

Abstract
Aims: Increased fructose consumption predisposes the liver to nonalcoholic fatty liver disease (NAFLD), but the mechanisms are elusive. Thioredoxin-interacting protein (TXNIP) links oxidative stress to NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome activation and this signaling axis may be involved in fructose-induced NAFLD. Here, we explore the role of reactive oxygen species (ROS)-induced TXNIP overexpression in fructose-mediated hepatic NLRP3 inflammasome activation, inflammation, and lipid accumulation. Results: Rats were fed a 10% fructose diet for 8 weeks and treated with allopurinol and quercetin during the last 4 weeks. Five millimolars of fructose-exposed hepatocytes (primary rat hepatocytes, rat hepatic parenchymal cells [RHPCs], HLO2, HepG2) were co-incubated with antioxidants or caspase-1 inhibitor or subjected to TXNIP or NLRP3 siRNA interference. Fructose induced NLRP3 inflammasome activation and pro-inflammatory cytokine secretion, janus-activated kinase 2/signal transducers and activators of transcription 3-mediated inflammatory signaling, and expression alteration of lipid metabolism-related genes in cultured hepatocytes and rat livers. NLRP3 silencing and caspase-1 suppression blocked these effects in primary rat hepatocytes and RHPCs, confirming that inflammasome activation alters hepatocyte lipid metabolism. Hepatocellular ROS and TXNIP were increased in animal and cell models. TXNIP silencing blocked NLRP3 inflammasome activation, inflammation, and lipid metabolism perturbations but not ROS induction in fructose-exposed hepatocytes, whereas antioxidants addition abrogated TXNIP induction and diminished the detrimental effects in fructose-exposed hepatocytes and rat livers. Innovation and Conclusions: This study provides a novel mechanism for fructose-induced NAFLD pathogenesis by which the ROS-TXNIP pathway mediates hepatocellular NLRP3 inflammasome activation, inflammation and lipid accumulation. Antioxidant-based interventions can inhibit the ROS-TXNIP pathway. Antioxid. Redox Signal. 22, 848–870.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a global medical problem (7, 24, 45). Clinical and experimental evidence indicates that excessive fructose consumption is involved in the pathogenesis of metabolic syndrome and associated NAFLD (7, 22, 24, 33, 45, 47–49, 59). The direct effect of fructose on hepatic lipid metabolism, which is characterized by intracellular triglyceride (TG) accumulation (hepatic steatosis), is considered the first hit in the pathogenesis of NAFLD. Hepatocellular oxidative stress as a result of excessive reactive oxygen species (ROS) production due to inflammation or endogenous toxins constitutes the second hit in the pathogenesis of NAFLD and its progression to nonalcoholic steatohepatitis (NASH) (24, 47–49). However, the molecular mechanisms that underlie fructose-mediated healthy liver→NAFLD→NASH transition are incompletely understood.
Innovation.
This study provides both in vitro and in vivo evidence for the induction of the reactive oxygen species (ROS)-thioredoxin-interacting protein (TXNIP) signaling axis in hepatocytes by fructose. Induction of this signaling axis leads to NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome activation, sterile inflammation, and alterations in the expression levels of lipid metabolism-related genes. Induction of the ROS-TXNIP signaling axis and downstream inflammatory and metabolic effects can be prevented by antioxidants, altogether providing a novel mechanism for the fructose-mediated pathogenesis of nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH) as well as potential sites for pharmacological intervention.
Kupffer cells seem to occupy a central role in the pathogenesis of fructose-induced NAFLD, given that Kupffer cells mediate the interplay between inflammasome activation, inflammatory signaling, and fat metabolism. The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is composed of NLRP3, apoptosis-associated speck-like protein (ASC) and caspase-1 and is present in innate immune cells, including Kupffer cells (34). NLRP3 interacts with ASC to cleave caspase-1, leading to maturation and secretion of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 (19, 55), which has been demonstrated in Kupffer cells during conditions of oxidative stress, sterile inflammation, and liver injury (15, 18). These cytokines are able to induce dyslipidemia and lipid accumulation in hepatocytes (32, 54). Moreover, genetic NLRP3 inflammasome deficiency in innate immune cells predisposes animals to develop NAFLD through alterations in gut microbiota (12), and gut-derived endotoxin-dependent activation of Kupffer cells in mice has been suggested to mediate the onset of fructose-induced hepatic steatosis via inducible nitric oxide synthase (iNOS) and Toll-like receptor 4 signaling (47–49). Lastly, this inflammasome activation in Kupffer cells has been observed in choline-deficient amino-acid-defined (CDAA) diet-induced NASH in mice (30), altogether attesting to the paracrine effects of immune cell-mediated inflammasome activation and inflammatory signaling on hepaocellular lipid metabolism.
Nevertheless, relatively little is known regarding the role of fructose in the inflammasome-inflammation-lipid metabolism signaling axis. Fructose is directly utilized and metabolized by hepatocytes, which express the fructose transporters glucose transporter 2 (GLUT2), GLUT5, and GLUT8 (16). Previous studies have revealed that a high fructose diet in rodents is associated with oxidative stress, inflammation, and fat accumulation (22, 36), underscoring an instigator role of fructose in the aforementioned signaling axis. Accordingly, fructose may directly interfere in the hepatocellular redox processes that lie at the basis of NAFLD/NASH and the metabolic syndrome.
Thioredoxin-interacting protein (TXNIP) has been implicated in inflammatory signaling in response to oxidative stress. In resting cells, TXNIP is bound and kept in inactive state by its intrinsic binding partner thioredoxin, a ubiquitous protein that reduces thiols (particularly insulin disulfides), and controls intracellular ROS levels to suppress the manifestation of oxidative stress (26). Under conditions of high ROS levels and oxidative stress, TXNIP dissociates from thiroredoxin to associate with NLRP3 (63), activating the inflammasome to mediate inflammatory signaling (62). The antioxidant transcription factor nuclear factor (erythroid-derived-2)-like 2 (Nrf2) represses TXNIP expression in response to oxidative stress (11). However, Nrf2, which is activated by oxidative stress in livers of NASH patients (52), and upregulated in livers of male Wistar rats with fructose-induced steatohepatitis (60), can also activate NLRP3 inflammasome (61). It is currently unknown whether the ROS-TXNIP pathway or the ROS-Nrf2 pathway is responsible for NLRP3 inflammasome activation in hepatocytes in the context of fructose-induced NAFLD.
Antioxidants have shown some promise in reducing hepatocellular inflammation and injury in patients with NASH (45). Allopurinol, a clinically approved xanthine oxidase (XO) inhibitor, suppresses ROS formation and ameliorates hepatic ischemia reperfusion injury in various animal models (40), ameliorates thioacetamide-induced decrease in liver Nrf2 in a rat model of acute liver failure (5), and prevents excess lysine-induced fatty liver development in growing rats (56). Furthermore, allopurinol reduces hemozoin-induced NLRP3 inflammasome activation (10) and controls IL-1β production in inflammasome-deficient mice (9). Quercetin is one of the most important dietary flavonoids with potentially beneficial properties for human health (44). It reduces H2O2-induced oxidative stress and ROS generation in human hepatocellular carcinoma (HepG2) cells (17) and ameliorates hepatic inflammation and fibrosis in mice with NASH (28). Quercetin also induces Nrf2 nuclear translocation and expression in ochratoxin A-exposed HepG2 cells (42) and rats with high fructose-, sucrose-, and fat diet-induced NAFLD (38). In our previous studies, allopurinol and quercetin reduced serum and renal TG and total cholesterol (TC) levels and suppressed renal NLRP3 inflammasome activation in fructose-fed rats (13, 14).
Based on what has been stated earlier, two main hypotheses were tested. First, we assessed whether ROS-induced TXNIP expression plays a fundamental role in fructose-mediated hepatic NLRP3 inflammasome activation, inflammatory signaling, and lipid accumulation in hepatocytes. Second, we tested the hypothesis that allopurinol and quercetin affect hepatic oxidative stress and TXNIP expression and consequently reduce the extent of NLRP3 inflammasome activation, resulting in protection from fructose-induced NAFLD.
Results
High fructose consumption induces NLRP3 inflammasome activation, inflammation, and lipid accumulation in rat livers
Previous studies in rats (22, 33) have demonstrated that a 10% fructose diet (in drinking water) during 8 weeks induced several features of human NAFLD, including hepatic inflammation (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/ars) and lipid accumulation (Supplementary Fig. S1B) characterized by increased levels of TG and TC (Supplementary Table S1). Increased body weight and decreased food intake were observed in fructose-fed rats, which were not accompanied by significant changes in total caloric intake compared with the control group (Supplementary Table S1).
Intrahepatic levels of NLRP3, ASC, and caspase-1, which are components of the NLRP3 inflammasome (Fig. 1A), as well as IL-1β and IL-18 (Fig. 1B) were elevated in fructose-fed rats versus control animals. Immunohistochemical (IHC) staining of cytokeratin 18 (CK18, hepatocytes) and CD163 (Kupffer cells) revealed that NLRP3 expression was weak in Kupffer cells and absent in hepatocytes in control rats. After 8-week fructose feeding, the expression of NLRP3 and co-localization of NLRP3 with CK18 and CD163 were significantly increased, indicating that fructose-induced NAFLD in rats is associated with augmented NLRP3 signaling in Kupffer cells and hepatocytes (Fig. 1C).
FIG. 1.
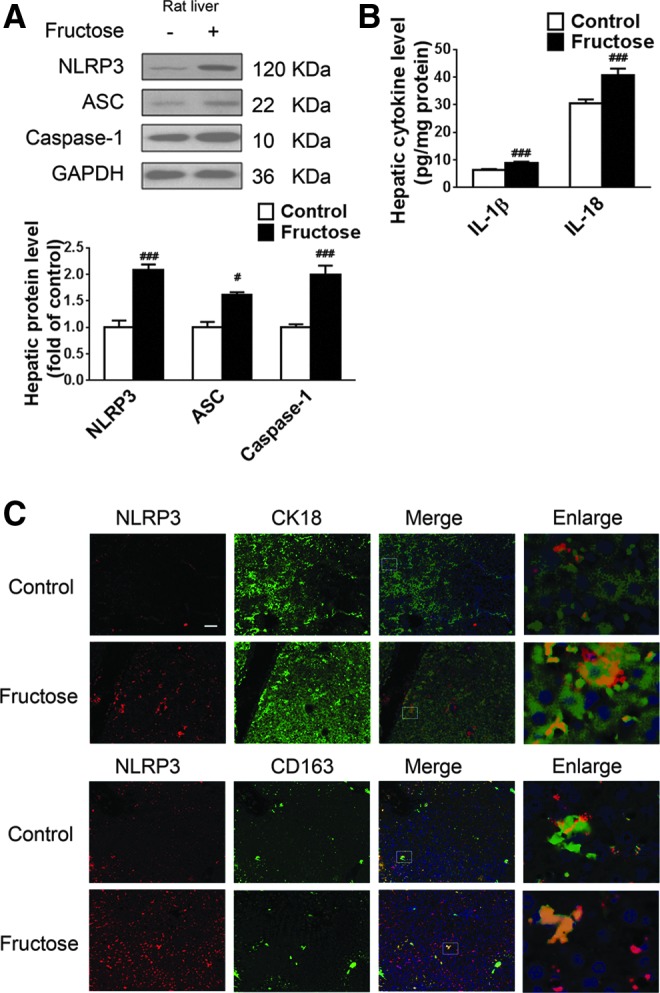
Excess fructose intake induces NLRP3 inflammasome activation and IL-1β and IL-18 production in rat livers. Rats were given drinking water with or without 10% fructose for 8 weeks, after which liver biopsies were obtained for analysis. (A) Western blot analysis of NLRP3, ASC, and caspase-1 protein levels (n=8/group). (B) ELISA-based quantification of IL-1β and IL-18 levels (n=8/group). (C) Immunohistochemistry of NLRP3 (red) in liver sections co-stained with fluorescently labeled antibodies against CK18 (hepatocytes, green) or against glucocorticoid-inducible member of the scavenger receptor cysteine-rich family (CD163, Kupffer cells, green). Scale bar=100 μm. Results are expressed as mean±SEM. #p<0.05, ###p<0.001 versus control group. ASC, apoptosis-associated speck-like protein; CK18, cytokeratin 18; IL, interleukin; NLRP3, NOD-like receptor family, pyrin domain containing 3. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Accordingly, we tested whether fructose directly activates NLRP3 inflammasome in hepatic parenchymal cells and Kupffer cells using primary hepatocytes and Kupffer cells isolated from rat livers. The results were validated in liver-pertinent rat and human cell lines, including rat hepatic parenchymal cells (RHPCs), human hepatic (HLO2) cells, and HepG2 cells.
Fructose exposure stimulates NLRP3 inflammasome activation, pro-inflammatory cytokine production, and secretion in cultured hepatocytes
Hepatocytes and Kupffer cells were incubated with 5 mM fructose for 72 h. Compared with the control group, protein levels of NLRP3, ASC, and caspase-1 were significantly upregulated in fructose-exposed primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 2A). Moreover, IL-1β and IL-18 levels were increased in the cell lysates and supernatants of these cells (Fig. 2B). However, 5 mM fructose did not significantly affect NLRP3, ASC, caspase-1, IL-1β, and IL-18 in Kupffer cells (data not shown). These data strongly suggest that 5 mM fructose directly induces NLRP3 inflammasome activation in hepatic parenchymal cells but not in innate immune cells.
FIG. 2.

Fructose exposure stimulates NLRP3 inflammasome activation and IL-1β and IL-18 secretion in cultured hepatocytes. (A) Primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were cultured with or without 5 mM fructose for 72 h and analyzed for NLRP3, ASC, and caspase-1 protein levels by Western blot (n=8/group). (B) ELISA-based quantification of IL-1β and IL-18 levels in cell lysate (top) and supernatant (bottom) (n=8/group). (C) Primary rat hepatocytes and RHPCs were co-incubated with or without 5 mM fructose or/and the caspase-1-specific inhibitor Ac-YVAD-CMK for 72 h. Caspase-1 activity and IL-1β and IL-18 levels (ELISA) were determined in cell lysate (top) and supernatant (bottom) (n=8/group). (D) Primary rat hepatocytes and RHPCs were transfected with or without NLRP3-specific siRNA for 48 h, after which NLRP3 was assayed by RT-PCR (n=4/group). (E) Primary rat hepatocytes and RHPCs were transfected with NLRP3-specific siRNA for 48 h and co-incubated with 5 mM fructose for another 72 h. Western blot analysis of caspase-1 protein levels and ELISA-based determination of IL-1β and IL-18 levels in cell lysate (top) and supernatant (bottom) are shown (n=8/group). Results are expressed as mean±SEM. #p<0.05, ##p<0.01, ###p<0.001 versus control group. Ac-YVAD-CMK, Ac-Tyr-Val-Ala-Asp-chloromethylketone; RHPCs, rat hepatic parenchymal cells; RT-PCR, reverse transcription–polymerase chain reaction.
To confirm that fructose-induced IL-1β and IL-18 production and secretion were a result of inflammasome activation, 5 mM fructose-exposed primary rat hepatocytes and RHPCs were co-incubated with the caspase-1 specific inhibitor Ac-YVAD-CMK (Ac-Tyr-Val-Ala-Asp-chloromethylketone). Ac-YVAD-CMK reduced caspase-1 activation (Fig. 2C) and diminished fructose-induced IL-1β and IL-18 induction (Fig. 2C). Furthermore, gene silencing of NLRP3 was performed with rat NLRP3-specific siRNA in primary rat hepatocytes and RHPCs and verified by reverse transcription–polymerase chain reaction (RT-PCR) (Fig. 2D). Transfection of primary rat hepatocytes and RHPCs with NLRP3 siRNA for 48 h before 5 mM fructose exposure also blocked inducible caspase-1 cleavage (Fig. 2E) and pro-inflammatory cytokine secretion (Fig. 2E). These results demonstrate that fructose directly activates NLRP3 inflammasome and corollary production and secretion of IL-1β and IL-18 in primary rat hepatocytes and RHPCs.
NLRP3 inflammasome activation alters lipid metabolism-related gene expression in fructose-exposed cultured hepatocytes
Inflammation is linked to lipid metabolism. Pro-inflammatory cytokines such as IL-1β are able to disturb lipid metabolism-related gene expression through the janus-activated kinase 2-signal transducers and activators of transcription 3 (JAK2-STAT3) inflammatory signaling pathway and its negative feedback regulator, suppressors of cytokine signaling 3 (SOCS3). Indeed, downregulation of JAK2 phosphorylation and upregulation of STAT3 phosphorylation as well as SOCS3 protein levels were detected in fructose-exposed primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 3A). Peroxisome proliferator-activated receptor-α (PPAR-α) functions as a sensor for fatty acid β-oxidation and controls sterol regulatory element binding protein 1 (SREBP1) and stearoyl-CoA desaturase 1 (SCD1) to reduce liver TG synthesis. In parallel, 5 mM fructose decreased PPAR-α protein levels and increased SREBP1 and SCD1 protein levels in these hepatocytes (Fig. 3B).
FIG. 3.

NLRP3 inflammasome activation alters lipid metabolism-related gene expression in fructose-exposed cultured hepatocytes. (A) Primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were cultured with or without 5 mM fructose for 72 h, after which p-JAK2, JAK2, p-STAT3, STAT3, and SOCS3 protein levels were assayed by Western blot (n=8/group). (B) Western blot analysis of PPAR-α, SREBP1, and SCD1 protein levels (n=8/group). (C) Primary rat hepatocytes and RHPCs were co-incubated with or without 5 mM fructose and caspase-1-specific inhibitor Ac-YVAD-CMK for 72 h, after which p-JAK2, JAK2, p-STAT3, STAT3, SOCS3, PPAR-α, SREBP1, and SCD1 protein levels were analyzed by Western blot. (D) Primary rat hepatocytes and RHPCs were transfected with NLRP3-specific siRNA for 48 h and co-incubated with or without 5 mM fructose for another 72 h, followed by Western blot analysis of p-JAK2, JAK2, p-STAT3, STAT3, SOCS3, PPAR-α, SREBP1, and SCD1 protein levels. Results are expressed as mean±SEM. #p<0.05, ##p<0.01, ###p<0.001 versus control group. JAK2, janus-activated kinase 2; PPAR-α, peroxisome proliferator-activated receptor-α; SCD1, stearoyl-CoA desaturase 1; SOCS3, suppressors of cytokine signaling 3; SREBP1, sterol regulatory element binding protein 1; STAT3, signal transducer and activator of transcription 3.
In addition, inhibition of caspase-1 activation with Ac-YVAD-CMK in primary rat hepatocytes and RHPCs significantly restored fructose-induced changes in JAK2 and STAT3 phosphorylation and SOCS3 expression (Fig. 3C), and in the expression levels of PPAR-α, SREBP1, and SCD1 (Fig. 3C). Moreover, RNAi silencing of NLRP3 in primary rat hepatocytes and RHPCs negated the fructose-mediated effects on the JAK2-STAT3 pathway (Fig. 3D) and lipid metabolism-related gene expression (Fig. 3D). These results indicate that fructose-induced inflammasome activation leads to the JAK2-STAT3 signaling, which, in turn, alters lipid metabolism-related gene expression in hepatocytes.
TXNIP mediates fructose-induced NLRP3 inflammasome activation and lipid metabolism-related gene deregulation in cultured hepatocytes
TXNIP participates in NLRP3 inflammasome activation (62). In agreement with the fructose-induced hepatic inflammation and lipid accumulation observed in rats, TXNIP protein levels were increased in rat livers (Fig. 4A). Immunohistochemistry confirmed the increased TXNIP levels in hepatocytes and Kupffer cells versus control animals (Fig. 4B). A significant increase in TXNIP protein levels was observed in 5 mM fructose-exposed primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 4C), but not in Kupffer cells (data not shown), indicating that TXNIP induction may be a specific response of these hepatic parenchymal cells to fructose stimulation.
FIG. 4.

Fructose induces TXNIP overexpression in rat livers and cultured hepatocytes. Rats were given drinking water with or without 10% fructose for 8 weeks, after which liver biopsies were obtained for analysis. (A) Hepatic TXNIP protein levels were determined by Western blot (n=8/group). (B) Representative immunohistological sections stained for TXNIP (red) and co-stained for CK18 (hepatocytes, green) or CD163 (Kupffer cells, green). Scale bar=100 μm. (C) Primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were cultured with or without 5 mM fructose for 72 h, followed by Western blot analysis of TXNIP protein levels (n=8/group). Results are expressed as mean±SEM. ##p<0.01, ###p<0.001 versus control group. TXNIP, thioredoxin-interacting protein. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
To demonstrate that TXNIP overexpression was a key factor in fructose-induced NLRP3 inflammasome activation in cultured hepatocytes, primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were transfected with TXNIP siRNA 24 h before fructose exposure. Gene silencing was verified by RT-PCR after 48 h (Fig. 5A). The silencing of TXNIP expression totally blocked fructose-induced NLRP3 and ASC upregulation (Fig. 5B), caspase-1 activation (Fig. 5B), and IL-1β and IL-18 secretion (Fig. 5C). In addition, TXNIP silencing almost completely blocked fructose-mediated PPAR-α downregulation, and SREBP1 and SCD1 upregulation (Fig. 5D) in primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells. Therefore, TXNIP may play a fundamental role in mediating hepatocellular lipid metabolism in response to fructose exposure.
FIG. 5.

TXNIP mediates fructose-induced NLRP3 inflammasome activation and lipid metabolism-related gene deregulation in cultured hepatocytes. (A) Primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were transfected with or without TXNIP siRNA for 48 h. TXNIP expression levels were analyzed by RT-PCR (n=4/group). (B) Primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were transfected with TXNIP siRNA for 48 h and co-incubated with or without 5 mM fructose for another 72 h. NLRP3, ASC, and caspase-1 protein levels were quantitated by Western blot (n=8/group). (C) IL-1β and IL-18 concentrations in cell lysate (top) and supernatant (bottom) were determined by ELISA (n=8/group). (D) Western blot analysis of PPAR-α, SREBP1, and SCD1 protein levels (n=8/group). (E) Primary rat hepatocytes and RHPCs were transfected with NLRP3 siRNA for 48 h and co-incubated with or without 5 mM fructose for another 72 h. TXNIP protein levels were measured by Western blot (n=8/group). (F) Primary rat hepatocytes and RHPCs were co-incubated with or without 5 mM fructose and the caspase-1 specific inhibitor Ac-YVAD-CMK for 72 h, after which TXNIP protein levels were analyzed by Western blot (n=8/group). Results are expressed as mean±SEM. #p<0.05, ##p<0.01, ###p<0.001 versus control group.
To confirm that TXNIP overexpression was upstream of hepatic NLRP3 inflammasome activation in response to fructose exposure, this study showed that neither silencing NLRP3 (Fig. 5E) nor repressing caspase-1 activation (Fig. 5F) affected fructose-induced TXNIP overexpression in primary rat hepatocytes and RHPCs.
Fructose induces TXNIP overexpression through ROS production in cultured hepatocytes
TXNIP is a well-known oxidative stress sensor and promoter (26). Consistent with the fructose-induced increase in hepatic TXNIP protein levels, elevation of total ROS, H2O2, O2•−, malondialdehyde (MDA), XO, and iNOS level and activity as well as a reduction in superoxide dismutase (SOD) activity and total antioxidant capacity (TAC) were found in livers of fructose-fed rats (Table 1), demonstrating that high fructose intake is associated with elevated hepatic ROS production and oxidative stress.
Table 1.
Total ROS, H2O2, O2•−, MDA, XO, iNOS, SOD, and TAC in Fructose-Fed Rats
| Group | Dosage (mg/kg) | Total ROS (intensity[a.u.]/μg protein) | H2O2 (μmol/g protein) | O2•− (intensity[a.u.]/mg protein) | MDA (nmol/mg protein) | XO (U/mg protein) | iNOS (U/mg protein) |
|---|---|---|---|---|---|---|---|
| Control | 15.31±0.75 | 6.32±0.69 | 16.58±1.29 | 5.03±0.54 | 0.52±0.08 | 0.16±0.01 | |
| Fructose | 37.68±4.91a | 20.13±0.93a | 29.96±1.29b | 7.72±0.48a | 9.52±0.33a | 0.63±0.07a | |
| +Allopurinol | 5 | 23.06±1.55c | 7.72±0.31d | 17.02±1.29d | 5.57±0.36e | 0.31±0.04c | 0.22±0.02d |
| +Quercetin | 50 | 32.03±2.56e | 10.44±0.96e | 23.19±1.29e | 5.97±0.23e | 4.86±0.24e | 0.25±0.03e |
| 100 | 29.08±2.02e | 8.26±0.72e | 23.21±1.29e | 5.33±0.47d | 0.77±0.13d | 0.20±0.01d |
| SOD (U/mg protein) | TAC (mmol Trolox/mg protein) | ||
|---|---|---|---|
| Control | 1.53±0.05 | 1.32±0.08 | |
| Fructose | 0.62±0.14f | 0.32±0.07a | |
| +Allopurinol | 5 | 1.57±0.09e | 1.14±0.09d |
| +Quercetin | 50 | 1.71±0.08d | 0.74±0.07e |
| 100 | 1.90±0.19d | 1.15±0.05d |
All data are expressed as mean±SEM (n=8/group).
p<0.001 versus control group.
p<0.01 versus control group.
<0.001 versus fructose vehicle group.
p<0.01 versus fructose vehicle group.
p<0.05 versus fructose vehicle group.
p<0.05 versus control group.
iNOS, inducible nitric oxide synthase; MDA, malondialdehyde; ROS, reactive oxygen species; SOD, superoxide dismutase; TAC, total antioxidant capacity; XO, xanthine oxidase.
Incubation of primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells with 5 mM fructose resulted in a direct increase in total ROS levels (Fig. 6A). However, 5 mM fructose did not induce alterations in total ROS in Kupffer cells (data not shown). Two known antioxidants, allopurinol (1–5 μM) and quercetin (10–40 μM), were dose dependently able to restore fructose-induced elevation of total ROS level and TXNIP expression in primary rat hepatocytes and RHPCs (Supplementary Fig. S2A), showing a positive correlation between total ROS and TXNIP under high fructose induction (linear regression analysis, Pearson's r=0.7183 in primary rat hepatocytes and r=0.7356 in RHPCs, Supplementary Fig. S2A). In addition, allopurinol (2 μM) and quercetin (20 μM) also reduced total ROS levels in 5 mM fructose-exposed RHPCs, HLO2, and HepG2 cells (Fig 6A).
FIG. 6.


Fructose induces TXNIP overexpression through ROS production in hepatocytes. (A) Primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were cultured with or without 5 mM fructose for 48 h and co-incubated with 2 μM allopurinol or 20 μM quercetin for another 24 h. Total ROS, H2O2, O2•−, MDA, iNOS, XO, SOD, and TAC level or activity were analyzed (n=8/group). (B) TXNIP protein levels were determined in these hepatocytes by Western blot (n=8/group). (C) Primary rat hepatocytes and RHPCs were cultured with or without 5 mM fructose for 48 h and co-incubated with 100 μM tempol, 50 U/ml catalase, 100 μM vitamin E, 2 μM allopurinol, 20 μM quercetin, 100 μM apocynin, or 100 μM aminoguanidine for another 24 h. Total ROS, MMP, and NOX level or activity were analyzed (n=8/group). (D) Primary rat hepatocytes and RHPCs were transfected with TXNIP siRNA for 48 h, co-incubated with or without 5 mM fructose for 48 h, and treated with 2 μM allopurinol or 20 μM quercetin for another 24 h. Total ROS levels were measured (n=8/group). (E) Primary rat hepatocytes and RHPCs were transfected with NLRP3 siRNA for 48 h, co-incubated with or without 5 mM fructose for 48 h, and treated with 2 μM allopurinol or 20 μM quercetin for another 24 h. Total ROS levels were measured (n=8/group). (F) Primary rat hepatocytes and RHPCs were transfected with NLRP3 siRNA for 48 h, co-incubated with or without 5 mM fructose for another 48 h, and treated with 2 μM allopurinol or 20 μM quercetin for another 24 h. TXNIP protein levels were measured by Western blot (n=8/group). (G) Primary rat hepatocytes and RHPCs were co-incubated with or without 5 mM fructose and the caspase-1 specific inhibitor Ac-YVAD-CMK for 48 h and treated with 2 μM allopurinol or 20 μM quercetin for another 24 h. TXNIP protein levels were determined by Western blot (n=8/group). (H) Primary rat hepatocytes and RHPCs were cultured with or without 5 mM fructose for 48 h and treated with 2 μM allopurinol or 20 μM quercetin for another 24 h. Total and nuclear Nrf2 protein levels were assayed by Western blot (n=8/group). (I) Rats were given drinking water containing 10% fructose for 4 weeks and then given 5 mg/kg allopurinol or 50 or 100 mg/kg quercetin for another 4 weeks. Total and nuclear Nrf2 protein levels were determined in liver biopsies by Western blot (n=8/group). Results are expressed as mean±SEM. #p<0.05, ##p<0.01, ###p<0.001 versus control group; *p<0.05, **p<0.01, ***p<0.001 versus fructose vehicle group. iNOS, inducible nitric oxide synthase; MDA, malondialdehyde; MMP, mitochondrial membrane potential; Nrf2, nuclear factor (erythroid-derived-2)-like 2; NOX, NADPH oxidase; ROS, reactive oxygen species; SOD, superoxide dismutase; TAC, total antioxidant capacity; XO, xanthine oxidase.
In addition, 5 mM fructose directly increased H2O2, O2•−, and MDA levels in primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 6A) but not in Kupffer cells (data not shown), which was blocked by allopurinol (2 μM) and quercetin (20 μM) (Fig. 6A). Allopurinol and quercetin completely inhibited fructose-induced TXNIP overexpression in these hepatocytes (Fig. 6B).
Fructose metabolism is associated with increased ROS production and may therefore lead to oxidative stress (20). Tempol (an ROS scavenger, 100 μM), catalase (an enzyme that dismutates hydrogen peroxide, 50 U/ml), and vitamin E (a lipophilic antioxidant, 100 μM) also significantly ameliorated fructose-induced total ROS overproduction in primary rat hepatocytes and RHPCs (Fig. 6C).
It is known that mitochondria are the major source of ROS production in most cells, including hepatocytes. Consequently, the involvement of mitochondria in fructose-induced hepatocellular ROS production was investigated. First, we observed that 5 mM fructose induced a reduction in mitochondrial membrane potential (MMP) and an increase in NADPH oxidase (NOX) activity in primary rat hepatocytes and RHPCs (Fig. 6C), which were restored by allopurinol (2 μM) and quercetin (20 μM) (Fig. 6C). In addition, the NOX inhibitor apocynin (100 μM) decreased total ROS levels in fructose-exposed primary rat hepatocytes and RHPCs (Fig. 6C). These data suggest an involvement of mitochondria in fructose-stimulated ROS production in hepatocytes.
iNOS occupies a key role in the onset of fructose-induced hepatic steatosis (48). Allopurinol (2 μM) and quercetin (20 μM) decreased iNOS activity in fructose-exposed primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 6A). The iNOS inhibitor aminoguanidine (100 μM, Fig. 6C) was able to reduce fructose-induced ROS production in primary rat hepatocytes and RHPCs. XO is also an important oxidoreductase that contributes to intracellular ROS production. The XO inhibitors allopurinol (2 μM) and quercetin (20 μM) were found to block fructose-induced XO hyperactivity in primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 6A). A reduction in TAC was also observed in these cells, which was in line with the decreased SOD activity. The reduction in TAC was restored by allopurinol (2 μM) and quercetin (20 μM) (Fig. 6A). Therefore, a dysfunctional intracellular antioxidant mechanism may lie at the basis of fructose-induced ROS production.
It should be noted that 5 mM fructose, 2 μM allopurinol, and 20 μM quercetin did not induce apoptosis (Supplementary Fig. S2B), and caspase-3 and caspase-9 activity (data not shown) in primary rat hepatocytes and RHPCs, indicating that apoptosis does not play an instrumental role in fructose-induced ROS production and inflammation.
To address whether TXNIP overexpression was a downstream or an upstream event in fructose-induced ROS production, we first detected the effect of TXNIP gene silencing on ROS production in fructose-exposed primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells. As shown in Figure 6D, TXNIP depletion did not affect 5 mM fructose-induced ROS production in these hepatocytes. In contrast, co-incubation with allopurinol (2 μM) and quercetin (20 μM) reduced total ROS levels in TXNIP-silenced primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 6D). NLRP3 depletion also did not alter fructose-induced ROS production in primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells (Fig. 6E). Allopurinol (2 μM) and quercetin (20 μM) were found to reduce total ROS levels in these NLRP3-silenced cells (Fig. 6E). Furthermore, allopurinol (2 μM) and quercetin (20 μM) blocked fructose-induced TXNIP overexpression in NLRP3-silenced primary rat hepatocytes and RHPCs (Fig. 6F) and in Ac-YVAD-CMK-incubated primary rat hepatocytes and RHPCs (Fig. 6G). Therefore, TXNIP overexpression is a response to fructose-induced ROS production, which probably emanates from hepatic fructose metabolism.
The transcription factor Nrf2 is a critical regulator of cell stress responses and suppresses the basal expression of TXNIP (11). Overproduction of ROS inhibits Nrf2 expression (41). To address these events in the context of high fructose exposure, we examined Nrf2 protein levels in fructose-fed rat livers and fructose-exposed hepatocytes. Activation of Nrf2-mediated gene transcription depends on redox-mediated dissociation from its binding partner KEAP1 (Kelch-like ECH-associated protein 1) in the cytosol and subsequent nuclear translocation. In this study, 5 mM fructose decreased protein levels of nuclear Nrf2 but not total Nrf2 in primary rat hepatocytes and RHPCs (Fig. 6H), which was attenuated by allopurinol (2 μM) and quercetin (20 μM) (Fig. 6H). Similar results were observed in livers of fructose-fed rats (Fig. 6I). These data suggest that fructose may inhibit Nrf2 transcriptional activity in hepatocytes. Fructose-induced overproduction of ROS may increase TXNIP expression by suppressing transcriptional Nrf2 activity in the liver.
Inhibition of ROS production diminishes the detrimental effects of fructose in hepatocytes both in vitro and in vivo
Quenching ROS blocks TXNIP induction, suggesting that antioxidants may be effective therapeutics for fructose-induced NAFLD development. Indeed, co-incubation of primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells with 2 μM allopurinol or 20 μM quercetin effectively inhibited fructose-induced NLRP3 inflammasome activation (Fig. 7A), IL-1β and IL-18 secretion (Fig. 7B), the JAK2/STAT3 pathway (Fig. 7C), and lipid metabolism-related gene expression dysregulation (Fig. 7D).
FIG. 7.


Antioxidants allopurinol and quercetin block NLRP3 inflammasome activation, IL-1β and IL-18 secretion, the JAK2/STAT3 pathway, and deregulated lipid metabolism in fructose-exposed cultured hepatocytes. Primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells were exposed to 5 mM fructose or vehicle and co-incubated with 2 μM allopurinol or 20 μM quercetin for another 24 h. (A) NLRP3, ASC, and caspase-1 protein levels were analyzed by Western blot (n=8/group). (B) IL-1β and IL-18 levels were determined in cell lysate (top) and supernatant (bottom) (n=8/group). (C) p-JAK2, JAK2, p-STAT3, STAT3, and SOCS3 protein levels were assayed by Western blot (n=8/group). (D) PPAR-α, SREBP1, and SCD1 protein levels were determined by Western blot (n=8/group). Results are expressed as mean±SEM. #p<0.05, ##p<0.01, ###p<0.001 versus control group; *p<0.05, **p<0.01, ***p<0.001 versus fructose vehicle group.
In line with the in vitro findings, administration of allopurinol (5 mg/kg) and quercetin (50 or 100 mg/kg) to fructose-fed rats significantly reduced total ROS, H2O2, O2•−, MDA, XO, and iNOS level and activity; increased SOD activity (Table 1), TAC level (Table 1), and nuclear Nrf2 protein levels (Fig. 6I); downregulated TXNIP protein levels (Fig. 8A); inhibited NLRP3 inflammasome activation (Fig. 8B); reduced IL-1β and IL-18 levels (Fig. 8C); and regulated the JAK2-STAT3 pathway (Fig. 8D) and lipid metabolism-related gene expression (Fig. 8E) in livers. These effects collectively led to a reduction in the extent of hepatic inflammation (Supplementary Fig. S1A) and lipid accumulation (Supplementary Fig. S1B), which was accompanied by a reduction in TG and TC levels (Supplementary Table S1). In this study, allopurinol and quercetin produced a slight but nonsignificant decrease in the body weight of fructose-fed rats, without change in food intake and total caloric intake (Supplementary Table S1).
FIG. 8.

Allopurinol and quercetin reduce TXNIP overexpression and NLRP3 inflammasome activation and ameliorate inflammation and the extent of lipid metabolism deregulation in livers of fructose-fed rats. Rats were given drinking water containing 10% fructose for 4 weeks and subsequently 5 mg/kg allopurinol or 50 or 100 mg/kg quercetin for another 4 weeks. (A) Hepatic TXNIP protein levels were determined by Western blot (n=8/group). (B) Hepatic NLRP3, ASC, and caspase-1 protein levels were measured by Western blot (n=8/group). (C) Hepatic IL-1β and IL-18 levels were determined by ELISA (n=8/group). (D) Western blot analysis of hepatic p-JAK2, JAK2, p-STAT3, STAT3, and SOCS3 protein levels (n=8/group). (E) Hepatic PPAR-α, SREBP1, and SCD1 protein levels were determined by Western blot (n=8/group). Results are expressed as mean±SEM. #p<0.05, ##p<0.01, ###p<0.001 versus control group; *p<0.05, **p<0.01, ***p<0.001 versus fructose vehicle group.
Oxidative stress- and inflammatory pathways induced by a high-fat diet differ from those induced by a high fructose diet in rats with NAFLD
High-fat intake is also considered a key dietary factor in the development of NAFLD (64). Whether similar mechanisms as described earlier play a role in high-fat diet-induced NAFLD has not yet been addressed. As expected, high-fat diet feeding for 8 weeks increased rat body weight, energy intake, hepatic TG and TC levels (Supplementary Table S2), and hepatic lipid accumulation (Supplementary Fig. S3A). Immunohistochemistry of liver sections revealed increased hepatic inflammation (Supplementary Fig. S3B) but unchanged NLRP3 expression in hepatocytes and Kupffer cells (Fig. 9A) compared with the control group, with elevation of hepatic IL-1β and IL-18 levels (Fig. 9B). In addition, high-fat diet feeding increased hepatic ASC and caspase-1 protein levels, but did not affect NLRP3 protein levels (Fig. 9C).
FIG. 9.

Allopurinol and quercetin reduce TXNIP overexpression and NLRP3 inflammasome activation and ameliorate inflammation and the extent of lipid metabolism deregulation in livers of high-fat diet-fed rats. Rats were fed a high-fat diet for 4 weeks and subsequently given 5 mg/kg allopurinol or 50 or 100 mg/kg quercetin for another 4 weeks. (A) Representative images of liver sections immunohistochemically stained for NLRP3 (red) and CK18 (hepatocytes, green) or CD163 (Kupffer cells, green). Scale bar=100 μm. (B) Hepatic IL-1β and IL-18 levels were determined by ELISA (n=8/group). (C) Western blot analysis of NLRP3, ASC, caspase-1, TXNIP, and total and nuclear Nrf2 protein levels in rat livers (n=8/group). (D) Representative images of liver sections immunohistochemically stained for TXNIP (red) and CK18 (hepatocytes, green) or CD163 (Kupffer cells, green). Scale bar=100 μm. Results are expressed as mean±SEM. #p<0.05, ##p<0.01 versus control group; *p<0.05, **p<0.01 versus high-fat diet vehicle group. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Moreover, high-fat diet-fed rats exhibited exacerbated ROS production and oxidative stress, as evidenced by the elevated total ROS, H2O2, O2•−, MDA, iNOS, and XO levels and activity (Table 2). Consistently, increased TXNIP protein levels in livers (Fig. 9C) and primarily in hepatocytes and Kupffer cells (Fig. 9D) were observed in this animal model. Of note, total and nuclear Nrf2 protein levels were significantly increased in livers of high-fat diet-fed rats compared with the control group (Fig. 9C). With the exception of energy intake and Nrf2, these parameters were significantly or slightly ameliorated after treatment with allopurinol (5 mg/kg) and quercetin (50 or 100 mg/kg) (Fig. 9, Table 2, and Supplementary Fig. S3, Supplementary Table S2). Allopurinol and quercetin did not affect nuclear Nrf2 protein levels but enhanced total Nrf2 protein levels in livers of high-fat diet-fed rats (Fig. 9C). These data indicate that intrahepatic oxidative stress- and inflammatory pathways induced by a high fructose diet are in some respects distinct from those induced by a high-fat diet in NAFLD-affected livers.
Table 2.
Total ROS, H2O2, O2•−, MDA, iNOS, and XO in High-Fat Diet-Fed Rats
| Group | Dosage (mg/kg) | Total ROS (intensity [a.u.]/μg protein) | H2O2 (μmol/g protein) | O2•− (intensity [a.u.]/mg protein) | MDA (nmol/mg protein) | iNOS (U/mg protein) | XO (U/mg protein) |
|---|---|---|---|---|---|---|---|
| Control | 20.17±1.08 | 5.27±0.59 | 19.33±1.40 | 3.92±0.12 | 0.18±0.01 | 0.62±0.09 | |
| High fat diet | 33.66±3.15a | 12.91±1.92b | 35.04±3.92b | 4.55±0.08a | 0.49±0.04b | 5.98±0.64c | |
| +Allopurinol | 5 | 20.42±1.38d | 5.61±0.51d | 19.35±1.89e | 3.74±0.13e | 0.20±0.03e | 1.05±0.19f |
| +Quercetin | 50 | 19.89±1.82d | 5.82±0.82d | 25.69±0.97d | 3.91±0.16d | 0.19±0.02d | 4.67±0.37 |
| 100 | 20.29±2.13d | 5.59±0.53d | 24.18±1.34d | 3.86±0.09d | 0.19±0.02e | 2.54±0.28d |
All data are expressed as mean±SEM (n=8/group).
p<0.05 versus control group.
p<0.01 versus control group.
p<0.001 versus control group.
p<0.05 versus high-fat diet vehicle group.
p<0.01 versus high-fat diet vehicle group.
p<0.001 versus high-fat diet vehicle group.
Discussion
NAFLD encompasses a spectrum of chronic liver diseases characterized by hepatic inflammation and lipid accumulation. Increased fructose consumption is implicated in the pathogenesis and progression of NAFLD (7, 22, 24, 33, 45, 47–49, 59), but the molecular mechanisms involved have not been completely elucidated. Generally, Kupffer cells are the primary source of hepatic inflammatory cytokines such as IL-1β. Enhanced Kupffer cell activity driven by oxidative stress is observed in livers of NAFLD patients with steatohepatitis (25). The NLRP3 inflammasome constitutes the major immune sensor for cellular stress signals such as ROS, mediating secretion of its effectors IL-1β and IL-18 (19, 55). Activation of NLRP3 inflammasome in Kupffer cells through Toll-like receptor 2/palmitic acid is an important factor in the pathogenesis of NASH in CDAA diet-fed mice (30). TXNIP links oxidative stress to NLRP3 inflammasome activation (62). Similarly, our study showed that feeding rats a high fructose diet for 8 weeks increased NLRP3 and TXNIP levels in Kupffer cells and aggravated the extent of oxidative stress in livers. However, in vitro fructose exposure (5 mM) did not induce NLRP3 inflammasome activation, ROS overproduction, and TXNIP upregulation in Kupffer cells isolated from rat livers. In RAW 264.7 macrophages, which are commonly used as a model for Kupffer cells, fructose stimulation also did not induce iNOS mRNA expression, nitrite formation, and inflammatory cytokines (48).
A recent study showed that alterations in gut microbiota can cause animals that have a genetic NLRP3 inflammasome deficiency in innate immune cells to develop NAFLD (12). In our preliminary study, no significant or very weak expression of fructose transport proteins GLUT2, 5, and 8 was detected in cultured Kupffer cells isolated from rat livers (data not shown). Consequently, hepatic inflammation observed in fructose-fed rats may have resulted from increased intestinal translocation of endotoxin and subsequent activation of NLRP3 in Kupffer cells rather than from direct effects of fructose. In fact, we found that enhanced protein levels of NLRP3 were observed in liver parenchymal cells of fructose-fed rats. Consistently, 5 mM fructose directly activated NLRP3 inflammasome and its effectors IL-1β and IL-18 through the induction of oxidative stress using cultured hepatic parenchymal cells such as primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells.
More importantly, the ROS-TXNIP pathway was demonstrated to be involved in orchestrating fructose-induced hepatic inflammation and lipid accumulation, as evidenced by the amelioration of the inflammatory signaling and oxidative stress by antioxidants allopurinol and quercetin. The involvement of this pathway was further demonstrated by the TXNIP gene silencing assay, which diminished NLRP3 inflammasome activation, pro-inflammatory cytokine production, and lipid metabolism-related gene deregulation in cultured primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells. In sum, we suggest that fructose induces NAFLD by directly causing the activation of inflammatory signaling and lipid accumulation in hepatic parenchymal cells by triggering oxidative stress in these cells.
Oxidative stress is involved in the development of NAFLD. Fructose promotes ROS formation and oxidative stress (26). This study further confirmed the direct induction of oxidative stress by fructose in rat livers and primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells, as evidenced by increased total ROS, H2O2, O2•−, MDA, XO, and iNOS and decreased SOD and TAC, which were repressed by antioxidants allopurinol and quercetin. Moreover, fructose did not induce apoptosis but decreased MMP and antioxidant capacity in primary rat hepatocytes and RHPCs, indicating that fructose may inflict direct injury to hepatocyte mitochondria and antioxidant function by causing excessive ROS generation. This hypothesis is supported by the experiments with tempol, catalase, vitamin E, apocynin, and aminoguanidine, which blocked fructose-induced elevation of total ROS levels in hepatocytes.
TXNIP interconnects cellular nutritional and oxidative states with metabolic responses. Liver cells that overexpress TXNIP are primed for oxidative injury (35). This study found that TXNIP upregulation correlated with ROS overproduction in fructose-fed rat livers and fructose-exposed primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells. Of note, the suppression of total ROS production by allopurinol and quercetin completely abrogated TXNIP induction in hepatocytes in response to fructose exposure. These effects of antioxidants were confirmed in livers of fructose-fed rats. More importantly, allopurinol and quercetin were found to reduce total ROS levels and TXNIP protein levels in fructose-exposed primary rat hepatocytes and RHPCs, altogether confirming that the fructose-induced ROS-TXNIP signaling axis is under redox control in hepatocytes. Furthermore, TXNIP silencing failed to alter ROS production in primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells exposed to fructose. Therefore, the inducible TXNIP overexpression appears to be a downstream event in fructose-induced hepatocellular oxidative stress.
Nrf2 is an important regulator of cellular stress responses and mitochondrial function (27, 41). This transcription factor induces TXNIP expression. Our study revealed that nuclear levels of Nrf2 protein were downregulated in fructose-fed rat livers and fructose-exposed primary rat hepatocytes and RHPCs—an effect that was reversed by allopurinol and quercetin, demonstrating that fructose-induced ROS overproduction may enhance TXNIP expression, at least partly, by suppressing Nrf2 transcriptional activity in hepatocytes. Accordingly, these results support the hypothesis that nuclear Nrf2 is a key regulator of the ROS-TXNIP signaling axis in hepatocytes at high intracellular fructose concentrations. Inasmuch as our study did not explicitly focus on the role of Nrf2, its involvement in fructose-induced redox modulation requires additional experimental scrutiny.
Of note, in cultured rat hepatocytes, 20 mM fructose can effectively protect against cocaine or nitrofurantoin-induced oxidative injury, independent of the glycolytic metabolism, by suppressing the iron-catalyzed formation of ROS (57), and inhibit reoxygenation-induced apoptosis by decreasing ROS via stabilization of the glutathione pool (8). These observations indicate that a high level of fructose (20 mM) reduces oxidative stress in the in vitro models of hepatocytes. In this study, we indeed found increased ROS overproduction and oxidative stress in 10% fructose-fed rat livers and 5 mM fructose-exposed cultured hepatocytes, partly mediated by inhibition of Nrf2 transcriptional activity and the ROS-TXNIP signaling axis. Recently, more evidence demonstrates the importance of fructose in steatotic hepatocytes (43). These contradictory findings may be related to fructose concentration, its metabolism, and use of different experimental models. Further studies are needed to address these details.
Evidence from clinical studies has suggested that NLRP3 inflammasome activation in myeloid cells or macrophages that have infiltrated visceral adipose tissue is associated with many metabolic disorders (7, 21). Specifically, NLRP3 inflammasome activation and IL-18-mediated dysbiosis aggravate the progression of obesity and NAFLD/NASH (12). Moreover, hepatic TG levels are reduced in mice deficient in NLRP3 inflammasome components (50), and high liver levels of NLRP3 inflammasome, IL-1β, and IL-18 are observed in morbidly obese patients (31). SREBP1 hyperactivity and NLPR3 inflammasome activation in lesions are detected in atherosclerotic pig aortas with vascular inflammation (23). Studies by us and others showed that fructose induced NLRP3-dependent renal inflammation and steatosis-associated metabolic syndrome (1, 14). In this study, inhibition of NLRP3 expression and caspase-1 activity reverted fructose-induced transcriptomic dysregulation of PPAR-α, SREBP1, and SCD1 in hepatocytes, suggesting that NLRP3 inflammasome activation plays a role in fructose-induced aberrations in hepatocellular lipid metabolism.
In fact, TXNIP mediates hepatic lipogenesis. TBP-2 knockout mice develop fatty acid β-oxidation disorder. Lipopolysaccharide-challenged TBP-2 mice exhibit elevated serum TG and TC levels, liver fat deposition, and hepatorenal injury (37). TXNIP is elevated in livers of human NAFLD patients (39). In this study, TXNIP upregulation caused hepatocellular secretion of IL-1β and IL-18 as a result of NLRP3 inflammasome activation, initiating a hepatocyte-driven sterile immune response on fructose exposure, and possibly driving NASH progression. More importantly, silencing TXNIP almost completely blocked fructose-induced deregulation of lipid metabolism-related genes in primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells. Therefore, TXNIP overexpression-mediated inflammation is instrumental in lipid metabolism disorder, which is possibly a TXNIP-dependent process in the case of fructose-induced NAFLD/NASH. In support of this, TXNIP was overexpressed in HLO2 and HepG2 cells after in-frame insertion into a pCDH-CMV-MCS vector and subsequent transfection, which resulted in NLRP3 inflammasome activation, pro-inflammatory cytokine secretion, and lipid metabolism disorder (data not shown). Thus, TXNIP has an essential role in activating NLRP3 inflammasome that, in turn, alters lipid metabolism in hepatocytes exposed to high fructose levels. These results suggest that excess fructose intake impairs the hepatic anti-oxidant system, contributing to the second hit of hepatic injury via ROS-TXNIP pathway-mediated hepatocyte NLRP3 inflammasome activation, and the switch from NAFLD to NASH in subjects with hepatic steatosis. Moreover, this study may help find a new approach in treating NAFLD/NASH with antioxidants. However, further studies are warranted to investigate the underlying mechanisms.
A high-fat diet is an important risk factor in the development of NAFLD (64). In this study, high-fat diet-induced NAFLD in rats was accompanied by increased ROS, oxidative stress, and TXNIP overexpression, which were restored by allopurinol and quercetin, underscoring the link between ROS production and TXNIP expression. In contrast to the fructose-fed animals, total and nuclear Nrf2 protein levels were significantly elevated in livers of high-fat diet-fed rats, which were consistent with recent reports (64). Allopurinol enhanced total but not nuclear Nrf2 levels in this animal model. Ablation of NLRP3 partially deters high-fat diet-induced caspase-1 auto-activation in mouse livers (58). In this study, high-fat diet feeding increased hepatic ASC and caspase-1 expression as well as IL-1β and IL-18 levels, which were restored by allopurinol and quercetin. Of note, there was no significant change in hepatic NLRP3 expression in high-fat diet-fed rats.
Hepatocytes utilize different metabolic routes for fat and fructose, whereby the resulting metabolic products/factors mediate similar and/or different oxidative stress and inflammatory pathways. For example, ROS at low to modest levels can increase Nrf2 expression, whereas its overproduction inhibits Nrf2 expression (41), which may explain the inconsistent molecular mechanisms underlying the pathogenesis of high-fat or fructose-associated NAFLD.
In the final analysis, this study revealed a novel mechanism underlying the pathogenesis of fructose-induced NAFLD. Induction of ROS by fructose in hepatocytes decreased nuclear Nrf2 and increased TXNIP expression, which, in turn, triggered NLRP3 inflammasome activation and consequent IL-1β and IL-18 secretion as a part of a sterile inflammatory response. Meanwhile, the inflammatory response triggered changes in lipid metabolism-related gene expression to promote lipid accumulation (Fig. 10). Induction of TXNIP overexpression through an as yet unidentified oxidative stress signaling pathway in hepatocytes may comprise a switch-on mechanism for NAFLD pathogenesis at high fructose levels. Moreover, the antioxidants allopurinol and quercetin suppressed hepatocellular ROS production, as a result of which nuclear Nrf2 level was increased and the extent of TXNIP expression and NLRP3 inflammasome activation was reduced. These results pave the way for the development and use of therapeutics that interfere with the pathogenesis of fructose-induced NAFLD. In that respect, the high fructose diet in rats has made it possible to investigate hepatopathological changes that also occur in humans with NAFLD (46). The potential clinical indications for allopurinol include metabolic syndrome, NAFLD, and NASH (51). Quercetin can prevent human obesity-related diseases (2), which exemplifies its potentially beneficial health effects (44). Future studies should therefore focus on further elucidating the ramifications of the fructose-induced ROS-TXNIP signaling axis on NAFLD/NASH development and on determining to what extent the findings regarding the effects of high fructose consumption and the efficacy of antioxidants translate to the human situation.
FIG. 10.

Induction of TXNIP overexpression through ROS production underlies fructose-induced hepatic inflammation and lipid accumulation. Fructose increased TXNIP expression through ROS production and nuclear Nrf2 suppression in hepatocytes, which, in turn, triggered NLRP3 inflammasome activation and secretion of IL-1β and IL-18. The consequent inflammatory signaling caused deregulation of lipid metabolism-related gene expression and lipid accumulation. These fructose-induced hepatic disturbances were blocked by the treatment of cells and livers with the antioxidants allopurinol and quercetin, which suppressed ROS levels.
Materials and Methods
Detailed Supplementary Materials and Methods section are provided in the Supplementary Data.
Animals, high fructose diet, and treatments
All animal experiments were approved by the animal welfare and ethics committee of Nanjing University, and all animals were treated in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male Sprague–Dawley (SD) rats weighing between 180 and 200 g were purchased from the Laboratory Animal Center (Zhejiang Province, P.R. China) and housed under standard laboratory conditions with a 12 h light-dark cycle and a constant temperature of 22°C–24°C.
High fructose-induced NAFLD in rats was achieved as previously described (22). Rats were given either tap water (control group, n=8) or water containing 10% fructose (∼60 ml/day) ad libitum (fructose-fed rats, n=32) with standard chow for 8 weeks. Fresh drinking water was provided every 2 days. After 4 weeks, the animals were randomized into 4 sub-groups (n=8/group) to receive water (vehicle), allopurinol (5 mg/kg; Sigma-Aldrich, St. Louis, MO), or quercetin (50 or 100 mg/kg) (98%; Sigma-Aldrich) while continuing the fructose supplementation during 4 weeks. All antioxidants/vehicle were administered per gavage once daily between 2:00 p.m. and 3:00 p.m.
High-fat diet-induced NAFLD in rats was achieved as previously described (6). Rats were given regular standard chow (control group, n=8) or a high-fat diet (D12492, with 60% of the calories from fat; Research Diets, New Brunswick, NJ) (high-fat diet-fed rats, n=32) with fresh drinking water ad libitum for 8 weeks. After 4 weeks, the animals were randomized into 4 sub-groups (n=8) and received antioxidants/vehicle as described earlier while continuing their diet regimen.
Body weight and daily food and fluid intake were measured weekly during the 8 weeks of fructose- or high-fat diet feeding.
Animals were sacrificed by decapitation between 9:00 a.m. and 10:00 a.m. after a 16-h fast. Blood samples were collected and centrifuged to isolate serum for biochemical assays. Liver tissue was dissected and quickly placed on ice. Part of the biopsies were fixed immediately in 10% buffered formalin for histological processing, while the remainder of the biopsies was stored in liquid nitrogen for biochemical, molecular, and histological analyses.
Cell culture, siRNA transfection, and treatment
Primary hepatocytes and Kupffer cells were isolated from liver tissue of male SD rats (200–220 g) as previously described (3, 4). Briefly, after digestion with a medium containing 0.05% collagenase type IV and 1 mM CaCl2, parenchymal cells were separated from the sinusoidal cell fraction by sedimentation at 50 g for 25 s at 4°C. For Kupffer cell isolation, the supernatant was centrifuged at 600 g for 4 min at 4°C and the resulting pellet containing the nonparenchymal cells was collected. The method of selective adherence was used to further purify isolated Kupffer cells after the nonparenchymal cell separation by gradient centrifugation. Nonparenchymal cells were seeded in a six-well plate at a density of 1×107 per well and cultured in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY) under standard culture condition (95% O2 and 5% CO2, 37°C, humidified atmosphere) for 2 h. Nonadherent cells were then removed by gently washing with phosphate-buffered saline (PBS). The adherent cells were Kupffer cells. The purity of the Kupffer cell fraction was >87% determined by CD163 immunofluorescence staining and scoring. Primary rat hepatocytes and Kupffer cells were cultured in RPMI 1640 (Invitrogen) supplemented with 10% FBS (Gibco) under standard culture conditions.
RHPCs (Pricells Company, Hubei, P.R. China), HLO2 cells, and HepG2 cells (maintained in our laboratory) were cultured in DMEM (Invitrogen) supplemented with 10% FBS under standard culture conditions.
Cells were seeded into six-well plates (primary rat hepatocytes, RHPCs, HLO2, and HepG2 at 1×106 cells/well; Kupffer cells at 5×105 cells/well) 24 h before experiments. For fructose stimulation, the medium was replaced with fresh medium containing no fructose (control) or 5 mM fructose (Sigma-Aldrich) and cells were incubated for 72 h. The medium was replaced every 24 h. Forty-eight hours after fructose supplementation, allopurinol (2 μM) or quercetin (20 μM) was added and the cells were incubated for an additional 24 h before harvesting and collection. The in vitro experimental setup is schematically outlined in Figure 11.
FIG. 11.

Schematic diagram of the cell culture experiments and treatment regimens.
To evaluate fructose-induced ROS production, the antioxidants tempol (100 μM) and vitamin E (100 μM), the antioxidant enzyme catalase (50 U/ml), the dicarbonyl scavenger aminoguanidine (100 μM), and the NOX inhibitor apocynin (100 μM) were added to primary rat hepatocyte and RHPC cultures (Fig. 11).
Allopurinol, quercetin, caspase-1-specific inhibitor Ac-YVAD-CMK (Taize, Beijing, P.R. China), tempol, vitamin E, apocynin, and aminoguanidine were dissolved in dimethyl sulfoxide (DMSO) while catalase was dissolved in PBS, respectively. The final DMSO concentration was <0.1% in all cases.
In primary rat hepatocytes and RHPCs, caspase-1 was inhibited with 20 μM of Ac-YVAD-CMK. Ac-YVAD-CMK was added to medium either lacking or containing 5 mM of fructose. After 48 h, allopurinol (2 μM) or quercetin (20 μM) was added and cells were incubated for an additional 24 h.
To knockdown NLRP3 or TXNIP, primary rat hepatocytes and RHPCs were transfected with 40 pM of rat NLRP3- or TXNIP-specific siRNA (rat siRNA for NLRP3: 5′-CCUGUCUUUGCCGTAGAUUACCGUAAG-3′, anti-sense; rat siRNA for TXNIP: 5′-AUCCCUUCUAUAUAGGCACCUGUGUC-3′, anti-sense; Invitrogen) or respective nontargeting control for Stealth RNAi siRNA duplexes (siControl, cat# 12935-300; Invitrogen) using RNAiMAX (4 μl; Invitrogen) according to the manufacturer's instructions. The same transfection method was used to knockdown TXNIP in HLO2 and HepG2 cells (human siRNA for TXNIP: 5′-GAGGUGGUCUUUAACGACCCUGAAA-3′, anti-sense; Invitrogen). The transfections were performed 48 h before the start of experiments. Silencing of NLRP3 and TXNIP was confirmed by RT-PCR as described in the Reverse Transcription–Polymerase Chain Reaction section. After transfection, the cells were incubated in 5 mM fructose-containing medium for an additional 48 h and subsequently treated with allopurinol (2 μM) or quercetin (20 μM) or respective vehicle controls for 24 h.
For all in vitro experiments, the dosage and treatment period were determined on the basis of preliminary experiments and other reports for every reagent. Moreover, cells, cell lysate, culture supernatant, total RNA, and total and nuclear protein samples were collected in every experimental group. Experiments were repeated at least four times.
Determination of TG and TC
Lipids were extracted from rat liver biopsies by liquid phase extraction using chloroform/methanol (2:1 v/v) as previously described (22). TG and TC levels in the extracts were determined with standard diagnostic kits (Jiancheng Biotech, Nanjing, P.R. China).
Histology
Formalin-fixed, paraffin-embedded liver sections (7 μm thick) were stained with hematoxylin and eosin (Sigma-Aldrich) according to standard protocol. Histological changes, especially the histopathological inflammation, were quantified by an experienced hepatopathologist.
Oil-red O staining
Cryostat sections of liver tissue were stained with Oil-red O solution (Sigma-Aldrich). After washing with 60% isopropyl alcohol, the sections were counterstained with hematoxylin.
Immunohistochemical staining
IHC staining of NLRP3 and TXNIP was performed on rat liver tissue sections as previously described (10). Briefly, the sections were incubated with primary anti-NLRP3 (dilution 1:100 cat# nbp2-12446; Novus Biologicals, Littleton, CO) or anti-TXNIP (dilution 1:100, cat# ab86983; Abcam, Cambridge, United Kingdom) antibodies or negative control (PBS and rabbit IgG Supplementary Fig. S4) at 4°C overnight. Rat hepatocytes and Kupffer cells were stained with anti-CK18 (cat# ab82254, dilution 1:100; Abcam) and anti-CD163 (cat# ab111250, dilution 1:100; Abcam), respectively. After threefold washing with PBS, the sections were incubated with Alexa-594 anti-rabbit secondary antibodies (dilution 1:1000) and Alexa-488 anti-mouse secondary antibodies (dilution 1:1000; Invitrogen) at room temperature for 2 h. The sections were stained with 2,6-diisopropylaniline (DAPI) for 5 min. Five random fields of view from each section were imaged with a confocal laser scanning microscope (FLUOVIEW FV1000; Olympus, Tokyo, Japan). All settings were kept constant during imaging. Optical density was analyzed with ImageJ (National Institutes of Health, Bethesda, MD).
Determination of IL-1β and IL-18 levels
Rat liver tissue was homogenized on ice in 0.9% sodium chloride solution (1:10 w/v) and centrifuged (10,000 g) for 15 min at 4°C. The cultured cells were lysed on ice in 0.9% sodium chloride solution and then centrifuged (12,000 g) for 10 min at 4°C. IL-1β and IL-18 levels in the supernatant of homogenized liver tissue, cell lysates, and culture supernatant of primary rat hepatocytes, Kupffer cells, RHPCs, HLO2, and HepG2 cells were determined by ELISA (commercial kits from IBL International, Hamburg, Germany for rats and R&D Systems, Minneapolis, MN for cells).
Determination of ROS
Different ROS were determined as specified in Supplementary Table S3 (some assays, as addressed next). Liver tissue was homogenized in PBS and centrifuged (10,000 g) for 15 min at 4°C. All cultured cells were detached from the wells by 0.25% trypsin digestion. The total ROS, MMP, and superoxide anion assays were performed with cell-containing aliquots (transferred to 96-well plate at 1×104 cells/well), while the other assays were performed on cell lysates after cell lysis using a buffer without enzyme inhibitors (P0013J; Beyotime Biotech, Jiangsu, P.R. China) or the lysis buffer provided in assay kits.
Total ROS were quantified with the fluorogenic probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA). On DCFH2-DA entry into the cell, the acetate groups are hydrolyzed by intracellular esterases to yield the nonfluorescent DCFH2. DCFH2 subsequently reacts with ROS to form the highly fluorescent dichlorofluorescein (DCF), which is largely retained in the cell. Trypsinized cells were incubated with 5 μM DCFH2-DA at 37°C for 20 min. DCF fluorescence was measured at λex=488 nm and λem=525 nm with a microplate reader (TECAN A-5082; TECAN Group, Salzburg, Austria).
MMP was determined by using JC-1, which preferentially accumulates in intact mitochondria. Trypsinized cells were transferred to a 96-well plate at 1×104 cells per well. JC-1 was added at a final concentration of 10 μM, and the cells were incubated at 37°C for 10 min during gentle shaking. Next, the fluorescence was measured at λex=490 nm and λem=530 nm (JC-1 monomers, signifying perturbed MMP), and λex=525 nm and λem=590 nm (JC-1 aggregates, signifying intact MMP) with a microplate reader (TECAN A-5082; TECAN Group). The capacity of mitochondria to take up and retain JC-1 was calculated as the difference (between control and treated cells) in green and red fluorescence.
NOX activity was analyzed as previously described (53). Briefly, 250 μM NADPH (NOX substrate) was added to cells and NADPH oxidation was monitored by the decrease in absorbance at 340 nm during 10 min. For analysis of specific oxidase activity, the rate of NADPH oxidation was measured in the presence of the NOX inhibitor apocynin, which was added 30 min before the assay at a final concentration of 10 μM. An aliquot of the cell lysate was assayed for protein content (bicinchoninic acid assay). The extinction coefficient used to calculate the amount of NADPH consumed was 6.22 mM−1cm−1 (53). Results were expressed as picomoles per liter of substrate per minute per milligram of protein.
Detection of apoptosis
Apoptosis was assessed by annexin V-FITC/propidium iodide staining of cells (29). Primary rat hepatocytes and RHPCs were suspended in PBS at 1×105 cells/ml, fixed by the addition of ice-cold 1% formaldehyde solution for 30 min, and permeabilized by the addition of Triton-X 100 (0.1%; Sigma-Aldrich). Next, aliquots of the cell suspension (100 μl) were stained with annexin V-FITC and propidium iodide (apoptosis assay kit, KGA105; KeyGEN, Nanjing, P.R. China), incubated at room temperature for 30 min in the dark, and analyzed by flow cytometry (FACSCalibur with CellQuest software; BD Biosciences, San Jose, CA).
Caspase-3 and caspase-9 activity in primary rat hepatocytes and RHPCs was assayed by commercial kits (C1116 or C1158; Beyotime Biotech). Absorbance was measured at 405 nm.
Reverse transcription–polymerase chain reaction
Total RNA was extracted from cells by using the Trizol Reagent protocol (Invitrogen). Two micrograms of total RNA was reverse transcribed in a 20 μl reaction mixture using Moloney Murine Leukemia Virus Reverse Transcription kit for cDNA synthesis (Invitrogen) according to the manufacturer's instructions. The cDNA was amplified by using a Taq DNA Polymerase kit (Biomed, Beijing, P.R. China). The following primer sequences were used: rat NLRP3, 5′-AGCCTCAGGGCACCAA-3′, 5′-GGGATGAAGCACATAGTAAACA-3′; human TXNIP, 5′-ACTGAAGCGTTGAGTAGTACA-3′, 5′-TGGCGTGGCAAGACAGTCA-3′; rat TXNIP, 5′-AAGCGTTGAGTAGTACAGATGAG-3′, 5′-ATGGCGTGGCAAGAGTC-3′; and rat β-actin, 5′-CACCCGCGAGTACAACCTTC-3′, 5′-CCCATACCCACCATCACACC-3′. All primer sequences were checked in GenBank to avoid inadvertent sequence homology. The PCR program was run on a Biometra system (Biometra, Gottingen, Germany) as follows: 95°C for 5 min; 94°C for 30 s; 58°C for 35 s; 72°C for 40 s (30 cycles); and 72°C for 10 min. PCR products (molecular weight of end-products) were validated by gel electrophoresis on 1% agarose gels. Gels were visualized with the Bio-Rad ChemiDoc XRS Gel Documentation system (BioRad Laboratories, Hercules, CA) and quantified using Bio-Rad Quantity One 1-D analysis software. The mRNA expression levels of each target gene were normalized to those of β-actin.
Immunoblotting
Equal amounts of protein extracted from liver homogenates and primary rat hepatocytes, Kupffer cells, RHPCs, HLO2, and HepG2 cell lysates were loaded on 8%–12% sodium dodecyl sulfate–polyacrylamide gels and transferred to PVDF membrane (Millipore, Billerica, MA). Nuclear protein was extracted by using a commercial kit (KGP150; KeyGEN). Membranes were probed with primary antibodies, including: anti-NLRP3 (cat# nbp2-12446, dilution 1:1000, Novus Biologicals), anti-ASC (cat# ab64808, dilution 1:1000; Abcam), anti-caspase-1 (cat# ab17820, dilution 1:1000; Abcam), anti-TXNIP (cat# ab86983, dilution 1:1000; Abcam), anti-pJAK2 (cat# ab68268, dilution 1:1000; Abcam), anti-JAK2 (cat# ab39636, dilution 1:1000; Abcam), anti-SOCS3 (cat# ab16030, dilution 1:1000; Abcam), anti-PPAR-α (cat# ab8934, dilution 1:1000; Abcam), anti-SCD1 (cat# ab19862, dilution 1:1000; Abcam), anti-p-STAT3 (cat# 9131, dilution 1:1000; Cell Signaling Technology, Danvers, MA), anti-STAT3 (cat# 4904, dilution 1:1000; Cell Signaling Technology), anti-lamin A/C (cat# 2032, dilution 1:1000; Cell Signaling Technology), anti-SREBP1 (cat# bs-1402R, dilution 1:1000; BioSS, Dundee, United Kingdom), anti-GAPDH (cat# 101M4777, dilution 1:5000; Sigma-Aldrich), anti-Nrf2 (cat# sc-722, dilution 1:300; Santa Cruz Biotechnology, Santa Cruz, CA), anti-GLUT2 (cat# sc-9117, dilution 1:500; Santa Cruz), anti-GLUT5 (cat# ab41533, dilution 1:1000; Abcam), and anti-GLUT8 (cat# ab169779, dilution 1:1000; Abcam). After incubating with horseradish peroxidase-conjugated secondary antibody (Jingmei Biotech, Shanghai, P.R. China), immunoreactive bands from rat liver homogenates and cell lysates were visualized by TMB reagent (KPL, Gaithersburg, MD) or ECL kit (Kangwei, Beijing, P.R. China) and X-ray film (Kodak, Fujian, P.R. China). Densitometric analysis of the bands was performed with Bio-Rad Quantity One 1-D analysis software.
Statistical analysis
All data are expressed as mean±SEM. Statistical analysis was performed in GraphPad Prism (GraphPad Software, San Diego, CA) using two-tailed Student's t-test or one-way analysis of variance followed by a least significant difference post hoc test. The Pearson's correlation coefficient was used to determine the linear relationship between total ROS production and TXNIP expression levels. A p-value of ≤0.05 was considered statistically significant.
Supplementary Material
Abbreviations Used
- Ac-YVAD-CMK
Ac-Tyr-Val-Ala-Asp-chloromethylketone
- ASC
apoptosis-associated speck-like protein
- CD163
glucocorticoid-inducible member of the scavenger receptor cysteine-rich family
- CDAA
choline-deficient amino acid-defined
- CK18
cytokeratin 18
- DCF
dichlorofluorescein
- DCFH2-DA
dichlorodihydrofluorescein diacetate
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GLUT
glucose transporter
- IHC
immunohistochemical
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- JAK2
janus-activated kinase 2
- MDA
malondialdehyde
- MMP
mitochondrial membrane potential
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NLRP3
NOD-like receptor family, pyrin domain containing 3
- NOX
NADPH oxidase
- Nrf2
nuclear factor (erythroid-derived-2)-like 2
- PBS
phosphate-buffered saline
- PPAR-α
peroxisome proliferator activated receptor-α
- PVDF
polyvinylidene fluoride
- RHPCs
rat hepatic parenchymal cells
- ROS
reactive oxygen species
- RT-PCR
reverse transcription–polymerase chain reaction
- SCD1
stearoyl-CoA desaturase 1
- SD
Sprague–Dawley
- SEM
standard error of the mean
- SOCS3
suppressors of cytokine signaling 3
- SREBP1
sterol regulatory element binding protein 1
- STAT3
signal transducer and activator of transcription 3
- SOD
superoxide dismutase
- TAC
total antioxidant capacity
- TC
total cholesterol
- TG
triglyceride
- TXNIP
thioredoxin-interacting protein
- XO
xanthine oxidase
Acknowledgments
This work has been supported by grants from the National Basic Research Program of China 973 Program No. 2012CB517600 (No. 2012CB517602) and the Natural Science Foundation of China (No. 81025025, 81373788, and J1103512). Support was also obtained from the PhD Programs Foundation of the Ministry of Education of China (20120091110039).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Bakker PJ, Butter LM, Kors L, Teske GJ, Aten J, Sutterwala FS, Florquin S, and Leemans JC. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int 85: 1112–1122, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Bischoff SC. Quercetin: potentials in the prevention and therapy of disease. Curr Opin Clin Nutr Metab Care 11: 733–740, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Cooper SR, Taylor JK, Miraglia LJ, and Dean NM. Pharmacology of antisense oligonucleotide inhibitors of protein expression. Pharmacol Ther 82: 427–435, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Crooke ST. Basic principles of antisense therapeutics. In: Antisense Research and Application. edited by Crooke ST. Berlin: Springer, 1998, pp. 1–50 [Google Scholar]
- 5.Demirel U, Yalniz M, Aygun C, Orhan C, Tuzcu M, Sahin K, Ozercan IH, and Bahcecioglu IH. Allopurinol ameliorates thioacetamide-induced acute liver failure by regulating cellular redox-sensitive transcription factors in rats. Inflammation 35: 1549–1557, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Deng XQ, Chen LL, and Li NX. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int 27: 708–715, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Esser N, L'Homme L, De Roover A, Kohnen L, Scheen AJ, Moutschen M, Piette J, Legrand-Poels S, and Paquot N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 56: 2487–2497, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Frenzel J, Richter J, and Eschrich K. Fructose inhibits apoptosis induced by reoxygenation in rat hepatocytes by decreasing reactive oxygen species via stabilization of the glutathione pool. Biochim Biophys Acta 1542: 82–94, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, and Couillin I. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 179: 903–913, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Griffith JW, Sun T, McIntosh MT, and Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol 183: 5208–5220, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He X. and Ma Q. Redox regulation by nuclear factor erythroid 2-related factor 2: gatekeeping for the basal and diabetes-induced expression of thioredoxin-interacting protein. Mol Pharmacol 82: 887–897, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, and Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–185, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu QH, Wang C, Li JM, Zhang DM, and Kong LD. Allopurinol, rutin, and quercetin attenuate hyperuricemia and renal dysfunction in rats induced by fructose intake: renal organic ion transporter involvement. Am J Physiol Renal Physiol 297: F1080–F1091, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Hu QH, Zhang X, Pan Y, Li YC, and Kong LD. Allopurinol, quercetin and rutin ameliorate renal NLRP3 inflammasome activation and lipid accumulation in fructose-fed rats. Biochem Pharmacol 84: 113–125, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, Ding Q, Loughran P, Beer-Stolz D, Billiar TR, Esmon CT, and Tsung A. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol 191: 2665–2679, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karim S, Adams DH, and Lalor PF. Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol 18: 6771–6781, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim GN. and Jang HD. Protective mechanism of quercetin and rutin using glutathione metabolism on HO-induced oxidative stress in HepG2 cells. Ann N Y Acad Sci 1171: 530–537, 2009 [DOI] [PubMed] [Google Scholar]
- 18.Kim HY, Kim SJ, and Lee SM. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J 282: 259–270, 2015 [DOI] [PubMed] [Google Scholar]
- 19.Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol 11: 213–220, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Le KA. and Tappy L. Metabolic effects of fructose. Curr Opin Clin Nutr Metab Care 9: 469–475, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, and Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62: 194–204, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li JM, Li YC, Kong LD, and Hu QH. Curcumin inhibits hepatic protein-tyrosine phosphatase 1B and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology 51: 1555–1566, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Xu S, Jiang B, Cohen RA, and Zang M. Activation of sterol regulatory element binding protein and NLRP3 inflammasome in atherosclerotic lesion development in diabetic pigs. PLoS One 8: e67532, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, and Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol 7: 251–264, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Lotowska JM, Sobaniec-Lotowska ME, and Lebensztejn DM. The role of Kupffer cells in the morphogenesis of nonalcoholic steatohepatitis—ultrastructural findings. The first report in pediatric patients. Scand J Gastroenterol 48: 352–357, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Lu J. and Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med 66: 75–87, 2014 [DOI] [PubMed] [Google Scholar]
- 27.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401–426, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcolin E, San-Miguel B, Vallejo D, Tieppo J, Marroni N, Gonzalez-Gallego J, and Tunon MJ. Quercetin treatment ameliorates inflammation and fibrosis in mice with nonalcoholic steatohepatitis. J Nutr 142: 1821–1828, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Milovanova T, Manevich Y, Haddad A, Chatterjee S, Moore JS, and Fisher AB. Endothelial cell proliferation associated with abrupt reduction in shear stress is dependent on reactive oxygen species. Antioxid Redox Signal 6: 245–258, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, and Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 57: 577–589, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moschen AR, Molnar C, Enrich B, Geiger S, Ebenbichler CF, and Tilg H. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol Med 17: 840–845, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouralidarane A, Soeda J, Visconti-Pugmire C, Samuelsson AM, Pombo J, Maragkoudaki X, Butt A, Saraswati R, Novelli M, Fusai G, Poston L, Taylor PD, and Oben JA. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 58: 128–138, 2013 [DOI] [PubMed] [Google Scholar]
- 33.Nagai Y, Yonemitsu S, Erion DM, Iwasaki T, Stark R, Weismann D, Dong J, Zhang D, Jurczak MJ, Loffler MG, Cresswell J, Yu XX, Murray SF, Bhanot S, Monia BP, Bogan JS, Samuel V, and Shulman GI. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab 9: 252–264, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, Delker DA, Jo J, Bertoletti A, Hagedorn CH, and Gale M., Jr IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog 9: e1003330, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nivet-Antoine V, Cottart CH, Lemarechal H, Vamy M, Margaill I, Beaudeux JL, Bonnefont-Rousselot D, and Borderie D. Trans-Resveratrol downregulates Txnip overexpression occurring during liver ischemia-reperfusion. Biochimie 92: 1766–1771, 2010 [DOI] [PubMed] [Google Scholar]
- 36.Nomura K. and Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem 23: 203–208, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Oka S, Liu W, Yoshihara E, Ahsan MK, Ramos DA, Son A, Okuyama H, Zhang L, Masutani H, Nakamura H, and Yodoi J. Thioredoxin binding protein-2 mediates metabolic adaptation in response to lipopolysaccharide in vivo. Crit Care Med 38: 2345–2351, 2010 [DOI] [PubMed] [Google Scholar]
- 38.Panchal SK, Poudyal H, and Brown L. Quercetin ameliorates cardiovascular, hepatic, and metabolic changes in diet-induced metabolic syndrome in rats. J Nutr 142: 1026–1032, 2012 [DOI] [PubMed] [Google Scholar]
- 39.Park MJ, Kim DI, Lim SK, Choi JH, Kim JC, Yoon KC, Lee JB, Lee JH, Han HJ, Choi IP, Kim HC, and Park SH. Thioredoxin-interacting protein mediates hepatic lipogenesis and inflammation via PRMT1 and PGC-1alpha regulation in vitro and in vivo. J Hepatol 61: 1151–1157, 2014 [DOI] [PubMed] [Google Scholar]
- 40.Peglow S, Toledo AH, Anaya-Prado R, Lopez-Neblina F, Toledo-Pereyra LH. Allopurinol and xanthine oxidase inhibition in liver ischemia reperfusion. J Hepatobiliary Pancreat Sci 18: 137–146, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Pejznochova M, Tesarova M, Hansikova H, Magner M, Honzik T, Vinsova K, Hajkova Z, Havlickova V, and Zeman J. Mitochondrial DNA content and expression of genes involved in mtDNA transcription, regulation and maintenance during human fetal development. Mitochondrion 10: 321–329, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Ramyaa P, Krishnaswamy R, and Padma VV. Quercetin modulates OTA-induced oxidative stress and redox signalling in HepG2 cells—up regulation of Nrf2 expression and down regulation of NF-kappaB and COX-2. Biochim Biophys Acta 1840: 681–692, 2014 [DOI] [PubMed] [Google Scholar]
- 43.Reiniers MJ, van Golen RF, van Gulik TM, and Heger M. Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver. Antioxid Redox Signal 21: 1119–1142, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russo M, Spagnuolo C, Tedesco I, Bilotto S, and Russo GL. The flavonoid quercetin in disease prevention and therapy: facts and fancies. Biochem Pharmacol 83: 6–15, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Smith BW. and Adams LA. Non-alcoholic fatty liver disease. Crit Rev Clin Lab Sci 48: 97–113, 2011 [DOI] [PubMed] [Google Scholar]
- 46.Spruss A. and Bergheim I. Dietary fructose and intestinal barrier: potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J Nutr Biochem 20: 657–662, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Spruss A, Kanuri G, Stahl C, Bischoff SC, and Bergheim I. Metformin protects against the development of fructose-induced steatosis in mice: role of the intestinal barrier function. Lab Invest 92: 1020–1032, 2012 [DOI] [PubMed] [Google Scholar]
- 48.Spruss A, Kanuri G, Uebel K, Bischoff SC, and Bergheim I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxid Redox Signal 14: 2121–2135, 2011 [DOI] [PubMed] [Google Scholar]
- 49.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, and Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 50: 1094–1104, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, Neale GA, Hooiveld GJ, Hijmans A, Vroegrijk I, van den Berg S, Romijn J, Rensen PC, Joosten LA, Netea MG, and Kanneganti TD. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A 108: 15324–15329, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki I, Yamauchi T, Onuma M, and Nozaki S. Allopurinol, an inhibitor of uric acid synthesis—can it be used for the treatment of metabolic syndrome and related disorders? Drugs Today (Barc) 45: 363–378, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Takahashi Y, Kobayashi Y, Kawata K, Kawamura K, Sumiyoshi S, Noritake H, Watanabe S, Chida T, