

Abstract
Objectives and Background
We evaluated the ability of 23 genetic variants to provide prognostic information in patients enrolled in the Genotype Sub-studies of the Surgical Treatment for Ischemic Heart Failure (STICH) trials.
Methods
Patients in STICH Hypothesis 1 were randomized to medical therapy with or without CABG (Coronary Artery Bypass Grafting). Those in STICH Hypothesis 2 were randomized to CABG or CABG with left ventricular reconstruction.
Results
In patients assigned to STICH Hypothesis 2 (n=714), no genetic variant met the pre-specified Bonferroni-adjusted threshold for statistical significance (p<0.002); however, several met nominal prognostic significance: variants in the β2-adrenergic receptor gene (β2-AR Gln27Glu) and in the A1-adenosine receptor gene (A1-717 T/G) were associated with an increased risk of a subject dying or being hospitalized for a cardiac problem (p=0.027 and 0.031, respectively). These relationships remained nominally significant even after multivariable adjustment for prognostic clinical variables. However, none of the 23 genetic variants influenced all-cause mortality or the combination of death or cardiovascular hospitalization in the STICH Hypothesis 1 population (n=532) by either univariate or multivariable analysis.
Conclusion
We were unable to identify the predictive genotypes in optimally treated patients in these two ischemic heart failure populations.
Keywords: heart failure, coronary artery disease, genotype
INTRODUCTION
Heart failure is a disease of epidemic proportions that affects over 5 million individuals in the U.S. and accounts for over 250,000 deaths and 1 million hospitalizations each year. [1] Heart failure is attributable to coronary artery disease in over 70% of these individuals. There is great variability in the progression of heart failure in different individuals as well as in their response to various therapies including drugs or devices. These differences have been attributable at least in part to genetic variation. [2,3]
Genetic variants in genes that encode proteins that effect cardiac remodeling and that encode proteins that are targets of pharmacologic therapy have been associated with the progression of heart failure. However, the results of studies that have assessed the association of these genetic variations with outcomes have often provided disparate results. For example, genetic variations in genes encoding the β1-adrenergic receptor [4,5], the β2-adrenergic receptor6, the angiotensin converting enzyme (ACE)[7,8,9], aldosterone synthase[10,11], matrix metalloproteinase type 9[12], tumor necrosis factor-α[13,14], Endothelial nitric oxide synthase [15,16] and adenosine monophosphate deaminase-1 [17,18] have all been associated with varying outcomes in patients with heart failure. The disparities across these studies have been attributed in part to the small size or genetic heterogeneity of the study populations, the inclusion of patients with both ischemic and idiopathic dilated cardiomyopathy, differences in baseline heart failure therapies, and/or statistical noise due to the absence of replicable results in a separate population with the same phenotype.
We evaluated the relationship between genotype and outcome in patients enrolled in the Genotype Sub-study of the Surgical Treatment for Ischemic Heart Failure trial (STICH) to assess the role of genetic variants in predicting outcome in a group of patients with heart failure secondary to ischemic heart disease.[19] Funded by the National Institutes of Health, this multi-center international study enrolled 2,136 patients with ischemic heart failure into one of two studies – STICH Hypothesis 1 and STICH Hypothesis 2 - thereby providing two independent studies in which to prospectively evaluate the ability of genotype to predict outcome. Hypothesis 1 assessed whether coronary artery bypass grafting with intensive medical therapy could improve long-term survival when compared with intensive medical therapy alone. There was not a statistically significant difference in the primary outcome of death from any cause between the two treatment groups; CABG relative to medical therapy alone led to a significant reduction in cardiovascular deaths and survival free of cardiovascular hospitalizations.[20] Hypothesis 2 evaluated the benefits of left ventricular surgical reconstruction (SVR) combined with coronary artery bypass grafting when compared with coronary artery bypass grafting alone. The addition of SVR had no effect on the primary outcome variable of death from any cause or hospitalization for cardiac cause.[21] Patients enrolled in STICH were carefully phenotyped, received optimal medical therapy including a β-blocker and an ACE inhibitor or an angiotensin receptor blocker, and were followed for a median of 48 months.
To test the hypothesis that genotype is associated with outcome in patients with ischemic heart failure being considered for surgical revascularization, we genotyped patients enrolled in the two STICH studies that participated in the STICH genotype sub-study. Genetic variants were chosen that had been found in other studies to be relevant in predicting either outcomes or response to therapy and that represented important neuro-pharmacologic targets including hormonal signaling pathways (β1-AB, β2-AR. Adenosine-R), 4-6; neurohormone levels (angiotensin converting enzyme, aldosterone synthase, adenosine monophosphate deaminase), 7-11, 17, 18; inflammatory mediators (tumor necrosis factor α, matrix metalloproteinases), 12-14; and vascular reactants (endothelial nitric oxide synthase), 15-16.
METHODS
Study Design
The rationale and design of the STICH trial as well as the results of both Hypothesis 1 and Hypothesis 2 were presented previously in detail.[19,20,21] In brief, STICH enrolled 2,136 patients with a left ventricular ejection fraction ≤35% and coronary artery disease that was amenable to coronary artery bypass grafting (CABG). Patients were excluded if they had a recent myocardial infarction, a need for aortic-valve replacement, a planned percutaneous coronary intervention, and coexisting non-cardiac disease that would shorten their life expectancy. All patients underwent cardiac imaging for assessment of left ventricular function and wall motion.
The enrolling physician assigned patients to one of three strata. Stratum A included patients who were eligible for either medical therapy alone or medical therapy plus CABG. Patients were eligible for medical therapy alone if they did not have significant left main coronary artery disease or Canadian Cardiovascular Society class III or IV angina. Stratum B included patients who were eligible for medical therapy alone, medical therapy plus CABG or medical therapy plus CABG and SVR. Patients were eligible for SVR if they had anterior left ventricular akinesia or dyskinesia. Stratum C patients were eligible for medical therapy plus CABG or medical therapy plus CABG and SVR. Patients were then randomly assigned to one of the treatment options for which they were eligible. All of the patients in stratum A and some of the patients in stratum B were randomly assigned to medical therapy or medical therapy plus CABG (STICH Hypothesis 1). All of the patients in stratum C and some of the patients in stratum B were randomly assigned to medical therapy and CABG or to medical therapy, CABG and SVR (STICH Hypothesis 2). Seventy-six patients randomized to CABG in Stratum B fit the criteria for assignment to either Hypothesis 1 or Hypothesis 2. Those patients and their genetic data were analyzed with the Hypothesis 2 cohort.
Patients received pharmacologic therapy based on consensus guideline recommendations; however, the use of device therapy was highly variable across different countries. [22] All countries and study centers were given the opportunity to participate in the STICH genotype sub-study; however, a number of centers declined because of local or national laws that prohibited them from transporting blood products or genetic material out of the country. Approval from the institutional review board was obtained from each institution participating in the genetic sub-study of the STICH trial and all patients provided written informed consent.
