

Abstract
Transforming growth factor (TGF)-beta is a central stimulus of the events leading to chronic progressive kidney disease, having been implicated in the regulation of cell proliferation, hypertrophy, apoptosis and fibrogenesis. The fact that it mediates these varied events suggests that multiple mechanisms play a role in determining the outcome of TGF-beta signaling. Regulation begins with the availability and activation of TGF-beta and continues through receptor expression and localization, control of the TGF-beta family-specific Smad signaling proteins, and interaction of the Smads with multiple signaling pathways extending into the nucleus. Studies of these mechanisms in kidney cells and in whole-animal experimental models, reviewed here, are beginning to provide insight into the role of TGF-beta in the pathogenesis of renal dysfunction and its potential treatment.
Keywords: TGF-beta, Fibrosis, Signal Transduction, Smad, Cross-Talk, Review
2. INTRODUCTION
Transforming growth factor (TGF)-beta is critical for normal kidney development and function. TGF-beta expression occurs in the mammalian metanephros during tubular development. Thompson et al identified TGF-beta protein within the cytoplasm of a subset of distal tubular epithelial cells and at the plasma membranes of cells in the corticomedullary junction in the embryonic murine kidney (1). In disease, TGF-beta affects multiple systems. It limits glomerular damage following acute injury by inhibiting macrophage function (2). Conversely, knockout of the TGF-beta1 gene causes mice to succumb to wasting and overwhelming inflammatory infiltration of abdominal viscera (3). In vitro, TGF-beta downregulates beta2 integrin (CR3), a receptor required by macrophages to adhere to areas of injury (4). In cultured mesangial and tubular epithelial cells, TGF-beta prevents mitosis through cyclin kinase inhibition (5), suppressing macrophage-derived IL-1 secretion.
Notwithstanding the importance of this growth factor for tissue homeostasis, TGF-beta is notorious for its role as a mediator of progressive kidney disease. Since TGF-beta has pleiotrophic effects, stimuli that normally would maintain homeostasis instead, when applied excessively or at the wrong time or location, may trigger a harmful response. Adverse effects of TGF-beta such as renal fibrosis occur under conditions of excessive or uncontrolled activation of all renal cell types. Following an insult, cells native to the kidney secrete chemotactic cytokines to summon monocytes and lymphocytes to the area of injury. TGF-beta stimulation also can activate a number of functions in the native cells that, unopposed, lead to apoptosis, extracellular matrix (ECM) accumulation or loss of function via cell dedifferentiation. As a result, the finely orchestrated structural and functional balance among the cells is disrupted.
3. TGF-BETA AND THE PATHOGENESIS OF CHRONIC KIDNEY DISEASE
Renal function is accomplished by a filtration apparatus, the glomerulus, that is comprised mainly of a vascular endothelium, a visceral epithelium, and a support structure provided by the mesenchymal-phenotype mesangial cells; and by a reabsorptive apparatus comprised of the renal tubular epithelium. In chronic kidney disease, damage to these structures impairs the ability of the kidney to purify the plasma; causes scarring and collapse of normal structures; and disrupts blood perfusion causing the structures to atrophy. These phenomena are characteristic of chronic, progressive kidney disease.
TGF-beta has been increasingly recognized as a central participant contributing to these events. Increased urinary TGF-beta is detected in patients with some forms of nephrotic syndrome (6), IgA nephropathy (7), and focal segmental glomerulosclerosis (FSGS) (7, 8). Interestingly, while detection of TGF-beta in the urine can differentiate between FSGS and the non-fibrotic process of minimal change disease (8), TGF-beta levels cannot be used predict response to corticosteroid therapy, despite the fact that some anti-fibrotic treatments may reduce these levels (9). In human FSGS (10) and in experimental models of renal fibrosis (11), TGF-beta is closely associated with ECM accumulation. Infusion of an expression plasmid encoding TGF-beta stimulates renal fibrosis (12), and mice transgenic for TGF-beta under the control of an albumin promoter (to stimulate hepatic synthesis of TGF-beta) develop glomerulosclerosis (13). In contrast, infusion of an oligonucleotide antisense to TGF-beta inhibits fibrosis in anti-Thy-1 nephritis (14) and repeated injection of an antibody against TGF-beta decreases renal scarring in a subtotal nephrectomy model (15). Administration of decorin, an ECM protein that binds TGF-beta (16), or infusion of a competing, soluble form of TGF-beta receptor (TbetaR) (17), also inhibits renal scarring.
Thus, understanding the role of TGF-beta in regulating cellular function will provide data that can address the molecular pathogenesis and treatment of progressive kidney failure. Here, we will present an overview of TGF-beta signaling and its relevance to progressive kidney disease. TGF-beta may affect four critical aspects of cell function in a manner that could mediate the development of renal failure. Proliferative changes may distort structures and render them nonfunctional; conversely, apoptosis or similar events may cause cytopenia that either directly or indirectly leads to atrophy and loss of structural or functional integrity. These events often are associated with phenotypic changes that alter the way the cell responds to various stimuli. Usually, these changes involve a relative de-differentiation of the cell in a process called epithelial-to-mesenchymal transition (EMT). Finally, ECM accumulation results in the typical scar that denotes a non-functional organ. This scar may be comprised of increased amounts of the basement membrane matrix proteins that are the usual substrate for the cells, such as certain laminins or type IV collagen; or the accumulated ECM may include alternative proteins such as different laminins or type I collagen.
It is important to remember that, in vivo, TGF-beta signaling mechanisms interact with a large number of additional hormones, mediators, ECM proteins and physical factors that influence the outcome of the TGF-beta signal. These interacting factors affect the four critical aspects of cell function mentioned above, in multiple ways. Here, we will address various levels of regulation of TGF-beta signaling, from outside the cell to the transcriptional complex. Some of the topics that we address will be considered in more detail elsewhere in this issue of Frontiers in Bioscience.
4. THE CANONICAL TGF-BETA SIGNALING PATHWAY
The TGF-beta superfamily consists of TGF-beta1, -beta2, and -beta3; activins; and bone morphogenic proteins (BMP). These proteins are expressed in virtually all mammalian cell types, as are their downstream signaling mediators, the Smad proteins. TGF-beta signaling is initiated when ligand-bound, type II TGF-beta receptor (TbetaRII) binds to, and phosphorylates, the type I TGF-beta receptor (TbetaRI) (18–20) at the TbetaRI cytoplasmic GS region. The resulting kinase activity phosphorylates serines at the carboxy-termini of the receptor-regulated Smads (R-Smads): Smad2 and Smad3 in the case of TGF-betas, and Smad1 and Smad5 with BMPs. The phosphorylated R-Smads form a multimeric complex with the common mediator (Co)-Smad (Smad4) and accumulate in the nucleus to regulate transcriptional responses. The inhibitory Smads, Smad6 and Smad7, compete with the R-Smads for binding to the activated receptors (20). R-Smads and Smad4 contain a MAD homology (MH)-1 domain (Figure 1) that interacts with DNA and with other transcriptional regulators; a hydrophilic linker region; and a cysteine-rich hydrophobic MH2 domain involved in protein-protein interactions with the TbetaR, transcriptional cofactors and other Smads. The MH2 domain also has transactivational activity (19, 20), and nuclear transport of the activated R-Smad depends at least in part on the MH2 region (21). The Smad pathway is present and active in kidney cells (22, 23), but its apparent simplicity raises an important question. Given the many cell functions that are regulated by TGF-beta, how can such a straightforward pathway determine which events occur upon TGF-beta stimulation? The answer lies in the interaction of Smad proteins with numerous other signaling molecules and pathways in the cell (Figure 2). The balance among these modifying inputs, addressed in this review, likely determines the signaling outcome.
Figure 1.
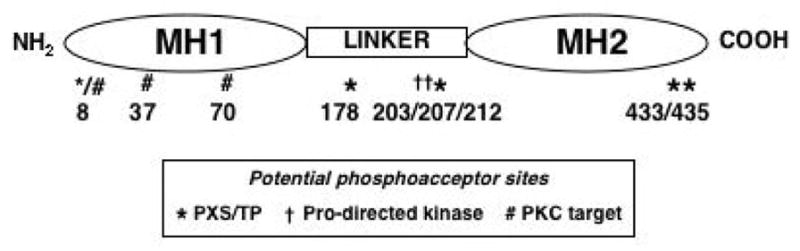
Domain structure of Smad3. Potential phosphoacceptor residues are indicated by the symbols; the corresponding numbers indicate the amino acid positions in the sequence.
Figure 2.

Signal transduction pathways from the cell membrane to the nucleus discussed in this article. Solid lines with arrows indicate positive effects; broken lines indicate inhibitory effects. Tsp, thrombospondin; HA, hyaluronan; 1, TGF-beta receptor (TbetaR) I; 2, TbetaR II; PI3K, Phosphatidylinositol-3-kinase; SARA, Smad anchor for receptor activation; Dab2, disabled-2; TAK, TGF-beta-activated kinase; FAK, focal adhesion kinase; PKC, protein kinase C; UBQ, ubiquitin; Smad3-cP, Smad3 phosphorylated on the C-terminus; Smad3-LP-cP, Smad3 phosphorylated at the linker region and at the C-terminus; CTGF, connective tissue growth factor; ECM, extracellular matrix. Three inhibitory pathways, involving (respectively) bone morphogenetic protein-7, hepatocyte growth factor and the nuclear receptor-associated hormones, are not shown because they act at multiple sites in the Smad2/Smad3-mediated pathway. The inclusion of arrows depicting both enhancing and inhibiting effects for Smad linker region phosphorylation indicate that the implications of these phosphorylation events remain poorly defined.
5. REGULATION OF TGF-BETA EXPRESSION AND ACTIVITY
TGF-beta synthesis is increased in several models of kidney disease. In addition to the increased urinary excretion cited above, increased renal immunostaining for TGF-beta has been noted in fibrotic kidney diseases (6). TGF-beta production may be a self-sustaining, autocrine event in injured renal cells (24), but it also is increased in these cells by a variety of events that initiate beyond the kidney such as diabetic nephropathy (25), where increased systemic glucose levels increase renal TGF-beta expression, probably mediated by enhanced protein kinase C (PKC)betaII activity and subsequently increased diacylglycerol (26). TGF-beta expression, or that of its receptor, may be mediated by activation of the AP-1 transcription factor, since AP-1 antagonists block an autocrine loop for TGF-beta expression in rat renal fibroblasts (27).
TGF-beta usually is present in latent form. It can be activated by interaction with integrins (28) and the renal cell response to TGF-beta may be enhanced by high ambient glucose levels (29) or integrin-ECM binding (30). Two ECM pre-receptor regulators of TGF-beta activity, thrombospondin-1 (Tsp-1) and hyaluronan (HA), are particularly noteworthy. Tsp-1 appears to enhance TGF-beta activity in several ways. It binds to TGF-beta, concentrating the effects of the growth factor at areas where cells are binding to the ECM. Tsp-1 expression is increased in diabetes (31), possibly through increased activity of the ERK and p38 MAP kinases (32). Both high glucose concentrations and elevated angiotensin II activity in diabetes increase Tsp-1 expression, amplifying TGF-beta signaling even in the absence of increased TGF-beta synthesis (33). In addition to diabetic nephropathy (34), Tsp-1 expression and activity are increased in complement-mediated nephritis (35) and mesangial proliferative glomerulonephritis (36). In the latter model, infusion of a soluble Tsp-blocking peptide ameliorated the extent of the disease, concomitant with a reduction in renal TGF-beta1 expression, further supporting a role for Tsp-1 in TGF-beta expression.
