rao C, et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27:397–412. doi: 10.1183/09031936.06.00025805. [DOI] [PubMed] [Google Scholar]
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS, Committee GS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445–2454. doi: 10.1056/NEJMra0804752. [DOI] [PubMed] [Google Scholar]
- Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378:1015–1026. doi: 10.1016/S0140-6736(11)60988-4. [DOI] [PubMed] [Google Scholar]
- Di Stefano A, Caramori G, Capelli A, et al. STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur Respir J. 2004;24:78–85. doi: 10.1183/09031936.04.00080303. [DOI] [PubMed] [Google Scholar]
- Motz GT, Eppert BL, Wesselkamper SC, Flury JL, Borchers MT. Chronic cigarette smoke exposure generates pathogenic T cells capable of driving COPD-like disease in Rag2–/– mice. Am J Respir Crit Care Med. 2010;181:1223–1233. doi: 10.1164/rccm.200910-1485OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan AK, Simonian PL, Falta MT, et al. Oligoclonal CD4+ T cells in the lungs of patients with severe emphysema. Am J Respir Crit Care Med. 2005;172:590–596. doi: 10.1164/rccm.200410-1332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairclough L, Urbanowicz RA, Corne J, Lamb JR. Killer cells in chronic obstructive pulmonary disease. Clin Sci (Lond) 2008;114:533–541. doi: 10.1042/CS20070356. [DOI] [PubMed] [Google Scholar]
- Urbanowicz RA, Lamb JR, Todd I, Corne JM, Fairclough LC. Enhanced effector function of cytotoxic cells in the induced sputum of COPD patients. Respir Res. 2010;11:76. doi: 10.1186/1465-9921-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Urbanowicz RA, Tighe PJ, Todd I, Corne JM, Fairclough LC. Differential activation of killer cells in the circulation and the lung: a study of current smoking status and chronic obstructive pulmonary disease (COPD) PLOS ONE. 2013;8:e58556. doi: 10.1371/journal.pone.0058556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosman MM, Willemse BW, Jansen DF, et al. Increased number of B-cells in bronchial biopsies in COPD. Eur Respir J. 2006;27:60–64. doi: 10.1183/09031936.06.00007005. [DOI] [PubMed] [Google Scholar]
- van der Strate BW, Postma DS, Brandsma CA, et al. Cigarette smoke-induced emphysema: a role for the B cell? Am J Respir Crit Care Med. 2006;173:751–758. doi: 10.1164/rccm.200504-594OC. [DOI] [PubMed] [Google Scholar]
- Litsiou E, Semitekolou M, Galani IE, et al. CXCL13 production in B cells via Toll-like receptor/lymphotoxin receptor signaling is involved in lymphoid neogenesis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187:1194–1202. doi: 10.1164/rccm.201208-1543OC. [DOI] [PubMed] [Google Scholar]
- Bracke KR, Verhamme FM, Seys LJ, et al. Role of CXCL13 in cigarette smoke-induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:343–355. doi: 10.1164/rccm.201211-2055OC. [DOI] [PubMed] [Google Scholar]
- Stefanska AM, Walsh PT. Chronic obstructive pulmonary disease: evidence for an autoimmune component. Cell Mol Immunol. 2009;6:81–86. doi: 10.1038/cmi.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimohammadi M, Dubois N, Skoldberg F, et al. Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and identification of KCNRG as a bronchial autoantigen. Proc Natl Acad Sci USA. 2009;106:4396–4401. doi: 10.1073/pnas.0809986106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow L, Rosas IO, Gochuico BR, et al. Identification of citrullinated hsp90 isoforms as novel autoantigens in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheum. 2013;65:869–879. doi: 10.1002/art.37881. [DOI] [PubMed] [Google Scholar]
- Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34:J258–265. doi: 10.1016/j.jaut.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Dobashi N, Fujita J, Ohtsuki Y, et al. Detection of anti-cytokeratin 8 antibody in the serum of patients with cryptogenic fibrosing alveolitis and pulmonary fibrosis associated with collagen vascular disorders. Thorax. 1998;53:969–974. doi: 10.1136/thx.53.11.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita J, Takeuchi T, Dobashi N, Ohtsuki Y, Tokuda M, Takahara J. Detection of anti-ADAM 10 antibody in serum of a patient with pulmonary fibrosis associated with dermatomyositis. Ann Rheum Dis. 1999;58:770–772. doi: 10.1136/ard.58.12.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fujita J, Bandoh S, et al. Detection of antivimentin antibody in sera of patients with idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. Clin Exp Immunol. 2002;128:169–174. doi: 10.1046/j.1365-2249.2002.01811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraseviciene-Stewart L, Scerbavicius R, Choe KH, et al. An animal model of autoimmune emphysema. Am J Respir Crit Care Med. 2005;171:734–742. doi: 10.1164/rccm.200409-1275OC. [DOI] [PubMed] [Google Scholar]
- Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13:567–569. doi: 10.1038/nm1583. [DOI] [PubMed] [Google Scholar]
- Grumelli S, Lu B, Peterson L, Maeno T, Gerard C. CD46 protects against chronic obstructive pulmonary disease. PLOS ONE. 2011;6:e18785. doi: 10.1371/journal.pone.0018785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsma CA, Kerstjens HA, Geerlings M, et al. The search for autoantibodies against elastin, collagen and decorin in COPD. Eur Respir J. 2011;37:1289–1292. doi: 10.1183/09031936.00116710. [DOI] [PubMed] [Google Scholar]
- Cottin V, Fabien N, Khouatra C, Moreira A, Cordier JF. Anti-elastin autoantibodies are not present in combined pulmonary fibrosis and emphysema. Eur Respir J. 2009;33:219–221. doi: 10.1183/09031936.00140208. [DOI] [PubMed] [Google Scholar]
- Greene CM, Low TB, O'Neill SJ, McElvaney NG. Anti-proline–glycine-proline or antielastin autoantibodies are not evident in chronic inflammatory lung disease. Am J Respir Crit Care Med. 2010;181:31–35. doi: 10.1164/rccm.200904-0545OC. [DOI] [PubMed] [Google Scholar]
- Wood AM, de Pablo P, Buckley CD, Ahmad A, Stockley RA. Smoke exposure as a determinant of autoantibody titre in alpha(1)-antitrypsin deficiency and COPD. Eur Respir J. 2011;37:32–38. doi: 10.1183/09031936.00033710. [DOI] [PubMed] [Google Scholar]
- Kuo YB, Chang CA, Wu YK, et al. Identification and clinical association of anti-cytokeratin 18 autoantibody in COPD. Immunol Lett. 2010;128:131–136. doi: 10.1016/j.imlet.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Nahm DH, Lee YE, Yim EJ, et al. Identification of cytokeratin 18 as a bronchial epithelial autoantigen associated with nonallergic asthma. Am J Respir Crit Care Med. 2002;165:1536–1539. doi: 10.1164/rccm.200201-009OC. [DOI] [PubMed] [Google Scholar]
- Feghali-Bostwick CA, Gadgil AS, Otterbein LE, et al. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:156–163. doi: 10.1164/rccm.200701-014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez B, Sauleda J, Anto JM, et al. Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183:1025–1031. doi: 10.1164/rccm.201001-0029OC. [DOI] [PubMed] [Google Scholar]
- Karayama M, Inui N, Suda T, Nakamura Y, Nakamura H, Chida K. Antiendothelial cell antibodies in patients with COPD. Chest. 2010;138:1303–1308. doi: 10.1378/chest.10-0863. [DOI] [PubMed] [Google Scholar]
- Kirkham PA, Caramori G, Casolari P, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184:796–802. doi: 10.1164/rccm.201010-1605OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidinger P, Keller A, Heisel S, et al. Novel autoantigens immunogenic in COPD patients. Respir Res. 2009;10:20. doi: 10.1186/1465-9921-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard TA, Li QZ, Cosgrove GP, Bowler RP, Cambier JC. COPD is associated with production of autoantibodies to a broad spectrum of self-antigens, correlative with disease phenotype. Immunol Res. 2013;55:48–57. doi: 10.1007/s12026-012-8347-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix-Desmazes S, Kaveri SV, Mouthon L, et al. Self-reactive antibodies (natural autoantibodies) in healthy individuals. J Immunol Methods. 1998;216:117–137. doi: 10.1016/s0022-1759(98)00074-x. [DOI] [PubMed] [Google Scholar]
- Hurez V, Kaveri SV, Kazatchkine MD. Expression and control of the natural autoreactive IgG repertoire in normal human serum. Eur J Immunol. 1993;23:783–789. doi: 10.1002/eji.1830230402. [DOI] [PubMed] [Google Scholar]