Genotyping
The planning for the STICH trials began in 2000 and enrollment into the two studies began shortly after receipt of the notice of funding from the National Institutes of Health (NIH) in 2002. [19] Our ability to assess the effects of a limitless number of genetic variables on outcome in the STICH trial was restricted by the technology available and its attendant costs at the time the study was begun. Therefore, we used the following criteria to select the specific variants for study: 1) discrete genetic variants that altered the function of genes that modify cardiac remodeling; 2) variants in genes that encoded proteins that were targets of heart failure pharmacologic therapy; and 3) genetic variants that had previously been associated with changes in the risk of developing heart failure or in response to pharmacologic therapy. We did not pro-actively eliminate any genetic variants that met at least one of these criteria. In fact, we added several variants as new information became available prior to the start of genotyping. Once genotyping began we did not alter the panel of variants. Approximately 10 ml of blood was obtained from each subject prior to randomization and shipped to the core laboratory at Thomas Jefferson University within 7 days. The core laboratory was blinded to the treatment arm to which the individual patients were assigned. Total genomic DNA was extracted from these samples using a genomic DNA extraction kit (Promega, Madison, WI). We assessed the presence of genetic variants using the PCR-based restriction fragment length polymorphism method that had been reported previously by either our own laboratory or by others. As new technologies became available, several of the genetic variants were assessed using high-throughput analysis. In all cases, we confirmed the identity of each restriction enzyme-based product by sequence analysis prior to utilizing the technology on the sample population. Specific details regarding the technique and primers used for each genotype are found in the Supplemental materials.
Statistical Analysis
A unique feature of STICH is that it consisted of two different studies in patients that had similar severity and etiology of their heart failure (Hypothesis 1 and Hypothesis 2), thus enabling an examination of the relationships of the genetic markers with clinical outcomes in two separate studies. Because blood samples for genotyping could not be obtained in every randomized patient (as described above), we examined the baseline characteristics and outcomes of patients in the genetic sub-study compared to the patients where samples for genotyping could not be obtained. Data are descriptively summarized using the median and interquartile range for continuous variables and frequencies and percentages for categorical variables. The distributions of continuous variables and ordinal categorical variables were compared between groups using the Wilcoxon rank-sum test, and nominal categorical variables were compared using conventional chi-square statistics. The incidence of the primary endpoint in each trial (mortality in Hypothesis 1 and death or cardiovascular hospitalization in Hypothesis 2) was also compared between patients in the genetic sub-study compared to the patients who were not included. Because homozygous variants were rare, heterozygote and homozygote variants were combined as a single endpoint.
The relationships of each of the 23 genetic markers with the clinical outcomes of (a) death and (b) death or cardiac hospitalization were examined separately in the Hypothesis 1 and Hypothesis 2 cohorts using the Cox regression model.[23] We assessed the univariate relationship of each genetic marker with the clinical outcomes and also examined the extent to which any of the genotypes contributed significant independent prognostic information beyond the baseline clinical variables routinely available in these patients. The clinical variables were the prognostic variables identified through separate multivariable Cox regression analyses for death and for death or cardiovascular hospitalization in the Hypothesis 1 and Hypothesis 2 patient cohorts and included age, New York Heart Association heart failure classification, creatinine, hemoglobin, end systolic volume index (ESVI), mitral regurgitation, and history of myocardial infarction, stroke, and atrial fibrillation. The specific variables for each cohort and endpoint are listed in e-Tables 1A, 1B, 2A, and 2B in the supplementary appendix. In the Hypothesis 1 cohort, we examined whether CABG + medical therapy had a greater (or lesser) effect on clinical outcomes compared to medical therapy alone depending on the genetic variant. This assessment was performed by examining treatment by genetic marker interactions using the Cox model. Identical analyses for CABG vs. CABG+SVR were performed in the Hypothesis 2 cohort.
Hazard ratios, 95% confidence intervals, and p-values were generated in both univariate and multivariable analyses using the Cox model. Because of the number of genetic markers examined [23] and the inherent multiplicity of comparisons, we used a Bonferroni-corrected level of (0.05/23) = 0.002 as a guide for interpreting the statistical significance of the prognostic value of each genetic marker. Nominal p-values are reported, however, for each of the assessments.
RESULTS
A total of 1212 patients were enrolled in STICH Hypothesis 1 between July 24, 2002 and May 5, 2007 and randomly assigned to receive medical therapy alone or medical therapy plus CABG. Samples from 532 Hypothesis 1 patients were included in the genotype analysis. This number did not include the patients with genetic data who were enrolled in both the Hypothesis 1 and Hypothesis 2 studies. Those patients were included in the Hypothesis 2 analyses. Between September 12, 2002 and January 24, 2006, clinical sites randomized 1,000 patients to STICH Hypothesis 2: treatment with CABG alone (n=499) or CABG plus SVR (n=501). Follow-up continued through December 31, 2008. Samples from 714 of Hypothesis 2 patients were included in the genotype analysis. Because enrollment into STICH Hypothesis 2 was completed before enrollment into STICH Hypothesis 1, we completed genotyping and analysis of the Hypothesis 2 cohort before completion of the Hypothesis 1 cohort and the data are therefore presented in that order.
Overall, the majority of genotype sub-study participants in STICH were from North America (34%) or Europe (59%). A smaller percentage of individuals assigned to Hypothesis 1 participated in the genetic analysis sub-study than did patients assigned to STICH Hypothesis 2. This difference was attributable to the fact that a higher percentage of patients assigned to Hypothesis 1 came from countries that did not participate in the genetic sub-study.
STICH Hypothesis 2
As seen in Table 1, the baseline characteristics of the subjects enrolled in STICH Hypothesis 2 who participated in the genetic study were generally similar to the Hypothesis 2 patients who did not participate. In particular, there were no significant differences in terms of age, gender, left ventricular ejection fraction, left ventricular end-systolic volume index, or history of a previous myocardial infarction, hypertension or diabetes. The patients who participated in the genetic sub-study had a lower incidence of chronic renal disease, peripheral vascular disease, and less symptomatic heart failure. There was a modestly higher incidence of the primary composite endpoint of death or cardiovascular hospitalization among the patients in the genetic sub-study.
Table 1.
Comparison of STICH Hypothesis 2 Patients with vs. without Genetic Data
| Characteristic | No Genetic Data (n=286) | Genetic Data (n=714) | P-Value |
|---|---|---|---|
| Age, median (25th, 75th), yrs. | 60.5 (53.6, 68.0) | 62.1 (55.2, 69.3) | 0.108 |
| Female, no. (%) | 50 (17.5%) | 97 (13.6%) | 0.116 |
| White race | 250 (87.4%) | 661 (92.6%) | 0.010 |
| Minority (Hispanic or racial minority) | 56 (19.6%) | 68 (9.5%) | <0.001 |
| BMI | 27.4 (24.5, 30.5) | 27.0 (24.5, 30.0) | 0.358 |
| Previous MI, no. (%) | 253 (88.5%) | 619 (86.7%) | 0.450 |
| Previous stroke, no. (%) | 10 (3.5%) | 46 (6.4%) | 0.067 |
| Diabetes, no. (%) | 96 (33.6%) | 248 (34.7%) | 0.725 |
| Hypertension, no. (%) | 177 (61.9%) | 408 (57.1%) | 0.169 |
| Hyperlipidemia, no. (%) | 208 (73.0%) | 510 (71.6%) | 0.667 |
| Current smoker | 66 (23.1%) | 151 (21.1%) | 0.504 |
| Chronic renal insufficiency, no. (%) | 33(11.6%) | 52 (7.3%) | 0.029 |
| Peripheral vascular disease | 53 (18.5%) | 93 (13.0%) | 0.026 |
| Atrial fibrillation/flutter | 38 (13.3%) | 79(11.1%) | 0.323 |
| Previous PCI, no. (%) | 36 (12.6%) | 159 (22.3%) | <0.001 |
| Previous CABG, no. (%) | 7 (2.4%) | 17 (2.4%) | 0.950 |
| Current CCS angina class, no. (%) | 0.843 | ||
| No angina | 85 (29.7%) | 164 (23.0%) | |
| I | 15 (5.2%) | 56 (7.8%) | |
| II | 39 (13.6%) | 149 (20.9%) | |
| III | 114 (39.9%) | 294 (41.2%) | |
| IV | 33 (11.5%) | 51 (7.1%) | |
| Current NYHA heart failure class, no. (%) | 0.004 | ||
| I | 30 (10.5%) | 56 (7.8%) | |
| II | 90 (31.5%) | 339 (47.5%) | |
| III | 147 (51.4%) | 281 (39.4%) | |
| IV | 19 (6.6%) | 38 (5.3%) | |
| Creatinine | 1.1 (0.9, 1.3) | 1.1 (0.9, 1.3) | 0.656 |
| Risk at randomization | 12 (6, 21) | 12 (5, 21) | 0.488 |
| No. of diseased vessels (≥75% Stenosis), no. (%) | 0.231 | ||
| 0-1 | 49 (17.1%) | 156 (21.8%) | |
| 2 | 123 (43.0%) | 288 (40.3%) | |
| 3 | 114 (39.9%) | 270 (37.8%) | |
| Left main (≥50% stenosis), no. (%) | 57 (19.9%) | 140 (19.6%) | 0.908 |
| Proximal LAD (≥75% stenosis), no. (%) | 232 (81.4%) | 525 (73.5%) | 0.009 |
| LV ejection fraction, median (25th, 75th), % | 28.0 (23.0, 34.0) | 28.0 (22.1, 34.0) | 0.672 |
| ESVI, median (25th, 75th), mL/m2 | 80.2 (61.0, 100.2) | 77.5 (59.0, 98.4) | 0.227 |
| Mitral regurgitation | 0.371 | ||
| None or trace | 92 (32.7%) | 271 (38.2%) | |
| Mild (≤2+) | 141 (50.2%) | 308 (43.4%) | |
| Moderate or severe | 48 (17.0%) | 130 (18.3%) | |
| Region, no. (%) | <0.001 | ||
| Europe | 158 (55.2%) | 415 (58.1%) | |
| US | 49 (17.1%) | 151 (21.1%) | |
| Canada | 50 (17.5%) | 104 (14.6%) | |
| Other | 29 (10.1%) | 44 (6.2%) | |
| Cardiovascular Medications | |||
| Beta blocker | 221 (77.3%) | 637 (89.2%) | <0.001 |
| ACE inhibitor or ARB | 246 (86.0%) | 633 (88.7%) | 0.247 |
| Statin | 198 (69.2%) | 573 (80.3%) | <0.001 |
| Antiarrhythmic | 40 (14.0%) | 93 (13.0%) | 0.686 |
| Digoxin | 47 (16.4%) | 110 (15.4%) | 0.687 |
| Aspirin or Warfarin | 217 (75.9%) | 603 (84.5%) | 0.001 |
| Clopidogrel | 21 (7.3%) | 60 (8.4%) | 0.579 |
| Diuretic | 195 (68.2%) | 481 (67.4%) | 0.804 |
| Nitrate | 188 (65.7%) | 399 (55.9%) | 0.004 |
| Previous ICD | 9 (3.1%) | 25 (3.5%) | 0.780 |
| Pacemaker for heart rate | 6 (2.1%) | 11 (1.5%) | 0.590 |
| Pacemaker for resynchronization | 3 (1.0%) | 2 (0.3%) | 0.145 |
| Clinical Endpoints* | |||
| Death | 71 (24.8%) | 208 (29.1%) | 0.497 |
| Death or CV hospitalization | 148 (51.7%) | 433 (60.6%) | 0.031 |
Comparisons based on log-rank test
BMI = body mass index; MI = myocardial infarction; PCI = percutaneous coronary intervention; CABG = coronary artery bypass grafting; CCS = Canadian Cardiovascular Society; NYHA = New York Heart Association; LAD = left anterior descending; LV = left ventricular; ESVI = end systolic volume index; US = United States; CV = cardiovascular
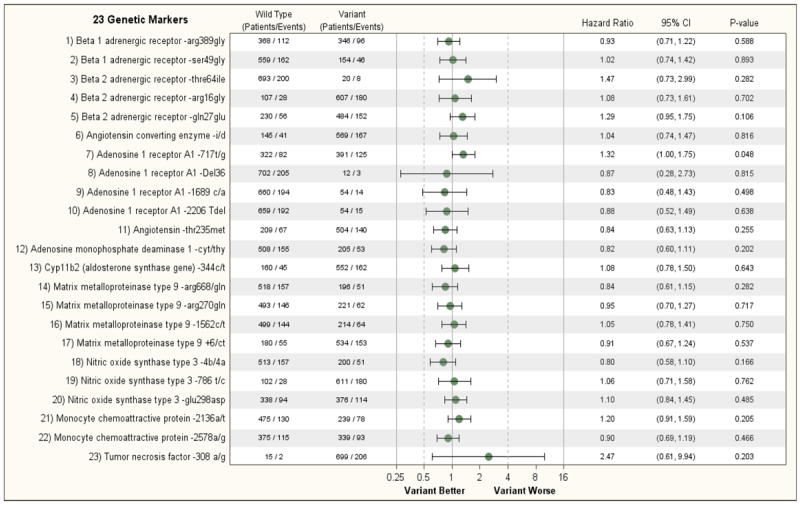
Figure 1A shows the relationship between the 23 genetic variants on one or both alleles of 11 cardiac genes and the primary endpoint of all-cause death or cardiovascular hospitalization of patients enrolled in Hypothesis 2 of the trial. No genetic variant met the threshold for statistical significance (p<0.002); however, several met nominal significance. The presence of an informative SNP on one or both alleles of the β2-AR gene that results in a shift of the amino acid at position 27 from a glutamine to a glutamic acid (β2-AR Gln27Glu) contributed nominal prognostic information in univariate analysis regarding a subject either dying or having a cardiovascular hospitalization during the course of the study (HR 1.27; 95% CI 1.03, 1.56, p = 0.027). This finding remained significant in multivariable analysis that examined the prognostic effect of this SNP after adjusting for prognostic clinical variables. (HR=1.24; 95% CI 1.01, 1.53, p = 0.045). The presence of a non-informative shift on one or both alleles in the nucleotide at position 717 in the coding region of the A1-adenosine receptor gene from a thymine to a guanine (A1-717 T/G) was associated with an increased risk of a subject either dying or being hospitalized for a cardiac problem by univariable analysis (HR = 1.23, 95% CI 1.02 – 1.49, p=0.031) and by multivariable analysis (HR=1.29, 95% CI 1.07, 1.57, p = 0.009); however, these associations only met nominal significance when assessed using the Bonferroni corrected significance level. The presence of a variant on one or both alleles of the A1 adenosine receptor gene was also nominally associated with the endpoint of all-cause mortality in both univariate (p=0.048) and multivariable (p=0.015) analysis, but again did not meet the Bonferroni-adjusted significance criterion. None of the other genetic variants provided significant predictive information with respect to the secondary end-point of mortality in the STICH Hypothesis 2 patients (Fig 1B and Supplemental Figure 1B). In addition, it is noteworthy that the treatment effect of CABG or the combination of CABG and ventricular reconstruction was not consistently modified by genotype. (Supplemental Figures 2A and 2B). Furthermore, the representation of each variant in the STICH population was consistent with earlier reports. Including the presence or absence of an ICD or the inclusion of the treatment itself in the adjusted model had no effect on the results.
Figure 1.
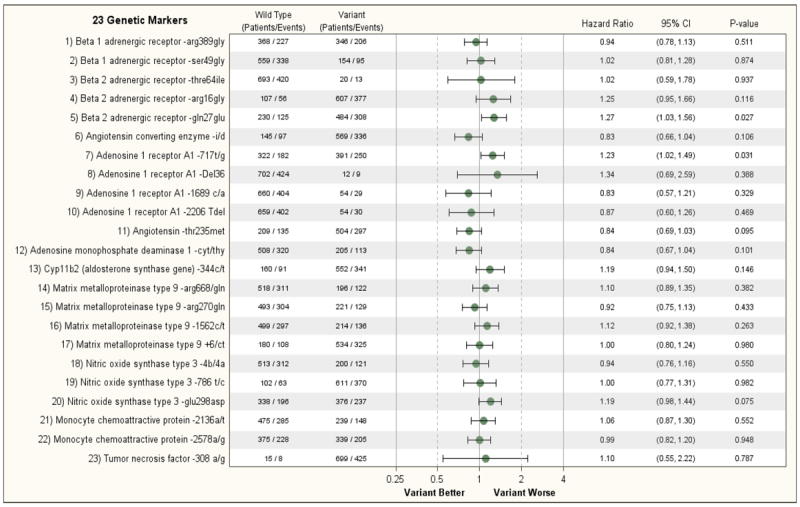
A: Relationship of 23 Genetic Markers and Clinical Outcomes (Non-adjusted) (Hypothesis 2, Endpoint= Death/CV Hospitalization)
B: Relationship of 23 Genetic Markers and Clinical Outcomes (Non-adjusted) (Hypothesis 2, Endpoint= Death)
STICH Hypothesis 1
Among the 1212 patients enrolled in Hypothesis 1, there were 76 patients who were also in Hypothesis 2. Those patients were analyzed with the Hypothesis 2 cohort, leaving a completely independent Hypothesis 1 cohort of 1136 patients. Table 2 shows the baseline characteristics of the 532 Hypothesis 1 patients who were included in the genetics sub-study cohort compared to the 604 patients who were not included. The patients with genetic data were older on average, had less racial diversity, had a higher frequency of history of stroke, hyperlipidemia, and atrial fibrillation, and higher average end systolic volume index, although a lower percentage with angina. The risk at randomization of the Hypothesis 1 genetic cohort was moderately higher than the patients who were not included, as evidenced by an increased incidence of death or cardiovascular hospitalization.
TABLE 2.
Comparison of STICH Hypothesis 1 Patients with vs. without Genetic Data
| Characteristic | No Genetic Data (n=604) | Genetic Data (n=532) | P Value |
|---|---|---|---|
| Age, median (25th, 75th), yrs. | 57.8 (52.5, 65.8) | 61.2 (54.8, 69.1) | <0.001 |
| Female, no. (%) | 67 (11.1%) | 72 (13.5%) | 0.210 |
| White race | 281 (46.5%) | 476 (89.5%) | <0.001 |
| Minority (Hispanic or racial minority) | 347 (57.5%) | 66 (12.4%) | <0.001 |
| BMI | 26.1 (23.3, 29.5) | 27.4 (24.5, 30.2) | <0.001 |
| Previous MI, no. (%) | 460 (76.2%) | 410 (77.1%) | 0.718 |
| Previous stroke, no. (%) | 29 (4.8%) | 56 (10.5%) | <0.001 |
| Diabetes, no. (%) | 229 (37.9%) | 226 (42.5%) | 0.117 |
| Hypertension, no. (%) | 353 (58.4%) | 338 (63.5%) | 0.080 |
| Hyperlipidemia, no. (%) | 313 (51.8%) | 361 (68.0%) | <0.001 |
| Current smoker | 128 (21.2%) | 100 (18.8%) | 0.308 |
| Chronic renal insufficiency, no. (%) | 39 (6.5%) | 48 (9.0%) | 0.105 |
| Peripheral vascular disease | 86 (14.2%) | 89 (16.7%) | 0.246 |
| Atrial fibrillation/flutter | 58 (9.6%) | 85 (16.0%) | 0.001 |
| Previous PCI, no. (%) | 55 (9.1%) | 82 (15.4%) | 0.001 |
| Previous CABG, no. (%) | 20 (3.3%) | 16 (3.0%) | 0.771 |
| Current CCS angina class, no. (%) | <0.001 | ||
| No angina | 175 (29.0%) | 242 (45.5%) | |
| I | 80 (13.2%) | 94 (17.7%) | |
| II | 322 (53.3%) | 170 (32.0%) | |
| III | 21 (3.5%) | 22 (4.1%) | |
| IV | 6 (1.0%) | 4 (0.8%) | |
| Current NYHA heart failure class, no. (%) | 0.012 | ||
| I | 58 (9.6%) | 73 (13.7%) | |
| II | 304 (50.3%) | 281 (52.8%) | |
| III | 230 (38.1%) | 161 (30.3%) | |
| IV | 12 (2.0%) | 17 (3.2%) | |
| Creatinine | 1.1 (1.0, 1.2) | 1.1 (0.9, 1.3) | 0.587 |
| Risk at randomization | 10.5 (5.0, 19.0) | 13.0 (5.0, 20.0) | 0.027 |
| No. of diseased vessels (≥75% stenosis), no. (%) | 0.151 | ||
| 1 | 136 (22.5%) | 143 (26.9%) | |
| 2 | 236 (39.1%) | 197 (37.1%) | |
| 3 | 232(38.4%) | 191 (36.0%) | |
| Left main (≥50% stenosis), no. (%) | 14 (2.3%) | 16 (3.0%) | 0.466 |
| Proximal LAD (≥75% stenosis), no. (%) | 432 (71.5%) | 333(62.7%) | 0.002 |
| LV ejection fraction, median (25th, 75th), % | 28.0 (22.9, 34.0) | 27.0 (22.0, 33.4) | 0.260 |
| ESVI, median (25th, 75th), mL/m2 | 76.6 (58.4, 97.0) | 81.8 (63.0, 105.3) | 0.005 |
| Mitral regurgitation | 0.326 | ||
| None or trace | 197 (32.7%) | 207 (39.1%) | |
| Mild (≤2+) | 306 (50.7%) | 218 (41.1%) | |
| Moderate or severe | 100 (16.6%) | 105 (19.8%) | |
| Region, no. (%) | <0.001 | ||
| Europe | 312 (51.7%) | 319 (60.0%) | |
| US | 33 (5.5%) | 74 (13.9%) | |
| Canada | 22 (3.6%) | 89 (16.7%) | |
| Other | 237 (39.2%) | 50 (9.4%) | |
| Cardiovascular Medications | |||
| Beta blocker | 497 (82.3%) | 471 (88.5%) | 0.003 |
| ACE inhibitor or ARB | 518 (85.8%) | 494 (92.9%) | <0.001 |
| Statin | 471 (78.0%) | 444 (83.5%) | 0.020 |
| Antiarrhythmic | 64 (10.6%) | 55 (10.3%) | 0.888 |
| Digoxin | 129 (21.4%) | 98 (18.4%) | 0.217 |
| Aspirin or Warfarin | 523 (86.6%) | 479 (90.0%) | 0.072 |
| Clopidogrel | 139 (23.0%) | 63(11.8%) | <0.001 |
| Diuretic | 419 (69.4%) | 434 (81.6%) | <0.001 |
| Nitrate | 364 (60.3%) | 245 (46.1%) | <0.001 |
| Previous ICD | 7 (1.2%) | 18 (3.4%) | 0.011 |
| Pacemaker for heart rate | 9 (1.5%) | 9 (1.7%) | 0.786 |
| Pacemaker for resynchronization | 4 (0.7%) | 2 (0.4%) | 0.690 |
| Clinical Endpoints* | |||
| Death | 226 (37.4%) | 215 (40.4%) | 0.234 |
| Death or CV hospitalization | 330 (54.6%) | 379 (71.2%) | <0.001 |
Comparisons based on log-rank test
BMI = body mass index; MI = myocardial infarction; PCI = percutaneous coronary intervention; CABG = coronary artery bypass grafting; CCS = Canadian Cardiovascular Society; NYHA = New York Heart Association; LAD = left anterior descending; LV = left ventricular; ESVI = end systolic volume index; United States; CV = cardiovascular
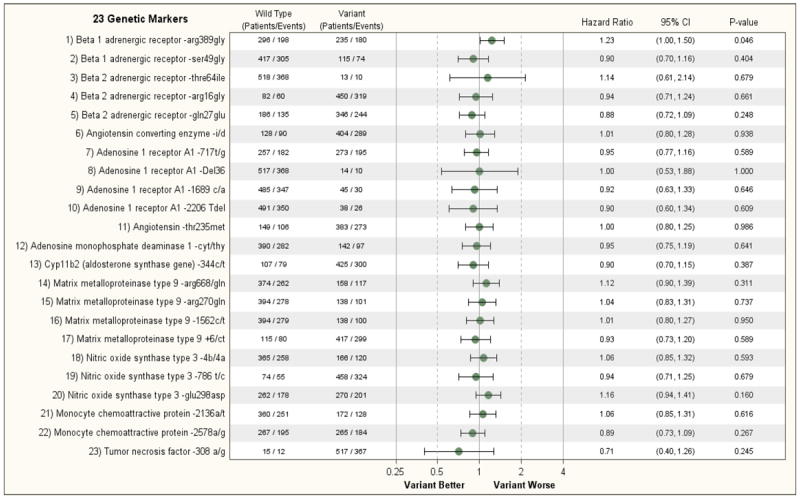
When assessing the relationship between the 23 genetic variants on one or both alleles of the 11 cardiac genes, no single genotype was even nominally associated with the primary outcome variable of all-cause mortality by either univariate or multivariable analysis (Figure 2A and Supplemental Figure 3A). Similar results were observed when assessing the secondary endpoint of death or cardiovascular hospitalization, that is, no single genetic variant was predictive of the secondary outcome in the STICH Hypothesis 1 population (Figure 2B and Supplemental Figure 3B). The treatment effect of CABG and optimal medical treatment vs. medical therapy alone was not significantly modified by the genotype with the exception of the A1-adensosine receptor gene (A1-717T/G) where there was a nominally significant interaction with treatment (p=0.032 for death and p=0.037 for death or cardiovascular hospitalization), with CABG having a greater effect in patients with no genetic variant present on either allele of the A1-adenosine receptor (Supplemental Figures 4A and 4B). However, given the number of comparisons performed, this result should be interpreted cautiously.
Figure 2.
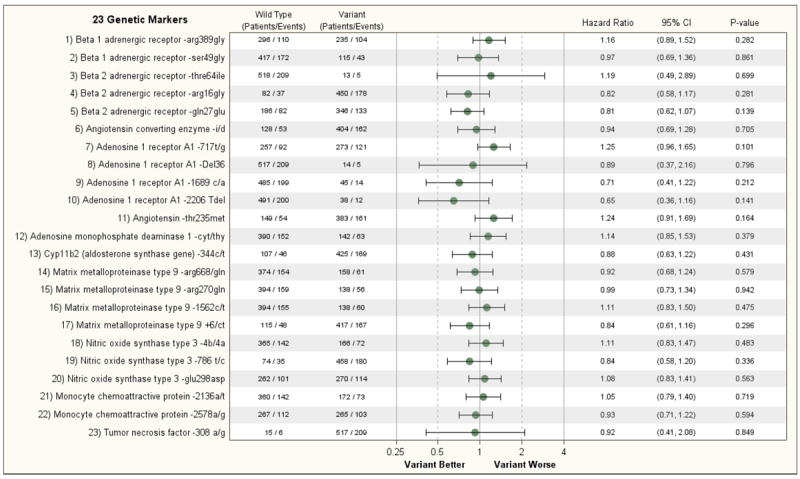
A: Relationship of 23 Genetic Markers and Clinical Outcomes (Non-adjusted) (Hypothesis 1, Endpoint= Death)
B: Relationship of 23 Genetic Markers and Clinical Outcomes (Non-adjusted) (Hypothesis 1, Endpoint= Death/CV Hospitalization)
DISCUSSION
Our finding that genotype was not consistently predictive of outcome in two distinct and heart failure populations is in conflict with some previous literature from single population studies. Polymorphisms in the beta-adrenergic receptor (β2-AR) gene have been extensively studied because of the critical role that the β2-AR plays in cardiac homeostasis.[24] Informative single nucleotide polymorphisms (SNPs) have been variably associated with changes in the function of the receptor, however, in most instances no relationship has been found. For example, deGroote et al.[25] found that neither the Arg16Gly, Gln27Glu or Thr16Ile polymorphism affected survival in a group of 444 consecutive patients with left ventricular systolic dysfunction although a significant effect was seen when looking at the haplotype. Similarly, Shin et al were not able to identify an association between a β2-AR genotype and outcome in a group of 227 patients followed at a single center. Furthermore, a study of 637 patients enrolled in 2 U.S. cardiovascular genetic registries with heart failure and left ventricular dysfunction and discharged on β-blockers, angiotensin converting enzyme inhibitors or angiotensin II receptor blockers and diuretics, failed to identify a relationship between β-AR genotypes and heart failure outcomes.[5] By contrast, a study of 122 patients demonstrated that those homozygous for the β2-AR Glu27 genotype were over five times more likely to have maladaptive ventricular remodeling after a myocardial infarction [26] and a study of 183 patients with heart failure demonstrated that this same group would have a more robust response to β-blocker therapy.[27]
Our study differed from the finding that a missense mutation at nucleotide 145 in the β1-AR gene was associated with decreased mortality in 184 patients with idiopathic dilated cardiomyopathy.[28] Our results are also disparate from earlier studies demonstrating that a SNP in the adenosine monophosphate deaminase 1gene predicted outcome in 132 patients with both ischemic and non-ischemic advanced heart failure [17] in 144 patients with heart failure post-myocardial infarction [28] and in 367 patients with coronary artery disease. [29] Our finding that the -1562 C/T variant in the Matrix metalloproteinase type 9 gene was not associated with outcome in patients with ischemic heart failure also conflicts with results of a previous study of 443 patients with both ischemic and non-ischemic heart failure, less than half of whom were receiving beta blockade, that suggested that the -1562 T/T genotype was an independent predictor of survival. [12]
It was also surprising that we were unable to demonstrate a relationship between the presence of the angiotensin converting enzyme (ACE) D/D or I/D genotypes and outcome in the STICH trial. The D/D allele has been associated with increased production of angiotensin II, a neurohormone widely associated with a poor prognosis in patients with heart failure.[8] While an early study in 99 patients with heart failure failed to identify an association between ACE genotype and outcome, [8] we found in relatively large populations of heart failure patients that the presence of the deletion on even one allele is associated with a significantly worse prognosis. [9] However, many of these patients were either receiving a low dose of an ACE inhibitor or no ACE inhibitor at all. Our results also differ from our recent finding that the C>T SNP in the aldosterone synthase gene may predict outcome in African-Americans with heart failure, although this group was poorly represented in the STICH trial.[10] The finding that a non-informative SNP in the adenosine receptor gene was nominally associated with a worse outcome in patients in STICH Hypothesis 2 was intriguing: however, we could not confirm this finding in the STICH Hypothesis 1 population suggesting that the finding was a statistical aberration.
Several important factors may explain the disparity between the present studies and earlier reports. First, and foremost we performed analysis of multiple genetic markers in two separate and distinct but very similar patient populations. This allowed us the opportunity to confirm or in our case refute findings from a single study in a comparable patient population. Second, by protocol, each of the patients enrolled in STICH Hypothesis 1 and STICH Hypothesis 2 was receiving optimal medical therapy. We have shown previously that optimal medical therapy can obviate differences seen in untreated or under-treated populations when assessing genetic variants in drug targets (β-blockers or ACE inhibitors).[30] Over 90% of patients enrolled in STICH were treated with both a β-blocker and an angiotensin converting enzyme inhibitor or an angiotensin receptor antagonist and pharmacologic dosages were optimized. Thus, the medical regimen of the patients was substantially more robust than in many of the earlier genotyping studies. The size of the trial was also of potential importance. The number of patients enrolled in the sub-study for both STICH Hypothesis 1 and STICH Hypothesis 2 was greater than the number of individuals enrolled in many earlier studies thereby reducing the risk of aberrant findings. And finally, the STICH population was relatively homogenous. All of the subjects had heart failure secondary to ischemic heart disease and the vast majority of the patients were Caucasian with a Northern-European ethnic background. This is in contrast to most earlier studies that assessed genotype in patients with idiopathic disease or with the idiopathic and ischemic cardiomyopathy.
A limitation of this study was that we took a reductionist approach to identifying genetic variants that could predict risk in patients with left ventricular dysfunction secondary to ischemic heart disease who were enrolled in the STICH trials. This approach was necessitated by the fact that the technology required for genome-wide association studies (GWAS) or whole exome or whole genome sequencing was not available during either the planning phase or the implementation stage of the STICH trials. Consistent with the present results, neither GWAS studies nor whole exome sequencing have identified the genetic variants that were evaluated in STICH despite the fact that many of these variants had been demonstrated to influence treatment outcome in large studies of patients with heart failure – the Arg389Gly variant in the β1-adrenergic receptor gene being a good example. [31] For example, Meder et al found a close association of genetic variants on chromosome 6p21 with the development of idiopathic dilated cardiomyopathy and an association of HLA-C gene expression with this locus: a finding that suggested a link between susceptibility to idiopathic dilated cardiomyopathy and auto-immune mechanisms that lead to myocardial inflammation. Similarly, GWAS identified a genetic susceptibility locus on chromosome 10q26 within the BCL2-associated athanogene 3 (BAG3) gene. Variants in this gene have been shown by our own group and by others to be a monogenic cause of dilated cardiomyopathy. [32,33,34] Because most GWAS and whole exome sequencing studies have focused on patients with idiopathic dilated cardiomyopathy, it is difficult to compare our results with those obtained from GWAS or other newer methodologies applied to individuals with idiopathic or familial heart failure. Further studies in patients with ischemic heart disease using GWAS or whole exome/genome sequencing will be useful in furthering our understanding of the linkage between genotype and outcomes in this group of patients.
Our study has several additional limitations. First, while the overwhelming majority of the patients were form North America and Europe, significant genetic differences and population stratification could have occurred in this international trial. Second, the study was begun in 2002. Therefore, we were not able to take advantage of new and less costly technology that might have allowed us to pursue genome wide association in haplotype identification. Third, since most of the patients in STICH were considered for cardiac surgery, the results may not be able to be extrapolated to the overall ischemic group or to the overall STICH group that differs in some baseline characteristics from the genetic sub-study population. Finally, because heart failure patients in general do well on optimal medical therapy, a longer period of follow-up might have revealed an association between genetic variants and outcomes that were not obvious in the present analysis.
Nonetheless, our failure to identify and confirm genetic markers of outcome in these two heart failure populations points out the need to confirm genotypic findings in a comparable population, the importance of study size in genetic analysis, the importance of optimizing medical therapy and the need to study populations that are homogenous. These lessons will be of particular importance as new technologies increase our ability to readily measure multiple genetic markers in populations as well as to sequence a subject’s entire exome or genome.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Heart, Lung and Blood Institutes, nos. HL69015, HL-069012, HL070011, HL072430, HL-069009, HL-069010, HL-069012, HL-069011, HL-069013, and HL-072683
Contributor Information
Arthur M. Feldman, Department of Medicine, Temple University School of Medicine, Philadelphia, PA
Lilin She, Duke Clinical Research Institute, Durham, NC
Dennis M. McNamara, Department of Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA
Douglas L. Mann, Department of Medicine, Washington University in St. Louis School of Medicine, St. Louis, MO
Michael R. Bristow, Department of Medicine, University of Colorado at Denver, Denver, CO
Alan S. Maisel, Department of Medicine, University of California, San Diego, San Diego, CA
Daniel R. Wagner, Laboratory of Cardiovascular Research, Luxembourg Heart Institute, Luxembourg, Luxembourg
Bert Andersson, Department of Cardiology, Sahigrenska University Hospital, Gothenberg, Sweden
Luigi Chiariello, Department of Cardiac Surgery, Policlinico Tor Vergate of Rome, Rome, Italy
Christopher S. Hayward, Department of Cardiology, St. Vincent’s Hospital, Sydney, AU
Paul Hendry, Department of Cardiac Surgery, Ottawa Heart Institute, Ottawa, Ontario, Canada
John D. Parker, Department of Medicine, Toronto General Hospital, Toronto, Ontario, Canada
Normand Racine, Department of Medicine, Montreal Heart Institute, Montreal, Quebec, Canada
Craig H. Selzman, Department of Surgery, University of Utah Hospital, Salt Lake City, UT
Michele Senni, Department of Medicine, Ospedali Riuniti, Bergamo, Italy
Janina Stepinska, Valvular Heart Department, National Institute of Cardiology, Warsaw, Poland
Marian Zembala, Department of Cardiac Surgery, Silesian Center for Heart Diseases, Zabrze, Poland
Jean Rouleau, Department of Medicine, Montreal Heart Institute, Montreal, Quebec, Canada
Eric J. Velazquez, Department of Medicine, Duke University School of Medicine, Durham, NC
Kerry L. Lee, Duke Clinical Research Institute, Durham, NC
References
- 1.Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y. Heart disease and stroke statistics--2008 update: A report from the american heart association statistics committee and stroke statistics subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 2.Evans WE, McLeod HL. Pharmacogenomics--drug disposition, drug targets, and side effects. N Engl J Med. 2003;348:538–549. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 3.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115:518–526. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, Weber SA, Greene SM, Hodne D, Nelson B, Morrison J, Domanski MJ, Wagoner LE, Abraham WT, Anderson JL, Carlquist JF, Krause-Steinrauf HJ, Lazzeroni LC, Port JD, Lavori PW, Bristow MR. A polymorphism within a conserved beta(1)-adrenergic receptor motif alters cardiac function and beta-blocker response in human heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11288–11293. doi: 10.1073/pnas.0509937103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sehnert AJ, Daniels SE, Elashoff M, Wingrove JA, Burrow CR, Horne B, Muhlestein JB, Donahue M, Liggett SB, Anderson JL, Kraus WE. Lack of association between adrenergic receptor genotypes and survival in heart failure patients treated with carvedilol or metoprolol. Journal of the American College of Cardiology. 2008;52:644–651. doi: 10.1016/j.jacc.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 6.Shin J, Lobmeyer MT, Gong Y, Zineh I, Langaee TY, Yarandi H, Schofield RS, Aranda JM, Jr, Hill JA, Pauly DF, Johnson JA. Relation of beta(2)-adrenoceptor haplotype to risk of death and heart transplantation in patients with heart failure. Am J Cardiol. 2007;99:250–255. doi: 10.1016/j.amjcard.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Raynolds MV, Bristow MR, Bush EW, Abraham WT, Lowes BD, Zisman LS, Taft CS, Perryman MB. Angiotensin-converting enzyme dd genotype in patients with ischaemic or idiopathic dilated cardiomyopathy. Lancet. 1993;342:1073–1075. doi: 10.1016/0140-6736(93)92061-w. [DOI] [PubMed] [Google Scholar]
- 8.Montgomery HE, Keeling PJ, Goldman JH, Humphries SE, Talmud PJ, McKenna WJ. Lack of association between the insertion/deletion polymorphism of the angiotensin-converting enzyme gene and idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 1995;25:1627–1631. doi: 10.1016/0735-1097(95)00109-h. [DOI] [PubMed] [Google Scholar]
- 9.McNamara DM, Holubkov R, Postava L, Janosko K, MacGowan GA, Mathier M, Murali S, Feldman AM, London B. Pharmacogenetic interactions between angiotensin-converting enzyme inhibitor therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. J Am Coll Cardiol. 2004;44:2019–2026. doi: 10.1016/j.jacc.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 10.McNamara DM, Tam SW, Sabolinski ML, Tobelmann P, Janosko K, Taylor AL, Cohn JN, Feldman AM, Worcel M. Aldosterone synthase promoter polymorphism predicts outcome in african americans with heart failure: Results from the a-heft trial. J Am Coll Cardiol. 2006;48:1277–1282. doi: 10.1016/j.jacc.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 11.Hengstenberg C, Holmer SR, Mayer B, Lowel H, Engel S, Hense HW, Riegger GA, Schunkert H. Evaluation of the aldosterone synthase (cyp11b2) gene polymorphism in patients with myocardial infarction. Hypertension. 2000;35:704–709. doi: 10.1161/01.hyp.35.3.704. [DOI] [PubMed] [Google Scholar]
- 12.Mizon-Gerard F, de Groote P, Lamblin N, Hermant X, Dallongeville J, Amouyel P, Bauters C, Helbecque N. Prognostic impact of matrix metalloproteinase gene polymorphisms in patients with heart failure according to the aetiology of left ventricular systolic dysfunction. Eur Heart J. 2004;25:688–693. doi: 10.1016/j.ehj.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Drigo SA, Cunha-Neto E, Ianni B, Cardoso MR, Braga PE, Fae KC, Nunes VL, Buck P, Mady C, Kalil J, Goldberg AC. Tnf gene polymorphisms are associated with reduced survival in severe chagas’ disease cardiomyopathy patients. Microbes Infect. 2006;8:598–603. doi: 10.1016/j.micinf.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Kubota T, McNamara DM, Wang JJ, Trost M, McTiernan CF, Mann DL, Feldman AM. Effects of tumor necrosis factor gene polymorphisms on patients with congestive heart failure. Vest investigators for tnf genotype analysis Vesnarinone survival trial. Circulation. 1998;97:2499–2501. doi: 10.1161/01.cir.97.25.2499. [DOI] [PubMed] [Google Scholar]
- 15.McNamara DM, Holubkov R, Postava L, Ramani R, Janosko K, Mathier M, MacGowan GA, Murali S, Feldman AM, London B. Effect of the asp298 variant of endothelial nitric oxide synthase on survival for patients with congestive heart failure. Circulation. 2003;107:1598–1602. doi: 10.1161/01.CIR.0000060540.93836.AA. [DOI] [PubMed] [Google Scholar]
- 16.McNamara DM, Tam SW, Sabolinski ML, Tobelmann P, Janosko K, Venkitachalam L, Ofili E, Yancy C, Feldman AM, Ghali JK, Taylor AL, Cohn JN, Worcel M. Endothelial nitric oxide synthase (nos3) polymorphisms in african americans with heart failure: Results from the a-heft trial. Journal of cardiac failure. 2009;15:191–198. doi: 10.1016/j.cardfail.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 17.Loh E, Rebbeck TR, Mahoney PD, DeNofrio D, Swain JL, Holmes EW. Common variant in ampd1 gene predicts improved clinical outcome in patients with heart failure. Circulation. 1999;99:1422–1425. doi: 10.1161/01.cir.99.11.1422. [DOI] [PubMed] [Google Scholar]
- 18.de Groote P, Lamblin N, Helbecque N, Mouquet F, Hermant X, Amouyel P, Dallongeville J, Bauters C. The impact of the ampd1 gene polymorphism on exercise capacity, other prognostic parameters, and survival in patients with stable congestive heart failure: A study in 686 consecutive patients. Am Heart J. 2006;152:736–741. doi: 10.1016/j.ahj.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Velazquez EJ, Lee KL, O’Connor CM, Oh JK, Bonow RO, Pohost GM, Feldman AM, Mark DB, Panza JA, Sopko G, Rouleau JL, Jones RH. The rationale and design of the surgical treatment for ischemic heart failure (stich) trial. The Journal of thoracic and cardiovascular surgery. 2007;134:1540–1547. doi: 10.1016/j.jtcvs.2007.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velazquez EJ, Lee KL, Deja MA, Jain A, Sopko G, Marchenko A, Ali IS, Pohost G, Gradinac S, Abraham WT, Yii M, Prabhakaran D, Szwed H, Ferrazzi P, Petrie MC, O’Connor CM, Panchavinnin P, She L, Bonow RO, Rankin GR, Jones RH, Rouleau JL. Coronary-artery bypass surgery in patients with left ventricular dysfunction. N Engl J Med. 364:1607–1616. doi: 10.1056/NEJMoa1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones RH, Velazquez EJ, Michler RE, Sopko G, Oh JK, O’Connor CM, Hill JA, Menicanti L, Sadowski Z, Desvigne-Nickens P, Rouleau JL, Lee KL. Coronary bypass surgery with or without surgical ventricular reconstruction. The New England journal of medicine. 2009;360:1705–1717. doi: 10.1056/NEJMoa0900559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC, Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. Acc/aha 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: A report of the american college of cardiology/american heart association task force on practice guidelines (writing committee to update the 2001 guidelines for the evaluation and management of heart failure): Developed in collaboration with the american college of chest physicians and the international society for heart and lung transplantation: Endorsed by the heart rhythm society. Circulation. 2005;112:e154–235. doi: 10.1161/CIRCULATIONAHA.105.167586. [DOI] [PubMed] [Google Scholar]
- 23.Cox DR. Regression models and life-tables. J Royal Statistical Society Series B (Methodological) 1972;34:187–220. [Google Scholar]
- 24.Dorn GW, 2nd, L S. Pharmacogenmoics in beta-adrenergic receptors and their acccessory signaling proteins in heart failure. Clin Transl Sci. 2008;1:255–262. doi: 10.1111/j.1752-8062.2008.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Groote P, Lamblin N, Helbecque N, Mouquet F, Mc Fadden E, Hermant X, Amouyel P, Dallongeville J, Bauters C. The impact of beta-adrenoreceptor gene polymorphisms on survival in patients with congestive heart failure. Eur J Heart Fail. 2005;7:966–973. doi: 10.1016/j.ejheart.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 26.McLean RC, Hirsch GA, Becker LC, Kasch-Semenza L, Gerstenblith G, Schulman SP. Polymorphisms of the beta adrenergic receptor predict left ventricular remodeling following acute myocardial infarction. Cardiovascular drugs and therapy / sponsored by the International Society of Cardiovascular Pharmacotherapy. 2011;25:251–258. doi: 10.1007/s10557-011-6307-7. [DOI] [PubMed] [Google Scholar]
- 27.Metra M, Covolo L, Pezzali N, Zaca V, Bugatti S, Lombardi C, Bettari L, Romeo A, Gelatti U, Giubbini R, Donato F, Dei Cas L. Role of beta-adrenergic receptor gene polymorphisms in the long-term effects of beta-blockade with carvedilol in patients with chronic heart failure. Cardiovascular drugs and therapy / sponsored by the International Society of Cardiovascular Pharmacotherapy. 2010;24:49–60. doi: 10.1007/s10557-010-6220-5. [DOI] [PubMed] [Google Scholar]
- 28.Collins RP, Palmer BR, Pilbrow AP, Frampton CM, Troughton RW, Yandle TG, Skelton L, Richards AM, Cameron VA. Evaluation of ampd1 c34t genotype as a predictor of mortality in heart failure and post-myocardial infarction patients. Am Heart J. 2006;152:312–320. doi: 10.1016/j.ahj.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 29.Anderson JL, Habashi J, Carlquist JF, Muhlestein JB, Horne BD, Bair TL, Pearson RR, Hart N. A common variant of the ampd1 gene predicts improved cardiovascular survival in patients with coronary artery disease. J Am Coll Cardiol. 2000;36:1248–1252. doi: 10.1016/s0735-1097(00)00850-0. [DOI] [PubMed] [Google Scholar]
- 30.McNamara DM, Holubkov R, Janosko K, Palmer A, Wang JJ, MacGowan GA, Murali S, Rosenblum WD, London B, Feldman AM. Pharmacogenetic interactions between beta-blocker therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. Circulation. 2001;103:1644–1648. doi: 10.1161/01.cir.103.12.1644. [DOI] [PubMed] [Google Scholar]
- 31.Bristow MR. Treatment of chronic heart failure with β-adrenergic receptor antagonists: a convergence of receptor pharmacology and clinical cardiology. Circ Res. 2011;109(10):1176–94. doi: 10.1161/CIRCRESAHA.111.245092. [DOI] [PubMed] [Google Scholar]
- 32.Villard E, Perrret C, Gary F, Proust C, Dilanian G, Hengstenberg C, Ruppert V, Arbustini E, Wichter T, Germain M, Dubourg O, Tavazzi L, Aumont MD, DeGroote p, Fauchier L, Trochu JN, Gibelin P, Aupetit JF, Stark K, Erdmann J, Hetzer R, Roberts AM, Barton PJ, Regitz-Zagrosek V, Aslam U, Duboscq-Bidot L, Meyborg M, Maisch B, Madeira H, Waldenstrom A, Galve E, Cleland JG, Dorent R, Roizes G, Zeller T, Blankenberg S, Goodall AH, Cook S, Tregouet DA, Tiret L, Isnard R, Komajda M, Charron P, Cambien F, Cariogenics Consortium A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. 2011 May;32(9):1065–76. doi: 10.1093/eurheartj/ehr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldman AM, Begay RL, Knezevic T, Myers VD, Slavov DB, Zhu W, Gowan K, Graw SL, Jones KL, Tilley DG, Coleman RC, Walinsky P, Cheung JY, Mestroni L, Khalili K, Taylor MR. Decreased levels of BAG3 in a Family with a Rare Variant and in Idiopathic Dilated Cardiomyopathy. J Cell Physiol. 2014 Nov;229(11):1697–702. doi: 10.1002/jcp.24615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA, Hershberger RE. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011 Mar 11;88(3):273–82. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.