Another pre-receptor molecule that modifies TGF-beta activity, HA, is an extracellular polysaccharide that interacts with its cognate receptor, CD44, and prevents the TbetaR from moving to early endosomes, instead driving it to caveolae (37). This altered TbetaR compartmentalization may promote its degradation (38). HA itself does not alter collagen synthesis, but it blocks TGF-beta-stimulated increased expression of type II and type IV collagen in cultured proximal tubular epithelial (PTE) cells (37).
6. REGULATION OF TGF-BETA SIGNALING
The TGF-beta/Smad signal transduction pathway is modulated by TbetaR expression and phosphorylation. The role of the Smads is critical, and many additional proteins have been found to promote Smad expression, stability, activation and assembly into transcription-regulatory complexes (23). In addition, the Smads engage in cross-talk with other signaling pathways, some activated by TGF-beta and others activated by additional cues present in the cellular microenvironment. Finally, TGF-beta may act through pathways that are not directly linked to Smad activation. Many of these moderating influences have been observed in the kidney.
6.1 Regulation at the level of the receptor
TbetaRI kinase activity usually results from interaction of TbetaRII with TbetaRI. Therefore, changes in cell surface expression of each TbetaR will alter the stoichiometry of this activity. In classical profibrotic TGF-beta signaling, the TbetaRI that is involved is the activin-linked kinase, ALK5. ALK5 and ALK4 typically activate Smad2 and Smad3 (39). In contrast, ALK1 (40) and ALK2 (41) primarily activate Smad1 and Smad5, and are usually associated with BMP signaling. A third receptor subgroup, TbetaRIII, lacks kinase activity but may participate in ligand binding, particularly in response to TGF-beta2, and in assembly of the signaling complex (42, 43). Glomerular expression of TbetaRIII varies with the nature, location and severity of disease (44, 45). Smad1 also is activated by endoglin, an auxiliary TbetaR protein that can participate in the receptor complex (46) and has anti-fibrotic effects in kidney cells (47). Endoglin may serve to recruit Smad1 to the Alk2 TbetaRI, enhancing BMP-7-related signaling (48). Although Smad1 typically is activated by BMPs, it can be activated by TGF-beta through endoglin, particularly in endothelial cells (49).
Since TbetaRI phosphorylation is important for transducing the ligand-induced signal to Smads, the level and duration of this phosphorylation is important. A complex of GADD34 and the phosphatase PP1c dephosphorylates the type I receptor (50). Another phosphatase, PP1alpha, dephosphorylates ALK1 (51). Both of these phosphatases are recruited to the activated TbetaRI by Smad7, suggesting a cascade of inhibitory actions induced by the involvement of Smad7 in the TbetaR complex.
Finally, signal propagation may depend on trafficking of the receptor. The TbetaR signaling complex is internalized by clathrin-mediated endocytosis to the early endosome (52–54). However, whether endocytosis is always required in TGF-beta signaling is unknown ((55–57) and references therein). As described above, the receptor may be internalized alternatively through a caveolin-1-lipid raft where it is degraded as part of a TbetaR/Smad7 complex with the ubiquitin E3 ligase, Smurf2 (38).
6.2. Regulation of Smad expression
Although Smads do not appear to be highly abundant molecules, they are present to some degree in most cells. Downregulation of Smads occurs by two mechanisms. They can be degraded through a ubiquitin/proteasome-mediated mechanism; phosphorylated Smads are particularly sensitive to this form of regulation. The E3 ubiquitin ligase Smurf family appears to play a role in regulating Smad expression (58), and these family members show increased activity in UUO (59) although the ultimate effect on progressive kidney disease is not entirely clear. Alternatively, TGF-beta specifically inhibits transcription of Smad3 by an as-yet unknown mechanism (60). It is noteworthy that in several systems, although total Smad3 expression is downregulated, renal levels of phosphorylated Smad3 may be maintained, at least for the duration of the studies that have been performed (30, 60). Thus, the intermediate and long-term implications of changes in Smad expression remain to be elucidated. An important consideration is not only the absolute expression of Smads, but their expression relative to each other, since the balance among Smad-mediated signals may have different effects on specific proteins. For example, Smad2 and Smad3 may play different roles in determining TGF-beta1 signaling outcome in fibrogenesis, with disproportionate Smad3 activity potentially being a critical factor in ECM accumulation (61–64) or epithelial-mesenchymal transition (EMT) (65) (see more extensive discussion of cell phenotypic changes, below). Cells with a mesenchymal phenotype have roughly equal amounts of Smad2 and Smad3, whereas epithelial cells express more Smad2 than Smad3 (unpublished observation). Interestingly, as renal tubular epithelial cells undergo EMT in response to TGF-beta, their levels of Smad3 decrease, a phenomenon also observed in the mouse kidney subjected to unilateral ureteral obstruction (UUO) (60), suggesting that EMT itself is conducive to Smad3 downregulation.
6.3. Post-translational modification of Smads
A critical event in the propagation of Smad activity is its phosphorylation at C-terminal serines in the MH2 domain, an event common to all R-Smads (20). This action is almost always dependent upon the kinase activity of the TbetaRI. A role for C-terminal Smad phosphorylation in renal cell fibrogenesis is well established (22). Subsequently, attention has focused on additional potential serine and threonine phosphorylation sites, mostly in the linker region but also at Thr8 in the MH1 region (Figure 1). These sites may be phosphorylated by the action of cyclin-dependent kinases, ERK, p38 MAP kinase and c-Jun-N-terminal kinase (summarized in (66)). Although it has been posited that linker region phosphorylation always inhibits Smad3 activity (67–70), subsequent work from a number of investigators has suggested that the effects of different Smad phosphorylation events are cell context-specific (71, 72); they could reflect tissue or cell phenotype, the cellular microenvironment, the activation of--and interaction with--other signaling molecules (73, 74), or the readout by which stimulation or inhibition is evaluated. It is generally accepted that some of the effects of linker region phosphorylation on cancer invasion and metastasis are inhibitory (70) but the effects of such phosphorylation on determination of non-neoplastic cell phenotype or on fibrogenic activity are less clear. In studies of human glomerular mesangial cells, events that promote linker region phosphorylation also promote TGF-beta1-stimulated collagen expression (30, 75, 76), although a causal connection between the two has not been established.
Smad phosphorylation also is regulated by a number of phosphatases that have preferential specificity for either the C-terminal or linker-region phosphorylations (66, 77, 78). These may have a significant impact on determining the outcome of TGF-beta signaling for different Smad-mediated events.
Another form of post-translational modification is acetylation. Trichostatin A (TSA), a histone deacetylase (HDAC) inhibitor, decreases the TGF-beta-stimulated collagen response or EMT in rat skin fibroblasts (79). It also decreases fibronectin and collagen expression, as well as Smad nuclear translocation, in fibroblasts from patients with scleroderma (80). Since histone deacetylation tends to generally decrease gene expression, it is intriguing that TSA, an HDAC inhibitor, decreases (rather than increases) collagen expression. One potential explanation is that the histone acetyl transferases (HATs) promote acetylation of critical lysines of Smad7, rendering it resistant to ubiquitination and thus stabilizing its potential inhibitory effects (81). These findings suggest that the balance between acetylation and deacetylation of Smads may be an important consideration in their stability and subsequent function, and that multiple, competing effects result from experimental maneuvers that alter this balance. The role of acetylation will be considered further below in the discussion of transcriptional regulation.
6.4. Adaptor molecules
One determinant of TbetaR function is the expression and activity of several adaptor molecules that appear to facilitate Smad interaction with the TbetaR (82). Because the adaptor molecules may have differential affinity for different Smads or respond under different conditions in the cell, Smad activation in response to TGF-beta may be modulated by the relative presence and activity of each of these proteins. One of these, Smad anchor for receptor activation (SARA), was initially described as a molecule that recruits R-Smads to the TbetaR (83). SARA contains both a Smad-binding domain (SBD) (84) and a TbetaR-interacting region. A third sequence, the FYVE domain, mediates interaction of SARA with the cell membrane or vesicles through its affinity for PI3P (83). Although the SBD of SARA appears capable of binding both Smad2 and Smad3, in activated kidney cells it appears to interact preferentially with Smad2 (54). Another adaptor molecule, disabled protein (Dab)2, binds with both Smad2 and Smad3. TGF-beta1 stimulates a transient increase in association of the phosphotyrosine-binding domain of Dab2 with the MH2 domains of either Smad. Ectopic expression of Dab2 restores Smad2 activity in a TGF-beta signaling-deficient cell line (85). In addition to its effects on Smad signaling, Dab2 appears to play a significant role in transport functions related to other proteins. It binds to megalin (86) and may have important functions related to protein reabsorption by the renal tubule (87); and in its signaling through MAP kinases (88) it could influence albumin-stimulated TGF-beta1 production (89). Axin and its functional homolog, Axil, are limiting components of Wnt signaling through interaction with beta-catenin, the adenomatous polyposis coli gene product (APC), and glycogen synthase kinase 3beta (GSK-3beta) (90–92). Axin binds to Smad3 (93) and with GSK3beta may regulate Smad3 stability (94). Two other adaptor molecules suggested to interact with the TbetaR are Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate--also called Hgs) (95) and cPML (96) (cytoplasmic promyleocytic leukemia protein), both proposed to facilitate the functions of SARA. While few studies have examined the role of these adaptor molecules in chronic kidney disease, their ability to modulate or direct TGF-beta/Smad signaling suggests that they could play an important role in determining cell phenotype and function in renal disorders.
6.5. Regulation of transcriptional activity
6.5.1. Enhancers
Smads participate in extensive transcription-regulatory complexes in the nucleus. These complexes have been characterized best in relation to fibrogenesis. A number of HATs, including p300, TCF/Lef and pCAF, interact with Smads in a manner that could involve the Smad linker region (97–100). TFE3 and the gut Kruppel-like factor (GKLF/KLF4) participate in TGF-beta-stimulated expression of the laminin gamma1 chain (101, 102), orchestrated in part by the human nuclear RNP K (103). The COL1A2 promoter contains binding sites for both Sp1 and Sp3 (104) and the Sp1 sites are essential for TGF-beta/Smad-mediated collagen expression in mesangial cells (105). Protein inhibitor of activated STAT3 (PIAS) interacts with Smad3 and p300 to stimulate Smad3-based responses (106). Additional regulators that enhance Smad-mediated transcription include members of the Forkhead/winged-helix transcription factor family, Fast-1 (FoxH1a) and Fast-3 (FoxH1b) (107). The inhibitor of growth (ING)2 interacts with Smads to promote TGF-beta-dependent activation of specific reporter constructs (108). Some of these molecules may have intranuclear effects. Smicl (Smad-interacting, CPSF-like factor, nuclear localized) regulates transcription but also could be part of an RNA-processing unit (109). Runx2 positively affects Smad signaling by regulating intranuclear targeting. It is phosphorylated by the ERK MAP kinase to promote its interaction with Smads (110).
6.5.2. Inhibitors
Interestingly, the HDAC antagonist TSA decreases Sp1 expression, and overexpression of Sp1 overcomes the TSA inhibition of TGF-beta-stimulated activation of the type I collagen gene in fibroblasts (111). This observation further supports the notion that, although TSA may affect HDACs, its inhibitory effects on Smad activity may occur through effects on the expression and function of other proteins, rather than directly through Smad/HDAC effects on transcription. Indeed, many of the known transcriptional repressors of TGF-beta signaling are HDACs or act by recruiting HDACs to the Smad signaling complex. For example, TGIF is a homeodomain protein that serves as a transcriptional co-repressor by recruiting HDACs to a target DNA (112, 113). Ski (also referred to as c-Ski) is a nuclear oncoprotein that interacts directly with Smads to recruit the nuclear co-repressor, N-CoR, and possibly its associated HDAC complex (114). In binding to Smad2 and Smad3, Ski interferes with binding of the HAT, p300, to the Smads (115). In comparison, Ski interacts only weakly with Smad1 and Smad5, and does not recruit HDACs to a Smad1/5 complex, suggesting that it is relatively specific for TGF-beta, as opposed to BMP, signaling (116). One report describes Ski inhibition of Smad2 or Smad3 phosphorylation, suggesting additional, non-transcriptional effect of Ski (117). A related protein, SnoN (118), participates in the same HDAC complex as Ski (119). SnoN is rapidly downregulated after cells are treated with TGF-beta (120), suggesting that such downregulation contributes to the ability of the Smad transcriptional complex to mediate target gene activation. Another oncoprotein that works through HDAC activity is Evi-1, a zinc finger protein that, when overexpressed in lymphocytes, may cause leukemia by blocking the anti-proliferative effects of Smad3 (121).
Several of these molecules have been found to play a role in kidney cells. TGIF appears to serve as a transcriptional repressor in mesangial cells (122), whereas SnoN appears to play a more significant role in renal tubular epithelial cells (123). Further, the I-Smad, Smad7, has been described as having direct inhibitory effects on transcription, and to block inflammatory signaling through increased IkappaB expression (124). In a complex pathway, TGF-beta increases the expression of connective tissue growth factor (CTGF/CCN2), which enhances the expression of a transcriptional repressor, TIEG-1, that in turn represses Smad7 expression; this process ultimately promotes collagen synthesis (125).
6.6. Cross-talk with other signaling pathways
Given the myriad effects of TGF-beta on the cell, and the marked differences with which cells respond to this ligand, it is likely that the cellular milieu is a significant determinant of the response. Structural and functional antecedents determine that milieu. These conditions are specific to each cell and tissue type and reflect the net effect of various signaling events that are occurring in the cell at any given time. In addition to these preconditioned states, other pathways may be activated by external stimuli, such as cell injury, to affect TGF-beta signaling. Thus, it is generally accepted that events both activated by, and independent of, TGF-beta may influence the outcome of TGF-beta signaling (126). For example, the stress-activated protein kinases p38 and JNK (69, 127), have been shown to play roles in TGF-beta signaling in a variety of systems. In renal cells, TGF-beta stimulates rapid phosphorylation of the TGF-beta-activated kinase (TAK)1 and TAK1-binding protein (128), in turn activating MKK3, which appears to function upstream of p38 (129). In murine PTE cells this series of events leads to fibronectin production by a TbetaR-dependent, Smad-independent mechanism, since it is blocked by p38 and ALK5 antagonists but not by Smad3 knockdown (130). The p38 MAP kinase also mediates vascular endothelial growth factor production by murine mesangial cells in response to TGF-beta (131).
As with the p38 MAP kinase, the ERK MAP kinase also requires the activation of a cascade, in this case from Ras through Raf-1 to MEK and finally to ERK. Expression of the Ras GTPases is increased in podocytes with various forms of injury (132). We have found that the ERK MAP kinase plays a critical role in TGF-beta-stimulated type I collagen expression by human mesangial cells and in collagen promoter activation in human PTE cells (75). As noted above, ERK may phosphorylate phosphoacceptor sites in the linker region of Smad3. This phosphorylation may be enhanced by glucose-stimulated PKCdelta activity (29), and also can result from the activation of cyclin-dependent kinases, p38, or JNK (summarized in (66)). PKCdelta may have additional positive effects on Smad3 transcriptional activity, independent of effects on ERK (133). Other molecules that may affect Smad signaling include phosphatidylinositol-3-kinase (PI3K) (134), calcium-calmodulin-dependent kinase (Cam kinase) II (135) and mTOR, which may act through effects on either inflammation or PI3K/Akt signaling (136).
Another form of crosstalk that may influence the outcome of TGF-beta signaling involves interaction of the cell with its microenvironment. Binding of mesangial and PTE cells to the ECM activates focal adhesion kinase (FAK) through adhesion-dependent pathways in a manner that activates ERK and could contribute to Smad3 linker-region phosphorylation. This integrin-mediated signaling appears to be essential for the collagen I response to TGF-beta (30). Attachment of mesangial cells to interstitial (type I) collagen, as opposed to basement membrane (type IV) collagen, promotes TGF-beta production and further collagen expression (137). Thus, as a cell makes more inappropriate or abnormal ECM, the resulting changes in cell-matrix interaction will stimulate further irregularities in cell function, and then more abnormal ECM, in a vicious cycle that is a negative example of what has been termed “dynamic reciprocity” (138), the means by which cells and the surrounding ECM exert mutual regulatory influences upon each other. These effects are likely mediated by the integrins. For example, the alphaVbeta6 integrin appears to play an important role in the renal ECM accumulation of mice with Alport disease (139).
7. INTERACTION OF TGF-BETA SIGNALING WITH OTHER GROWTH FACTORS/HORMONAL MEDIATORS
7.1. Bone morphogenetic protein (BMP)-7
An important delineator of Smad signaling pathways is the difference between outcomes after TGF-beta stimulation of Smad2 and Smad3 versus BMP-7 stimulation of Smad1 and Smad5. Whereas TGF-beta signaling favors fibrosis, BMP-7 signaling opposes fibrogenesis (140). Renal tubular expression of BMP-7, high-affinity BMP type II receptor and the Alk2 and Alk3 type I receptors is decreased in streptozotocin-induced diabetes (141), and overexpression of BMP-7 is protective against experimental diabetic nephropathy (142). TGF-beta-stimulated fibrogenesis is blocked by BMP-7 in mesangial cells (143), accompanied by a decrease in nuclear accumulation of Smad3 that is Smad5-dependent (144). Interestingly, while much attention has focused on competition between the two Smad signaling pathways, BMP-7 also likely mitigates events such as inflammation that engender fibrogenesis but occur prior to actual ECM expression. In addition, BMP-7 also may decrease inflammatory stimulation of PTE-cell TGF-beta1 promoter activity, further decreasing the TGF-beta signal (145).
7.2. Hepatocyte growth factor (HGF)
HGF ameliorates experimental fibrotic kidney disease by suppressing EMT, reducing ECM accumulation and decreasing expression of TGF-beta and TbetaRI (146). Prevention of fibrogenic changes is achieved by blocking Smad signal transduction through various means that are cell type-dependent (123). Most notably, HGF stabilizes the Smad transcriptional co-repressors TGIF (122) and SnoN (147).
7.3. Mechanical stress, glucose and angiotensin II
Elevated levels of glucose and the effects of angiotensin II can synergize to enhance fibrogenic signaling. Since angiotensin II leads to contraction by many cells of the kidney, it is worth considering how tensile forces may interact with TGF-beta signaling to affect cellular events in chronic kidney disease. Repeated stretching of cultured mesangial cells induces TGF-beta production and collagen accumulation (148). Cyclic stretch activates Raf-1 (149) and Akt-mediated collagen expression is stimulated by stretch (150). Both of these molecules are important for TGF-beta-stimulated collagen expression. Given the long-standing observation that renal hypertension is associated with progressive renal disease, these results raise the notion that at least part of the enhancing effect of hypertension on chronic kidney disease results from its amplification of the cross-talk mechanisms that we have described here.
High glucose in mesangial cells (151), stretch in podocytes (152), and angiotensin II in tubular cells (153) all stimulate TGF-beta expression and activity. Given that angiotensin II activates multiple signaling pathways in the cell (154), it may be impossible to distinguish the effects of strain/tension on the cells from hypertrophic stimuli that have been attributed to angiotensin II (155). Further synergy is achieved by interaction of glucose and angiotensin pathways with Tsp-1 (33). While most studies in renal cells have suggested that angiotensin-stimulated fibrogenic responses result from increased expression of TGF-beta, in vascular smooth muscle cells angiotensin II stimulates C-terminal Smad3 phosphorylation within 15 to 30 minutes in an ERK-dependent, TGF-beta-independent manner (156).
7.4. Connective tissue growth factor (CTGF/CCN2)
CTGF is a cytokine that promotes the expression of ECM proteins and is temporally and spatially associated with scarring in the kidney (157). Downstream from TGF-beta, it has been proposed to be an essential mediator of fibrogenic signaling (158, 159), in part by decreasing Smad7 expression as mentioned previously. However, Smads may directly stimulate collagen promoter activity independently of CTGF (22) and studies have suggested that blocking CTGF only partially blocks TGF-beta-stimulated ECM production by PTE cells (160). TGF-beta-stimulated CTGF expression requires both Smad and ERK activity (161), further supporting synergy between Smads and ERK.
7.5. Nuclear receptor-associated hormones
Several nuclear receptors have been associated with TGF-beta-stimulated kidney injury. Activation of the peroxisome proliferator-activated receptor (PPAR)gamma is protective against chronic renal injury, possibly through stimulation of HGF production (162). 9-cis retinoic acid suppresses TGF-beta-stimulated induction of smooth muscle alpha-actin (aSMA), fibronectin and the plasminogen activator inhibitor, PAI-1, but does not affect cell proliferation or survival. It also enhances HGF expression and subsequent TGIF expression. Because the retinoid receptors interact with the vitamin D receptor and the PPARs, these results suggest the possibility of crosstalk among the Smads and nuclear hormone receptors (163). Another nuclear-receptor hormone, 17 beta-estradiol, ameliorates renal injury in albumin/TGF-beta transgenic mice (164). The pathway(s) involved in this renoprotection may include MAP kinase activity (165) or inhibition of the protein kinase, casein kinase II (166).
8. REGULATION OF TISSUE FUNCTIONS IN THE KIDNEY
While all of the signaling actions described here have implications for the cellular response to injury in the kidney, it is worth considering how these pathways interact with specific structural and functional events that are observed in progressive kidney disease.
8.1. The podocyte and the glomerular filtration barrier
Diseases of glomerular scarring appear to initiate primarily with podocyte injury (167, 168). Although TGF-beta has been associated with proteinuria, the preponderance of the data suggest that this association represents the effects of chronic nephron injury, not an acute effect of TGF-beta on glomerular permselectivity. However, TGF-beta may indirectly affect permeability properties through apoptosis (169). Because podocytes are usually terminally differentiated, apoptosis results in podocytopenia and hypertrophy of the remaining cells, leading via a common pathway to glomerular dysfunction, hyperfiltration and scarring (170). Potential mechanisms are suggested by studies in other cell types. An immediate early gene that is activated by TGF-beta through Smads, GADD45b, has been identified as an effector of apoptosis in hepatocytes (171). In mesangial cells, glucose-stimulated apoptosis is blocked by an anti-TGF-beta antibody (171, 172). The mechanism attributed to TGF-beta in this case is an alteration in the Bcl-2:Bax ratio.
8.2. Hyperplasia and hypertrophy
Increases in cell number may result from either decreased apoptosis or increased proliferation (173). An anti-apoptotic pathway activated by CTGF could promote mesangial cell hypertrophy (174). CTGF-stimulated cell hypertrophy results from cell entry into the cell cycle but then subsequent arrest at G1, associated with increases in expression of the cyclin-dependent kinase inhibitors p15 (ink4), p21 (cip1) and p37 (kip1), with concomitant inactivation of cyclin D and cyclin-dependent kinases 4 and 6 (175). Knockout of p21 prevents podocytes from undergoing apoptosis when they are stimulated with TGF-beta (176). Deletion of the gene for p27-Kip ameliorated proteinuria and increased glomerular volume in experimental diabetes mellitus (177).