- Mouthon L, Haury M, Lacroix-Desmazes S, Barreau C, Coutinho A, Kazatchkine MD. Analysis of the normal human IgG antibody repertoire. Evidence that IgG autoantibodies of healthy adults recognize a limited and conserved set of protein antigens in homologous tissues. J Immunol. 1995;154:5769–5778. [PubMed] [Google Scholar]
- Iwata T, Philipovskiy A, Fisher AJ, et al. Anti-type V collagen humoral immunity in lung transplant primary graft dysfunction. J Immunol. 2008;181:5738–5747. doi: 10.4049/jimmunol.181.8.5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder CJ, Shaw PX, Chang MK, et al. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46:1353–1363. doi: 10.1194/jlr.R500005-JLR200. [DOI] [PubMed] [Google Scholar]
- Fleming SD, Tsokos GC. Complement, natural antibodies, autoantibodies and tissue injury. Autoimmun Rev. 2006;5:89–92. doi: 10.1016/j.autrev.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Zhang M, Austen WG, Jr, Chiu I, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2004;101:3886–3891. doi: 10.1073/pnas.0400347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussone G, Dib H, Dimitrov JD, et al. Identification of target antigens of self-reactive IgG in intravenous immunoglobulin preparations. Proteomics. 2009;9:2253–2262. doi: 10.1002/pmic.200800819. [DOI] [PubMed] [Google Scholar]
- Bruley-Rosset M, Mouthon L, Chanseaud Y, Dhainaut F, Lirochon J, Bourel D. Polyreactive autoantibodies purified from human intravenous immunoglobulins prevent the development of experimental autoimmune diseases. Lab Invest. 2003;83:1013–1023. doi: 10.1097/01.lab.0000077982.70800.02. [DOI] [PubMed] [Google Scholar]
- Siberil S, Elluru S, Graff-Dubois S, et al. Intravenous immunoglobulins in autoimmune and inflammatory diseases: a mechanistic perspective. Ann NY Acad Sci. 2007;1110:497–506. doi: 10.1196/annals.1423.052. [DOI] [PubMed] [Google Scholar]
- Richmond I, Pritchard GE, Ashcroft T, Avery A, Corris PA, Walters EH. Bronchus associated lymphoid tissue (BALT) in human lung: its distribution in smokers and non-smokers. Thorax. 1993;48:1130–1134. doi: 10.1136/thx.48.11.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahmer T, Heemann U. Anti-glomerular basement membrane antibody disease: a rare autoimmune disorder affecting the kidney and the lung. Autoimmun Rev. 2012;12:169–173. doi: 10.1016/j.autrev.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Bobadilla JL, Love RB, Jankowska-Gan E, et al. Th-17, monokines, collagen type V, and primary graft dysfunction in lung transplantation. Am J Respir Crit Care Med. 2008;177:660–668. doi: 10.1164/rccm.200612-1901OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117:3498–3506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mares DC, Heidler KM, Smith GN, et al. Type V collagen modulates alloantigen-induced pathology and immunology in the lung. Am J Respir Cell Mol Biol. 2000;23:62–70. doi: 10.1165/ajrcmb.23.1.3924. [DOI] [PubMed] [Google Scholar]
- Fernandez-Madrid F, Karvonen RL, Kraut MJ, Czelusniak B, Ager JW. Autoimmunity to collagen in human lung cancer. Cancer Res. 1996;56:121–126. [PubMed] [Google Scholar]
- Velosa AP, Teodoro WR, dos Anjos DM, et al. Collagen V-induced nasal tolerance downregulates pulmonary collagen mRNA gene and TGF-beta expression in experimental systemic sclerosis. Respir Res. 2010;11:1. doi: 10.1186/1465-9921-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima M, Chida K, Suda T, Sato M. Is autoimmunity really related to the pathogenesis of COPD? Am J Respir Crit Care Med. 2011;184:1212–1213. doi: 10.1164/ajrccm.184.10.1212a. author reply 3. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63:713–721. doi: 10.1002/art.30156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monin L, Khader SA. B cells produce CXCL13 in lymphoid neogenesis during chronic obstructive pulmonary disease. The new kid on the block? Am J Respir Crit Care Med. 2013;187:1162–1164. doi: 10.1164/rccm.201303-0552ED. [DOI] [PMC free article] [PubMed] [Google Scholar]