8.3. Epithelial-to-mesenchymal transition (EMT)
In response to tissue injury, renal cells de-differentiate into a more primordial form characterized by less anchorage, more migratory activity, altered intermediate filament and actin isoforms, loss of apical-basal polarity that is critical for epithelial membrane transport function, and increased ECM production. These events have been termed “epithelial-to-mesenchymal transition,” although all cells that are injured, regardless of whether they are epithelial in nature, appear to undergo some elements of this process. TGF-beta stimulates renal tubular epithelial cells to lose tight junctions, express less adherens junction proteins such as E-cadherin and ZO-1 (178), increase migratory activity (65) and express increased amounts of aSMA (179) and of the ECM proteases, MMP-2 and MMP-9 (180). Accompanying these changes is increased expression of a fibroblast marker, Fsp1 (181). There are many effectors of TGF-beta-stimulated EMT. Smad2 and Smad3 may mediate distinct events that contribute to EMT (65). Further, Dab2 is required for EMT to occur (182), and CTGF production may play a role in connection with the integrin-linked kinase, ILK (183), which has been shown to play a role in EMT. In a complex series of interactions illustrating the importance of crosstalk among pathways, TGF-beta stimulates Jagged1 and Hey1 to effect EMT in a series of events that are Smad3-dependent early in the response but Jagged1/Notch-dependent subsequently. Interfering with Jagged1, Hey1 or Notch expression prevents EMT in this model (184). In addition to the majority of studies that have been performed in PTE cells, recent reports describe TGF-beta-stimulated EMT in cortical collecting duct cells (185) and podocytes (186). Parallel changes in marker expression (aSMA expression, cell proliferation) also occur in mesangial cells, even though they already are mesenchymal in nature (187).
Several proteins that bind to Smads play roles in EMT. Smad-interacting protein (SIP)-1 is a transcriptional co-repressor encoded by the ZFHX1B gene (188) that inhibits Wnt signaling downstream of TGF-beta-family-stimulated signaling by increasing the expression of the Wnt antagonist, Sfrp1 (189). SIP-1 decreases E-cadherin expression and promotes cell migration/invasiveness (190), indicating that it simulates both changes in marker expression and enhanced cell functions associated with EMT. Of note, whereas under physiological conditions pancreatic cancer cells are adherent to collagen IV, these cells cultured on type I collagen show a SIP-1-dependent decrease in E-cadherin expression without TGF-beta treatment (191). Thus, SIP-1 may participate in the progressive changes of cell-matrix interactions that characterize EMT. SIP-1 effects are regulated by sumoylation (192), indicating that TGF-beta/Smad signaling can be inhibited by ubiquitin-mediated degradation at the level of transcription factors as well as Smad expression. SIP-1 is important in development, as its mutation results in Mowat-Wilson syndrome, a complex disorder characterized by a typical facies, epilepsy, mental retardation, Hirschsprung disease and agenesis of the corpus callosum (193). A related protein, deltaEF-1, is expressed by TGF-beta-treated, murine breast epithelial cells (194), but downregulated by TGF-beta in mesangial cells (195). It appears to synergize with SIP-1 to mediate a number of phenotypic changes stimulated by TGF-beta (194). It is noteworthy that a recent publication describes a micro-RNA, miR-192, that targets SIP-1 and is found in increased quantities in glomeruli of mice with diabetic nephropathy (195). This finding suggests that downregulation of SIP-1 could play a role in the pathogenesis of diabetic nephropathy.
Another EMT-associated protein, Snail, is induced by TGF-beta. Snail overexpression and activation is sufficient to induce EMT and kidney fibrosis in adult transgenic mice, and has been observed in kidney disease (196). Snail expression is enhanced in unilateral ureteral expression in vivo, and in renal tubular epithelial cells treated with TGF-beta in vitro. GSK-3beta, which degrades Snail1, is inactivated by phosphorylation either in UUO or after TGF-beta treatment, causing Snail to accumulate (197). Snail disrupts E-cadherin localization in cultured collecting duct epithelial cells (185) and P-cadherin and nephrin localization in podocytes (186). Expression of another cell-cell adhesion molecule, connexin43, has been reported to suppressed by Snail in non-kidney culture models of EMT (198).
BMP-7 ameliorates chronic renal injury at least in part by suppressing EMT (140). One mechanism by which this may occur is through induction of the inhibitors of differentiation (Id) family of molecules. Smad1 mediates Id1 and Id3 expression in vascular smooth muscle cells by a mechanism that also requires the participation of MAP kinases (199). Conversely, the expression of Id2 and Id3 in epithelial cells, although increased by BMPs, is antagonized by TGF-beta (200, 201).
Thus, EMT involves multiple steps, each of which may be differentially regulated by the various Smad-interacting molecules described here.
9. PERSPECTIVE
From the foregoing discussion, it is apparent that a significant amount of data has been generated relating various basic signaling mechanisms that are activated by TGF-beta. Together, the observations describe pleiotrophic effects of TGF-beta on cell function that can contribute to all the elements of the response to injury that characterize chronic kidney disease: excessive cell proliferation, apoptosis and loss of functional tissue, transition of the cells to a less differentiated phenotype, and ECM accumulation (Figure 3). However, significant issues remain. First, the data have been generated in different cell types from different species, and conclusions regarding pathways and effector mechanisms in one tissue or species cannot necessarily be extrapolated to others. Second, while we have gained insight into the mechanisms underlying pathophysiology, the hierarchy of events is not clear. Some investigators have taken reductionist approaches to signaling mechanisms. These studies cannot account for the complexity of actions among various hormones and signals, as illustrated by the crosstalk among pathways. Other investigators have pursued whole-animal experimental models, which better illustrate the disease state but are unlikely to permit analysis of the impact and timing of specific signaling events. The essential task is to find ways to relate in vitro and in vivo discoveries. Finally, because these signaling mechanisms largely exist inside the cell, and affect many biological functions, we have yet to determine how to focus the delivery and biochemical specificity of pathway inhibitors in order to safely and effectively prevent, or even reverse the course of, disease. Addressing the issue of therapy will be the ultimate challenge for investigators in the field.
Figure 3.
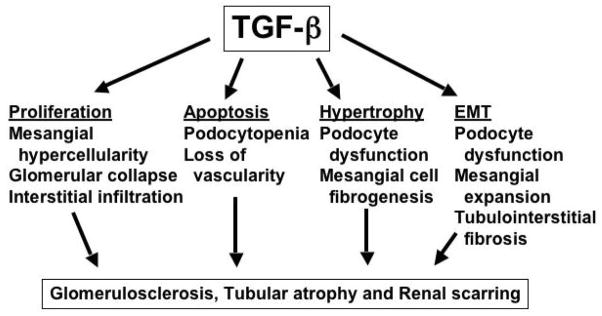
Mechanisms by which TGF-beta induces chronic kidney disease.
Acknowledgments
Supported in part by grants R01 DK49362 and R01 DK75663 from the NIDDK.
References
- 1.Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, Sporn MB. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol. 1989;108(2):661–9. doi: 10.1083/jcb.108.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitamura M, Suto T, Yokoo T, Shimizu F, Fine LG. Transforming growth factor-beta 1 is the predominant paracrine inhibitor of macrophage cytokine synthesis produced by glomerular mesangial cells. J Immunol. 1996;156(8):2964–71. [PubMed] [Google Scholar]
- 3.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359(6397):693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suto TS, Fine LG, Kitamura M. Mesangial cell-derived transforming growth factor-beta 1 reduces macrophage adhesiveness with consequent deactivation. Kidney Int. 1996;50(2):445–52. doi: 10.1038/ki.1996.335. [DOI] [PubMed] [Google Scholar]
- 5.Alexandrow MG, Moses HL. Transforming growth factor beta and cell cycle regulation. Cancer Res. 1995;55(7):1452–7. [PubMed] [Google Scholar]
- 6.Goumenos DS, Tsakas S, El Nahas AM, Alexandri S, Oldroyd S, Kalliakmani P, Vlachojannis JG. Transforming growth factor-beta (1) in the kidney and urine of patients with glomerular disease and proteinuria. Nephrol Dial Transplant. 2002;17(12):2145–52. doi: 10.1093/ndt/17.12.2145. [DOI] [PubMed] [Google Scholar]
- 7.Murakami K, Takemura T, Hino S, Yoshioka K. Urinary transforming growth factor-beta in patients with glomerular diseases. Pediatr Nephrol. 1997;11(3):334–6. doi: 10.1007/s004670050289. [DOI] [PubMed] [Google Scholar]
- 8.Woroniecki RP, Shatat IF, Supe K, Du Z, Kaskel FJ. Urinary cytokines and steroid responsiveness in idiopathic nephrotic syndrome of childhood. Am J Nephrol. 2008;28(1):83–90. doi: 10.1159/000109396. [DOI] [PubMed] [Google Scholar]
- 9.Wasilewska AM, Zoch-Zwierz WM. Transforming growth factor-beta1 in nephrotic syndrome treated with cyclosporine and ACE inhibitors. Pediatr Nephrol. 2004;19(12):1349–53. doi: 10.1007/s00467-004-1619-5. [DOI] [PubMed] [Google Scholar]
- 10.Kim JH, Kim BK, Moon KC, Hong HK, Lee HS. Activation of the TGF-beta/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 2003;64(5):1715–21. doi: 10.1046/j.1523-1755.2003.00288.x. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T, Noble N, Miller DE, Border WA. Sustained expression of TGF-beta1 underlies development of progressive kidney fibrosis. Kidney Int. 1994;45:916–927. doi: 10.1038/ki.1994.122. [DOI] [PubMed] [Google Scholar]
- 12.Isaka Y, Fujiwara Y, Ueda N, Kaneda Y, Kamada T, Imai E. Glomerulosclerosis induced by in vivo transfection of transforming growth factor-beta or platelet-derived growth factor gene into the rat kidney. J Clin Invest. 1993;92:2597–2601. doi: 10.1172/JCI116874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mozes MM, Bottinger EP, Jacot TA, Kopp JB. Renal expression of fibrotic matrix proteins and of transforming growth factor-beta (TGF-beta) isoforms in TGF-beta transgenic mice. J Am Soc Nephrol. 1999;10:271–280. doi: 10.1681/ASN.V102271. [DOI] [PubMed] [Google Scholar]
- 14.Akagi Y, Isaka Y, Arai M, Kaneko T, Takenaka M, Moriyama T, Kaneda Y, Ando A, Orita Y, Kamada T, Ueda N, Imai E. Inhibition of TGF-beta1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1996;50:148–155. doi: 10.1038/ki.1996.297. [DOI] [PubMed] [Google Scholar]
- 15.Lavoie P, Robitaille G, Agharazii M, Ledbetter S, Lebel M, Lariviere R. Neutralization of transforming growth factor-beta attenuates hypertension and prevents renal injury in uremic rats. J Hypertens. 2005;23(10):1895–903. doi: 10.1097/01.hjh.0000182521.44440.c5. [DOI] [PubMed] [Google Scholar]
- 16.Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, Ruoslahti E. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360(6402):361–4. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- 17.Russo LM, del Re E, Brown D, Lin HY. Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: amelioration by soluble TGF-beta type II receptor. Diabetes. 2007;56(2):380–8. doi: 10.2337/db06-1018. [DOI] [PubMed] [Google Scholar]
- 18.Roberts AB, Flanders KC, Heine UI, Jakowlew S, Kondaiah P, Kim SJ, Sporn MB. Transforming growth factor-beta: multifunctional regulator of differentiation and development. Philos Trans R Soc Lond B Biol Sci. 1990;327(1239):145–54. doi: 10.1098/rstb.1990.0050. [DOI] [PubMed] [Google Scholar]
- 19.Piek E, Heldin C-H, ten Dijke P. Specificity, diversity and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–2124. [PubMed] [Google Scholar]
- 20.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 21.Randall RA, Germain S, Inman GJ, Bates PA, Hill CS. Different Smad2 partners bind a common hydrophobic pocket in Smad2 via a defined proline-rich motif. Embo J. 2002;21(1–2):145–56. doi: 10.1093/emboj/21.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poncelet AC, de Caestecker MP, Schnaper HW. The TGF-beta/SMAD signaling pathway is present and functional in human mesangial cells. Kidney Int. 1999;56:1354–1365. doi: 10.1046/j.1523-1755.1999.00680.x. [DOI] [PubMed] [Google Scholar]
- 23.Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13(10):2600–10. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 24.Cheng J, Grande JP. Transforming growth factor-beta signal transduction and progressive renal disease. Exp Biol Med (Maywood) 2002;227(11):943–56. doi: 10.1177/153537020222701102. [DOI] [PubMed] [Google Scholar]
- 25.Sharma K, Ziyadeh FN, Alzahabi B, McGowan TA, Kapoor S, Kurnik BR, Kurnik PB, Weisberg LS. Increased renal production of transforming growth factor-beta1 in patients with type II diabetes. Diabetes. 1997;46:854–859. doi: 10.2337/diab.46.5.854. [DOI] [PubMed] [Google Scholar]
- 26.Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–126. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaedeke J, Noble NA, Border WA. Curcumin blocks multiple sites of the TGF-beta signaling cascade in renal cells. Kidney Int. 2004;66(1):112–20. doi: 10.1111/j.1523-1755.2004.00713.x. [DOI] [PubMed] [Google Scholar]
- 28.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 29.Hayashida T, Schnaper HW. High ambient glucose enhances sensitivity to TGF-beta1 via extracellular signal--regulated kinase and protein kinase Cdelta activities in human mesangial cells. J Am Soc Nephrol. 2004;15(8):2032–41. doi: 10.1097/01.ASN.0000133198.74973.60. [DOI] [PubMed] [Google Scholar]
- 30.Hayashida T, Wu MH, Pierce A, Poncelet AC, Varga J, Schnaper HW. MAP-kinase activity necessary for TGFbeta1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J Cell Sci. 2007;120(Pt 23):4230–40. doi: 10.1242/jcs.03492. [DOI] [PubMed] [Google Scholar]
- 31.Yevdokimova N, Wahab NA, Mason RM. Thrombospondin-1 is the key activator of TGF-beta1 in human mesangial cells exposed to high glucose. J Am Soc Nephrol. 2001;12(4):703–12. doi: 10.1681/ASN.V124703. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa T, Lan HY, Glushakova O, Zhu HJ, Kang DH, Schreiner GF, Bottinger EP, Johnson RJ, Sautin YY. Role of ERK1/2 and p38 mitogen-activated protein kinases in the regulation of thrombospondin-1 by TGF-beta1 in rat proximal tubular cells and mouse fibroblasts. J Am Soc Nephrol. 2005;16(4):899–904. doi: 10.1681/ASN.2004080689. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Y, Poczatek MH, Berecek KH, Murphy-Ullrich JE. Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem Biophys Res Commun. 2006;339(2):633–41. doi: 10.1016/j.bbrc.2005.11.060. [DOI] [PubMed] [Google Scholar]
- 34.Wahab NA, Schaefer L, Weston BS, Yiannikouris O, Wright A, Babelova A, Schaefer R, Mason RM. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia. 2005;48(12):2650–60. doi: 10.1007/s00125-005-0006-5. [DOI] [PubMed] [Google Scholar]
- 35.Gao L, Qiu W, Wang Y, Xu W, Xu J, Tong J. Sublytic complement C5b-9 complexes induce thrombospondin-1 production in rat glomerular mesangial cells via PI3-k/Akt: association with activation of latent transforming growth factor-beta1. Clin Exp Immunol. 2006;144(2):326–34. doi: 10.1111/j.1365-2249.2006.03069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniel C, Wiede J, Krutzsch HC, Ribeiro SM, Roberts DD, Murphy-Ullrich JE, Hugo C. Thrombospondin-1 is a major activator of TGF-beta in fibrotic renal disease in the rat in vivo. Kidney Int. 2004;65(2):459–68. doi: 10.1111/j.1523-1755.2004.00395.x. [DOI] [PubMed] [Google Scholar]
- 37.Ito T, Williams JD, Fraser DJ, Phillips AO. Hyaluronan regulates transforming growth factor-beta1 receptor compartmentalization. J Biol Chem. 2004;279(24):25326–32. doi: 10.1074/jbc.M403135200. [DOI] [PubMed] [Google Scholar]
- 38.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5(5):410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 39.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, Pten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12(4):817–28. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 40.Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. Embo J. 2004;23(20):4018–28. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Craft CS, Romero D, Vary CP, Bergan RC. Endoglin inhibits prostate cancer motility via activation of the ALK2-Smad1 pathway. Oncogene. 2007;26(51):7240–50. doi: 10.1038/sj.onc.1210533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rotzer D, Roth M, Lutz M, Lindemann D, Sebald W, Knaus P. Type III TGF-beta receptor-independent signalling of TGF-beta2 via TbetaRII-B, an alternatively spliced TGF-beta type II receptor. Embo J. 2001;20(3):480–90. doi: 10.1093/emboj/20.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taniguchi A, Matsuzaki K, Nakano K, Kan M, McKeehan WL. Ligand-dependent and -independent interactions with the transforming growth factor type II and I receptor subunits reside in the aminoterminal portion of the ectodomain of the type III subunit. In vitro Cell Dev Biol Anim. 1998;34(3):232–8. doi: 10.1007/s11626-998-0129-3. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe T, Yamamoto T, Ikegaya N, Fujigaki Y, Suzuki H, Togawa A, Fukasawa H, Nagase M, Hishida A. Transforming growth factor-beta receptors in self-limited vs. chronic progressive nephritis in rats. J Pathol. 2002;198(3):397–406. doi: 10.1002/path.1213. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto T, Watanabe T, Ikegaya N, Fujigaki Y, Matsui K, Masaoka H, Nagase M, Hishida A. Expression of types I, II, and III TGF-beta receptors in human glomerulonephritis. J Am Soc Nephrol. 1998;9(12):2253–61. doi: 10.1681/ASN.V9122253. [DOI] [PubMed] [Google Scholar]
- 46.Ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11(1):79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 47.Diez-Marques L, Ortega-Velazquez R, Langa C, Rodriguez-Barbero A, Lopez-Novoa JM, Lamas S, Bernabeu C. Expression of endoglin in human mesangial cells: modulation of extracellular matrix synthesis. Biochim Biophys Acta. 2002;1587(1):36–44. doi: 10.1016/s0925-4439(02)00051-0. [DOI] [PubMed] [Google Scholar]
- 48.Scherner O, Meurer SK, Tihaa L, Gressner AM, Weiskirchen R. Endoglin differentially modulates antagonistic transforming growth factor-beta1 and BMP-7 signaling. J Biol Chem. 2007;282(19):13934–43. doi: 10.1074/jbc.M611062200. [DOI] [PubMed] [Google Scholar]
- 49.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002;21(7):1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi W, Sun C, He B, Xiong W, Shi X, Yao D, Cao X. GADD34-PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J Cell Biol. 2004;164(2):291–300. doi: 10.1083/jcb.200307151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valdimarsdottir G, Goumans MJ, Itoh F, Itoh S, Heldin CH, ten Dijke P. Smad7 and protein phosphatase 1alpha are critical determinants in the duration of TGF-beta/ALK1 signaling in endothelial cells. BMC Cell Biol. 2006;7:16. doi: 10.1186/1471-2121-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayes S, Chawla A, Corvera S. TGFbeta receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158(7):1239–1249. doi: 10.1083/jcb.200204088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Penheiter SG, Mitchell H, Garamszegi N, Edens M, Dore JJE, Leof EB. Internalization-dependent and -independent requirements for transforming growth factor beta receptor signaling via the Smad pathway. Mol Cell Biol. 2002;22(13):4750–4759. doi: 10.1128/MCB.22.13.4750-4759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Runyan CE, Schnaper HW, Poncelet AC. The role of internalization in transforming growth factor beta1-induced Smad2 association with Smad anchor for receptor activation (SARA) and Smad2-dependent signaling in human mesangial cells. J Biol Chem. 2005;280(9):8300–8. doi: 10.1074/jbc.M407939200. [DOI] [PubMed] [Google Scholar]
- 55.Goto D, Nakajima H, Mori Y, Kurasawa K, Kitamura N, Iwamoto I. Interaction between Smad anchor for receptor activation and Smad3 is not essential for TGF-beta/Smad3-mediated signaling. Biochem Biophys Res Commun. 2001;281(5):1100–5. doi: 10.1006/bbrc.2001.4489. [DOI] [PubMed] [Google Scholar]
- 56.Lu Z, Murray JT, Luo W, Li H, Wu X, Xu H, Backer JM, Chen YG. Transforming growth factor beta activates Smad2 in the absence of receptor endocytosis. J Biol Chem. 2002;277(33):29363–29368. doi: 10.1074/jbc.M203495200. [DOI] [PubMed] [Google Scholar]
- 57.Zhou Y, Scolavino S, Funderburk SF, Ficociello LF, Zhang X, Klibanski A. Receptor internalization-independent activation of Smad2 in activin signaling. Mol Endocrinol. 2004;18(7):1818–1826. doi: 10.1210/me.2004-0079. [DOI] [PubMed] [Google Scholar]
- 58.Arora K, Warrior R. A new Smurf in the village. Dev Cell. 2001;1(4):441–2. doi: 10.1016/s1534-5807(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 59.Liu FY, Li XZ, Peng YM, Liu H, Liu YH. Arkadia-Smad7-mediated positive regulation of TGF-beta signaling in a rat model of tubulointerstitial fibrosis. Am J Nephrol. 2007;27(2):176–83. doi: 10.1159/000100518. [DOI] [PubMed] [Google Scholar]
- 60.Poncelet AC, Schnaper HW, Tan R, Liu Y, Runyan CE. Cell phenotype-specific down-regulation of Smad3 involves decreased gene activation as well as protein degradation. J Biol Chem. 2007;282(21):15534–40. doi: 10.1074/jbc.M701991200. [DOI] [PubMed] [Google Scholar]
- 61.Wang W, Koka V, Lan HY. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton) 2005;10(1):48–56. doi: 10.1111/j.1440-1797.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 62.Roberts AB, Russo A, Felici A, Flanders KC. Smad3: a key player in pathogenic mechanisms dependent on TGF-beta. Ann NY Acad Sci. 2003;995:1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x. [DOI] [PubMed] [Google Scholar]
- 63.Uemura M, Swenson ES, Gaca MD, Giordano FJ, Reiss M, Wells RG. Smad2 and Smad3 play different roles in hepatic stellate cell function and (alpha)-smooth muscle actin organization. Mol Biol Cell. 2005;16(9):4214–24. doi: 10.1091/mbc.E05-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poncelet AC, Schnaper HW. Sp1 and Smad proteins cooperate to mediate transforming growth factor-beta 1-induced alpha 2 (I) collagen expression in human glomerular mesangial cells. J Biol Chem. 2001;276(10):6983–92. doi: 10.1074/jbc.M006442200. [DOI] [PubMed] [Google Scholar]
- 65.Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J. 2006;393(Pt 2):601–7. doi: 10.1042/BJ20051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wrighton KH, Willis D, Long J, Liu F, Lin X, Feng XH. Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-beta signaling. J Biol Chem. 2006;281(50):38365–75. doi: 10.1074/jbc.M607246200. [DOI] [PubMed] [Google Scholar]
- 67.Massague J. Integration of Smad and MAPK pathways: a link and a linker revisited. Genes Dev. 2003;17(24):2993–7. doi: 10.1101/gad.1167003. [DOI] [PubMed] [Google Scholar]
- 68.Pierreux CE, Nicolas FJ, Hill CS. Transforming growth factor beta-independent shuttling of Smad4 between the cytoplasm and nucleus. Mol Cell Biol. 2000;20(23):9041–54. doi: 10.1128/mcb.20.23.9041-9054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem. 2002;277(17):14884–93. doi: 10.1074/jbc.M111718200. [DOI] [PubMed] [Google Scholar]
- 70.Xu L. Regulation of Smad activities. Biochim Biophys Acta. 2006;1759(11–12):503–13. doi: 10.1016/j.bbaexp.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Inman GJ. Linking Smads and transcriptional activation. Biochem J. 2005;386(Pt 1):e1–e3. doi: 10.1042/BJ20042133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rahimi RA, Leof EB. TGF-beta signaling: A tale of two responses. J Cell Biochem. 2007 doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- 73.Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, Fujisawa J, Okazaki K. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23(44):7416–29. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 74.Wilkes MC, Leof EB. Transforming growth factor beta activation of c-Abl is independent of receptor internalization and regulated by phosphatidylinositol 3-kinase and PAK2 in mesenchymal cultures. J Biol Chem. 2006;281(38):27846–54. doi: 10.1074/jbc.M603721200. [DOI] [PubMed] [Google Scholar]
- 75.Hayashida T, de Caestecker MP, Schnaper HW. Cross-talk between ERK MAP kinase and Smad-signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J. 2003;17:1576–78. doi: 10.1096/fj.03-0037fje. [DOI] [PubMed] [Google Scholar]
- 76.Hayashida T, Poncelet AC, Hubchak SC, Schnaper HW. TGF-beta1 activates MAP kinases in human mesangial cells: a possible role in collagen expression. Kidney Int. 1999;56:1710–20. doi: 10.1046/j.1523-1755.1999.00733.x. [DOI] [PubMed] [Google Scholar]
- 77.Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, Hu M, Davis CM, Wang J, Brunicardi FC, Shi Y, Chen YG, Meng A, Feng XH. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell. 2006;125(5):915–28. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sapkota G, Knockaert M, Alarcon C, Montalvo E, Brivanlou AH, Massague J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281(52):40412–9. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
- 79.Rombouts K, Niki T, Greenwel P, Vandermonde A, Wielant A, Hellemans K, De Bleser P, Yoshida M, Schuppan D, Rojkind M, Geerts A. Trichostatin A, a histone deacetylase inhibitor, suppresses collagen synthesis and prevents TGF-beta (1)-induced fibrogenesis in skin fibroblasts. Exp Cell Res. 2002;278(2):184–97. doi: 10.1006/excr.2002.5577. [DOI] [PubMed] [Google Scholar]
- 80.Huber LC, Distler JH, Moritz F, Hemmatazad H, Hauser T, Michel BA, Gay RE, Matucci-Cerinic M, Gay S, Distler O, Jungel A. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007;56(8):2755–64. doi: 10.1002/art.22759. [DOI] [PubMed] [Google Scholar]
- 81.Simonsson M, Heldin CH, Ericsson J, Gronroos E. The balance between acetylation and deacetylation controls Smad7 stability. J Biol Chem. 2005;280(23):21797–803. doi: 10.1074/jbc.M503134200. [DOI] [PubMed] [Google Scholar]
- 82.Runyan CE, Poncelet AC, Schnaper HW. TGF-beta receptor-binding proteins: Complex interactions. Cell Signal. 2006;18:2044–88. doi: 10.1016/j.cellsig.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 83.Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–91. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 84.Wu G, Chen Y, Ozdamar B, Gyuricza CA, Chong A, Wrana JL, Massague J, Shi Y. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science. 2000;287:92–7. doi: 10.1126/science.287.5450.92. [DOI] [PubMed] [Google Scholar]
- 85.Hocevar BA, Smine A, Xu XX, Howe PH. The adaptor molecule Disabled-2 links the transforming growth factor beta receptors to the Smad pathway. EMBO J. 2001;20:2789–2801. doi: 10.1093/emboj/20.11.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oleinikov AV, Zhao J, Makker SP. Cytosolic adaptor protein Dab2 is an intracellular ligand of endocytic receptor gp600/megalin. Biochem J. 2000;347(Pt 3):613–21. [PMC free article] [PubMed] [Google Scholar]
- 87.Morris SM, Tallquist MD, Rock CO, Cooper JA. Dual roles for the Dab2 adaptor protein in embryonic development and kidney transport. Embo J. 2002;21(7):1555–64. doi: 10.1093/emboj/21.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hocevar BA, Prunier C, Howe PH. Disabled-2 (Dab2) mediates transforming growth factor beta (TGFbeta)-stimulated fibronectin synthesis through TGFbeta-activated kinase 1 and activation of the JNK pathway. J Biol Chem. 2005;280(27):25920–7. doi: 10.1074/jbc.M501150200. [DOI] [PubMed] [Google Scholar]
- 89.Diwakar R, Pearson AL, Colville-Nash P, Baines DL, Dockrell ME. Role played by Disabled-2 in albumin induced MAP Kinase signalling. Biochem Biophys Res Commun. 2007 doi: 10.1016/j.bbrc.2007.11.171. [DOI] [PubMed] [Google Scholar]
- 90.Ikeda S, Kishida M, Matsuura Y, Usui H, Kikuchi A. GSK-3beta-dependent phosphorylation of adenomatous polyposis coli gene product can be modulated by beta-catenin and protein phosphatase 2A complexed with Axin. Oncogene. 2000;19(4):537–45. doi: 10.1038/sj.onc.1203359. [DOI] [PubMed] [Google Scholar]
- 91.Kikuchi A. Modulation of Wnt signaling by Axin and Axil. Cytokine Growth Factor Rev. 1999;10(3–4):255–65. doi: 10.1016/s1359-6101(99)00017-9. [DOI] [PubMed] [Google Scholar]
- 92.Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol Cell Biol. 1999;19(6):4414–22. doi: 10.1128/mcb.19.6.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Furuhashi M, Yagi K, Yamamoto H, Furukawa Y, Shimida S, Nakamura Y, Kikuchi A, Miyazono K. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol Cell Biol. 2001;21(15):5132–41. doi: 10.1128/MCB.21.15.5132-5141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. Axin and GSK3-beta control Smad3 protein stability and modulate TGF- signaling. Genes Dev. 2008;22(1):106–20. doi: 10.1101/gad.1590908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miura S, Takeshita T, Asao H, Kimura Y, Murata K, Sasaki Y, Hanai JI, Beppu H, Tsukazaki T, Wrana JL, Miyazono K, Sugamura K. Hgs (Hrs), a FYVE domain protein, is involved in Smad signaling through cooperation with SARA. Mol Cell Biol. 2000;20(24):9346–55. doi: 10.1128/mcb.20.24.9346-9355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin H, Bergmann S, Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature. 2004;431:205–211. doi: 10.1038/nature02783. [DOI] [PubMed] [Google Scholar]
- 97.Prokova V, Mavridou S, Papakosta P, Kardassis D. Characterization of a novel transcriptionally active domain in the transforming growth factor beta-regulated Smad3 protein. Nucleic Acids Res. 2005;33(12):3708–21. doi: 10.1093/nar/gki679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29(5):265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 99.Wang G, Long J, Matsuura I, He D, Liu F. The Smad3 linker region contains a transcriptional activation domain. Biochem J. 2005;386(Pt 1):29–34. doi: 10.1042/BJ20041820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cui Q, Lim SK, Zhao B, Hoffmann FM. Selective inhibition of TGF-beta responsive genes by Smad-interacting peptide aptamers from FoxH1, Lef1 and CBP. Oncogene. 2005;24(24):3864–74. doi: 10.1038/sj.onc.1208556. [DOI] [PubMed] [Google Scholar]
- 101.Higaki Y, Schullery D, Kawata Y, Shnyreva M, Abrass CK, Bomsztyk K. Synergistic activation of the rat laminin γ1 chain promoter by the gut-enriched Kruppel-like factor (GKLF/KLF4) and Sp1. Nucl Acids Res. 2002;30:2270–79. doi: 10.1093/nar/30.11.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kawata Y, Suzuki H, Higaki Y, Denisenko O, Schullery D, Abrass C, Bomsztyk K. bcn-1 Element-dependent activation of the laminin gamma 1 chain gene by the cooperative action of transcription factor E3 (TFE3) and Smad proteins. J Biol Chem. 2002;277(13):11375–84. doi: 10.1074/jbc.M111284200. [DOI] [PubMed] [Google Scholar]
- 103.Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004;26(6):629–38. doi: 10.1002/bies.20048. [DOI] [PubMed] [Google Scholar]
- 104.Zhang W, Ou J, Inagaki Y, Greenwel P, Ramirez F. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates TGFbeta1 stimulation of alpha2 (I) collagen (COL1A2) transcription. J Biol Chem. 2000;275:39237–39245. doi: 10.1074/jbc.M003339200. [DOI] [PubMed] [Google Scholar]
- 105.Poncelet A-CHW. Schnaper Sp1 and Smad proteins cooperate to mediate TGF-beta1-induced alpha2(I) collagen expression in human glomerular mesangial cells. J Biol Chem. 2001;276:6983–92. doi: 10.1074/jbc.M006442200. [DOI] [PubMed] [Google Scholar]
- 106.Long J, Wang G, Matsuura I, He D, Liu F. Activation of Smad transcriptional activity by protein inhibitor of activated STAT3 (PIAS3) Proc Natl Acad Sci U S A. 2004;101(1):99–104. doi: 10.1073/pnas.0307598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Randall RA, Howell M, Page CS, Daly A, Bates PA, Hill CS. Recognition of phosphorylated-Smad2-containing complexes by a novel Smad interaction motif. Mol Cell Biol. 2004;24(3):1106–21. doi: 10.1128/MCB.24.3.1106-1121.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sarker KP, Kataoka H, Chan A, Netherton SJ, Pot I, Huynh MA, Feng X, Bonni A, Riabowol K, Bonni S. ING2 as a novel mediator of TGF-beta -dependent responses in epithelial cells. J Biol Chem. 2008 doi: 10.1074/jbc.M708834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Collart C, Remacle JE, Barabino S, van Grunsven LA, Nelles L, Schellens A, Van de Putte T, Pype S, Huylebroeck D, Verschueren K. Smicl is a novel Smad interacting protein and cleavage and polyadenylation specificity factor associated protein. Genes Cells. 2005;10(9):897–906. doi: 10.1111/j.1365-2443.2005.00887.x. [DOI] [PubMed] [Google Scholar]
- 110.Afzal F, Pratap J, Ito K, Ito Y, Stein JL, van Wijnen AJ, Stein GS, Lian JB, Javed A. Smad function and intranuclear targeting share a Runx2 motif required for osteogenic lineage induction and BMP2 responsive transcription. J Cell Physiol. 2005;204(1):63–72. doi: 10.1002/jcp.20258. [DOI] [PubMed] [Google Scholar]
- 111.Ghosh AK, Mori Y, Dowling E, Varga J. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: involvement of Sp1. Biochem Biophys Res Commun. 2007;354(2):420–6. doi: 10.1016/j.bbrc.2006.12.204. [DOI] [PubMed] [Google Scholar]
- 112.Imoto I, Pimkhaokham A, Watanabe T, Saito-Ohara F, Soeda E, Inazawa J. Amplification and overexpression of TGIF2, a novel homeobox gene of the TALE superclass, in ovarian cancer cell lines. Biochem Biophys Res Commun. 2000;276(1):264–70. doi: 10.1006/bbrc.2000.3449. [DOI] [PubMed] [Google Scholar]
- 113.Wotton D, Lo RS, Lee S, Massague J. A Smad transcriptional corepressor. Cell. 1999;97(1):29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- 114.Luo K, Stroschein SL, Wang W, Chen D, Martens E, Zhou S, Zhou Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signaling. Genes Dev. 1999;13(17):2196–206. doi: 10.1101/gad.13.17.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Akiyoshi S, Inoue H, Hanai J, Kusanagi K, Nemoto N, Miyazono K, Kawabata M. c-Ski acts as a transcriptional co-repressor in transforming growth factor-beta signaling through interaction with smads. J Biol Chem. 1999;274(49):35269–77. doi: 10.1074/jbc.274.49.35269. [DOI] [PubMed] [Google Scholar]
- 116.Takeda M, Mizuide M, Oka M, Watabe T, Inoue H, Suzuki H, Fujita T, Imamura T, Miyazono K, Miyazawa K. Interaction with Smad4 is indispensable for suppression of BMP signaling by c-Ski. Mol Biol Cell. 2004;15(3):963–72. doi: 10.1091/mbc.E03-07-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Prunier C, Pessah M, Ferrand N, Seo SR, Howe P, Atfi A. The oncoprotein Ski acts as an antagonist of transforming growth factor-beta signaling by suppressing Smad2 phosphorylation. J Biol Chem. 2003;278(28):26249–57. doi: 10.1074/jbc.M304459200. [DOI] [PubMed] [Google Scholar]
- 118.Mizuide M, Hara T, Furuya T, Takeda M, Kusanagi K, Inada Y, Mori M, Imamura T, Miyazawa K, Miyazono K. Two short segments of Smad3 are important for specific interaction of Smad3 with c-Ski and SnoN. J Biol Chem. 2003;278(1):531–6. doi: 10.1074/jbc.C200596200. [DOI] [PubMed] [Google Scholar]
- 119.Shinagawa T, Dong HD, Xu M, Maekawa T, Ishii S. The sno gene, which encodes a component of the histone deacetylase complex, acts as a tumor suppressor in mice. Embo J. 2000;19(10):2280–91. doi: 10.1093/emboj/19.10.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science. 1999;286(5440):771–4. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- 121.Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood. 2001;97(9):2815–22. doi: 10.1182/blood.v97.9.2815. [DOI] [PubMed] [Google Scholar]
- 122.Dai C, Liu Y. Hepatocyte growth factor antagonizes the profibrotic action of TGF-beta1 in mesangial cells by stabilizing Smad transcriptional corepressor TGIF. J Am Soc Nephrol. 2004;15(6):1402–12. doi: 10.1097/01.asn.0000130568.53923.fd. [DOI] [PubMed] [Google Scholar]
- 123.Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol. 2004;287(1):F7–16. doi: 10.1152/ajprenal.00451.2003. [DOI] [PubMed] [Google Scholar]
- 124.Wang W, Huang XR, Li AG, Liu F, Li JH, Truong LD, Wang XJ, Lan HY. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol. 2005;16(5):1371–83. doi: 10.1681/ASN.2004121070. [DOI] [PubMed] [Google Scholar]
- 125.Wahab NA, Weston BS, Mason RM. Modulation of the TGFbeta/Smad signaling pathway in mesangial cells by CTGF/CCN2. Exp Cell Res. 2005;307(2):305–14. doi: 10.1016/j.yexcr.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 126.Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24(37):5742–50. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 127.Mulsow JJ, Watson RW, Fitzpatrick JM, O’Connell PR. Transforming growth factor-beta promotes pro-fibrotic behavior by serosal fibroblasts via PKC and ERK1/2 mitogen activated protein kinase cell signaling. Ann Surg. 2005;242(6):880–7. doi: 10.1097/01.sla.0000189606.58343.cd. discussion 887–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim SI, Kwak JH, Zachariah M, He Y, Wang L, Choi ME. TGF-beta-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-beta1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Renal Physiol. 2007;292(5):F1471–8. doi: 10.1152/ajprenal.00485.2006. [DOI] [PubMed] [Google Scholar]
- 129.Wang L, Ma R, Flavell RA, Choi ME. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF-beta 1 in murine mesangial cells. J Biol Chem. 2002;277(49):47257–62. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 130.Niculescu-Duvaz I, Phanish MK, Colville-Nash P, Dockrell ME. The TGFbeta1-induced fibronectin in human renal proximal tubular epithelial cells is p38 MAP kinase dependent and Smad independent. Nephron Exp Nephrol. 2007;105(4):e108–16. doi: 10.1159/000100492. [DOI] [PubMed] [Google Scholar]
- 131.Wang L, Kwak JH, Kim SI, He Y, Choi ME. Transforming growth factor-beta1 stimulates vascular endothelial growth factor 164 via mitogen-activated protein kinase kinase 3-p38alpha and p38delta mitogen-activated protein kinase-dependent pathway in murine mesangial cells. J Biol Chem. 2004;279(32):33213–9. doi: 10.1074/jbc.M403758200. [DOI] [PubMed] [Google Scholar]
- 132.Kocher HM, Moorhead J, Sharpe CC, Dockrell ME, Al-Nawab M, Hendry BM. Expression of Ras GTPases in normal kidney and in glomerulonephritis. Nephrol Dial Transplant. 2003;18(11):2284–92. doi: 10.1093/ndt/gfg375. [DOI] [PubMed] [Google Scholar]
- 133.Runyan CE, Schnaper HW, Poncelet AC. Smad3 and PKCdelta mediate TGF-beta1-induced type I collagen expression in human mesangial cells. Am J Physiol Renal Physiol. 2003;285:F413–F422. doi: 10.1152/ajprenal.00082.2003. [DOI] [PubMed] [Google Scholar]
- 134.Runyan CE, Schnaper HW, Poncelet AC. The PI3K-Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to TGF-beta1. J Biol Chem. 2004;279:2632–2639. doi: 10.1074/jbc.M310412200. [DOI] [PubMed] [Google Scholar]
- 135.Wicks SJ, Lui S, Abdel-Wahab N, Mason RM, Chantry A. Inactivation of smad-transforming growth factor beta signaling by Ca (2+)-calmodulin-dependent protein kinase II. Mol Cell Biol. 2000;20(21):8103–11. doi: 10.1128/mcb.20.21.8103-8111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G, Rama I, Vidal A, Grinyo JM. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol. 2006;17(5):1395–404. doi: 10.1681/ASN.2005050549. [DOI] [PubMed] [Google Scholar]
- 137.Ortega-Velazquez R, Gonzalez-Rubio M, Ruiz-Torres MP, Diez-Marques ML, Iglesias MC, Rodriguez-Puyol M, Rodriguez-Puyol D. Collagen I upregulates extracellular matrix gene expression and secretion of TGF-beta 1 by cultured human mesangial cells. Am J Physiol Cell Physiol. 2004;286(6):C1335–43. doi: 10.1152/ajpcell.00279.2003. [DOI] [PubMed] [Google Scholar]
- 138.Roskelley CD, Bissell MJ. Dynamic reciprocity revisited: a continuous, bidirectional flow of information between cells and the extracellular matrix regulates mammary epithelial cell function. Biochem Cell Biol. 1995;73(7–8):391–7. doi: 10.1139/o95-046. [DOI] [PubMed] [Google Scholar]
- 139.Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, Simon KJ, Chun Wang L, Leone DR, Lobb RR, McCrann DJ, Allaire NE, Horan GS, Fogo A, Kalluri R, Shield CF, 3rd, Sheppard D, Gardner HA, Violette SM. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170(1):110–25. doi: 10.2353/ajpath.2007.060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9(7):964–8. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 141.Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol. 2001;12(11):2392–9. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- 142.Wang S, de Caestecker M, Kopp J, Mitu G, Lapage J, Hirschberg R. Renal bone morphogenetic protein-7 protects against diabetic nephropathy. J Am Soc Nephrol. 2006;17(9):2504–12. doi: 10.1681/ASN.2006030278. [DOI] [PubMed] [Google Scholar]
- 143.Wang S, Hirschberg R. BMP7 antagonizes TGF-beta -dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol. 2003;284(5):F1006–13. doi: 10.1152/ajprenal.00382.2002. [DOI] [PubMed] [Google Scholar]
- 144.Wang S, Hirschberg R. Bone morphogenetic protein-7 signals opposing transforming growth factor beta in mesangial cells. J Biol Chem. 2004;279(22):23200–6. doi: 10.1074/jbc.M311998200. [DOI] [PubMed] [Google Scholar]
- 145.Zhang XL, Selbi W, de la Motte C, Hascall V, Phillips AO. Bone morphogenic protein-7 inhibits monocyte-stimulated TGF-beta1 generation in renal proximal tubular epithelial cells. J Am Soc Nephrol. 2005;16(1):79–89. doi: 10.1681/ASN.2004050395. [DOI] [PubMed] [Google Scholar]
- 146.Yang J, Liu Y. Delayed administration of hepatocyte growth factor reduces renal fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2003;284(2):F349–57. doi: 10.1152/ajprenal.00154.2002. [DOI] [PubMed] [Google Scholar]
- 147.Yang J, Dai C, Liu Y. A novel mechanism by which hepatocyte growth factor blocks tubular epithelial to mesenchymal transition. J Am Soc Nephrol. 2005;16(1):68–78. doi: 10.1681/ASN.2003090795. [DOI] [PubMed] [Google Scholar]
- 148.Riser BL, Cortes P, Heilig C, Grondin J, Ladson-Wofford S, Patterson D, Narins RG. Cyclic stretching force selectively up-regulates transforming growth factor-β isoforms in cultured rat mesangial cells. Am J Pathol. 1996;148:1915–1923. [PMC free article] [PubMed] [Google Scholar]
- 149.Krepinsky JC, Li Y, Tang D, Liu L, Scholey J, Ingram AJ. Stretch-induced Raf-1 activation in mesangial cells requires actin cytoskeletal integrity. Cell Signal. 2005;17(3):311–20. doi: 10.1016/j.cellsig.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 150.Krepinsky JC, Li Y, Chang Y, Liu L, Peng F, Wu D, Tang D, Scholey J, Ingram AJ. Akt mediates mechanical strain-induced collagen production by mesangial cells. J Am Soc Nephrol. 2005;16(6):1661–72. doi: 10.1681/ASN.2004100897. [DOI] [PubMed] [Google Scholar]
- 151.Isono M, Cruz MC, Chen S, Hong SW, Ziyadeh FN. Extracellular signal-regulated kinase mediates stimulation of TGF-beta1 and matrix by high glucose in mesangial cells. J Am Soc Nephrol. 2000;11:2222–2230. doi: 10.1681/ASN.V11122222. [DOI] [PubMed] [Google Scholar]
- 152.Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65(1):30–9. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 153.Wolf G, Ziyadeh FN, Zahner G, Stahl RA. Angiotensin II-stimulated expression of transforming growth factor beta in renal proximal tubular cells: attenuation after stable transfection with the c-mas oncogene. Kidney Int. 1995;48(6):1818–27. doi: 10.1038/ki.1995.480. [DOI] [PubMed] [Google Scholar]
- 154.Sayeski PP, Bernstein KE. Signal transduction mechanisms of the angiotensin II type AT (1)-receptor: looking beyond the heterotrimeric G protein paradigm. J Renin Angiotensin Aldosterone Syst. 2001;2(1):4–10. doi: 10.3317/jraas.2001.007. [DOI] [PubMed] [Google Scholar]
- 155.Chen J, Chen JK, Neilson EG, Harris RC. Role of EGF receptor activation in angiotensin II-induced renal epithelial cell hypertrophy. J Am Soc Nephrol. 2006;17(6):1615–23. doi: 10.1681/ASN.2005111163. [DOI] [PubMed] [Google Scholar]
- 156.Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, Bhowmick NA, Ju W, Bottinger EP, Lan HY. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res. 2006;98(8):1032–9. doi: 10.1161/01.RES.0000218782.52610.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, Goldschmeding R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998;53(4):853–61. doi: 10.1111/j.1523-1755.1998.00820.x. [DOI] [PubMed] [Google Scholar]
- 158.Wang S, Denichilo M, Brubaker C, Hirschberg R. Connective tissue growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney Int. 2001;60(1):96–105. doi: 10.1046/j.1523-1755.2001.00776.x. [DOI] [PubMed] [Google Scholar]
- 159.Yokoi H, Mukoyama M, Sugawara A, Mori K, Nagae T, Makino H, Suganami T, Yahata K, Fujinaga Y, Tanaka I, Nakao K. Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. Am J Physiol Renal Physiol. 2002;282(5):F933–42. doi: 10.1152/ajprenal.00122.2001. [DOI] [PubMed] [Google Scholar]
- 160.Qi W, Twigg S, Chen X, Polhill TS, Poronnik P, Gilbert RE, Pollock CA. Integrated actions of transforming growth factor-beta1 and connective tissue growth factor in renal fibrosis. Am J Physiol Renal Physiol. 2005;288(4):F800–9. doi: 10.1152/ajprenal.00179.2004. [DOI] [PubMed] [Google Scholar]
- 161.Phanish MK, Wahab NA, Hendry BM, Dockrell ME. TGF-beta1-induced connective tissue growth factor (CCN2) expression in human renal proximal tubule epithelial cells requires Ras/MEK/ERK and Smad signalling. Nephron Exp Nephrol. 2005;100(4):e156–65. doi: 10.1159/000085445. [DOI] [PubMed] [Google Scholar]
- 162.Li Y, Wen X, Spataro BC, Hu K, Dai C, Liu Y. hepatocyte growth factor is a downstream effector that mediates the antifibrotic action of peroxisome proliferator-activated receptor-gamma agonists. J Am Soc Nephrol. 2006;17(1):54–65. doi: 10.1681/ASN.2005030257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wen X, Li Y, Hu K, Dai C, Liu Y. Hepatocyte growth factor receptor signaling mediates the anti-fibrotic action of 9-cis-retinoic acid in glomerular mesangial cells. Am J Pathol. 2005;167(4):947–57. doi: 10.1016/S0002-9440(10)61185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Blush J, Lei J, Ju W, Silbiger S, Pullman J, Neugarten J. Estradiol reverses renal injury in Alb/TGF-beta1 transgenic mice. Kidney Int. 2004;66(6):2148–54. doi: 10.1111/j.1523-1755.2004.66005.x. [DOI] [PubMed] [Google Scholar]
- 165.Neugarten J, Medve I, Lei J, Silbiger SR. Estradiol suppresses mesangial cell type I collagen synthesis via activation of the MAP kinase cascade. Am J Physiol. 1999;277:F875–F881. doi: 10.1152/ajprenal.1999.277.6.F875. [DOI] [PubMed] [Google Scholar]
- 166.Zdunek M, Silbiger S, Lei J, Neugarten J. Protein kinase CK2 mediates TGF-beta1-stimulated type IV collagen gene transcription and its reversal by estradiol. Kidney Int. 2001;60:2097–2108. doi: 10.1046/j.1523-1755.2001.00041.x. [DOI] [PubMed] [Google Scholar]
- 167.Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol. 2007;2(3):529–42. doi: 10.2215/CJN.04121206. [DOI] [PubMed] [Google Scholar]
- 168.Pollak MR. Inherited podocytopathies: FSGS and nephrotic syndrome from a genetic viewpoint. J Am Soc Nephrol. 2002;13(12):3016–23. doi: 10.1097/01.asn.0000039569.34360.5e. [DOI] [PubMed] [Google Scholar]
- 169.Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Bottinger EP. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71(12):1205–14. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 171.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, Jr, Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem. 2003;278(44):43001–7. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 172.Khera T, Martin J, Riley S, Stedman R, Phillips AO. Glucose enhances mesangial cell apoptosis. Lab Invest. 2006;86(6):566–77. doi: 10.1038/labinvest.3700418. [DOI] [PubMed] [Google Scholar]
- 173.Shankland SJ, Eitner F, Hudkins KL, Goodpaster T, D’Agati V, Alpers CE. Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: role in podocyte proliferation and maturation. Kidney Int. 2000;58(2):674–83. doi: 10.1046/j.1523-1755.2000.00213.x. [DOI] [PubMed] [Google Scholar]
- 174.Wahab N, Cox D, Witherden A, Mason RM. Connective tissue growth factor (CTGF) promotes activated mesangial cell survival via up-regulation of mitogen-activated protein kinase phosphatase-1 (MKP-1) Biochem J. 2007;406(1):131–8. doi: 10.1042/BJ20061817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Abdel-Wahab N, Weston BS, Roberts T, Mason RM. Connective tissue growth factor and regulation of the mesangial cell cycle: role in cellular hypertrophy. J Am Soc Nephrol. 2002;13(10):2437–45. doi: 10.1097/01.asn.0000031828.58276.02. [DOI] [PubMed] [Google Scholar]
- 176.Wada T, Pippin JW, Terada Y, Shankland SJ. The cyclin-dependent kinase inhibitor p21 is required for TGF-beta1-induced podocyte apoptosis. Kidney Int. 2005;68(4):1618–29. doi: 10.1111/j.1523-1755.2005.00574.x. [DOI] [PubMed] [Google Scholar]
- 177.Wolf G, Schanze A, Stahl RA, Shankland SJ, Amann K. p27 (Kip1) Knockout mice are protected from diabetic nephropathy: evidence for p27 (Kip1) haplotype insufficiency. Kidney Int. 2005;68(4):1583–9. doi: 10.1111/j.1523-1755.2005.00570.x. [DOI] [PubMed] [Google Scholar]
- 178.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2002;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15(3–4):290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- 180.Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Muller GA, Neilson EG. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61(5):1714–28. doi: 10.1046/j.1523-1755.2002.00333.x. [DOI] [PubMed] [Google Scholar]
- 181.Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol. 1997;273(4 Pt 2):F563–74. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- 182.Prunier C, Howe PH. Disabled-2 (Dab2) is required for transforming growth factor beta-induced epithelial to mesenchymal transition (EMT) J Biol Chem. 2005;280(17):17540–8. doi: 10.1074/jbc.M500974200. [DOI] [PubMed] [Google Scholar]
- 183.Liu BC, Li MX, Zhang JD, Liu XC, Zhang XL, Phillips AO. Inhibition of integrin-linked kinase via a siRNA expression plasmid attenuates connective tissue growth factor-induced human proximal tubular epithelial cells to mesenchymal transition. Am J Nephrol. 2008;28(1):143–51. doi: 10.1159/000110019. [DOI] [PubMed] [Google Scholar]
- 184.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. Embo J. 2004;23(5):1155–65. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ivanova L, Butt MJ, Matsell DG. Mesenchymal transition in kidney collecting duct epithelial cells. Am J Physiol Renal Physiol. 2008 doi: 10.1152/ajprenal.00326.2007. [DOI] [PubMed] [Google Scholar]
- 186.Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172(2):299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Johnson RJ, Floege J, Yoshimura A, Iida H, Couser WG, Alpers CE. The activated mesangial cell: a glomerular “myofibroblast”? J Am Soc Nephrol. 1992;2(10 Suppl):S190–7. doi: 10.1681/ASN.V210s190. [DOI] [PubMed] [Google Scholar]
- 188.Bassez G, Camand OJ, Cacheux V, Kobetz A, Dastot-Le Moal F, Marchant D, Catala M, Abitbol M, Goossens M. Pleiotropic and diverse expression of ZFHX1B gene transcripts during mouse and human development supports the various clinical manifestations of the “Mowat-Wilson” syndrome. Neurobiol Dis. 2004;15(2):240–50. doi: 10.1016/j.nbd.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 189.Miquelajauregui A, Van de Putte T, Polyakov A, Nityanandam A, Boppana S, Seuntjens E, Karabinos A, Higashi Y, Huylebroeck D, Tarabykin V. Smad-interacting protein-1 (Zfhx1b) acts upstream of Wnt signaling in the mouse hippocampus and controls its formation. Proc Natl Acad Sci U S A. 2007;104(31):12919–24. doi: 10.1073/pnas.0609863104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267–78. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 191.Imamichi Y, Konig A, Gress T, Menke A. Collagen type I-induced Smad-interacting protein 1 expression downregulates E-cadherin in pancreatic cancer. Oncogene. 2007;26(16):2381–5. doi: 10.1038/sj.onc.1210012. [DOI] [PubMed] [Google Scholar]
- 192.Long J, Zuo D, Park M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J Biol Chem. 2005;280(42):35477–89. doi: 10.1074/jbc.M504477200. [DOI] [PubMed] [Google Scholar]
- 193.Dastot-Le Moal F, Wilson M, Mowat D, Collot N, Niel F, Goossens M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat. 2007;28(4):313–21. doi: 10.1002/humu.20452. [DOI] [PubMed] [Google Scholar]
- 194.Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol Biol Cell. 2007;18(9):3533–44. doi: 10.1091/mbc.E07-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi JJ, Natarajan R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci U S A. 2007;104(9):3432–7. doi: 10.1073/pnas.0611192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Boutet A, De Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. Embo J. 2006;25(23):5603–13. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yoshino J, Monkawa T, Tsuji M, Inukai M, Itoh H, Hayashi M. Snail1 is involved in the renal epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2007;362(1):63–8. doi: 10.1016/j.bbrc.2007.07.146. [DOI] [PubMed] [Google Scholar]
- 198.de Boer TP, van Veen TA, Bierhuizen MF, Kok B, Rook MB, Boonen KJ, Vos MA, Doevendans PA, de Bakker JM, van der Heyden MA. Connexin43 repression following epithelium-to-mesenchyme transition in embryonal carcinoma cells requires Snail1 transcription factor. Differentiation. 2007;75(3):208–18. doi: 10.1111/j.1432-0436.2006.00133.x. [DOI] [PubMed] [Google Scholar]
- 199.Chen X, Zankl A, Niroomand F, Liu Z, Katus HA, Jahn L, Tiefenbacher C. Upregulation of ID protein by growth and differentiation factor 5 (GDF5) through a smad-dependent and MAPK-independent pathway in HUVSMC. J Mol Cell Cardiol. 2006;41(1):26–33. doi: 10.1016/j.yjmcc.2006.03.421. [DOI] [PubMed] [Google Scholar]
- 200.Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11(4):915–26. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 201.Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol. 2004;24(10):4241–54. doi: 10.1128/MCB.24.10.4241-4254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]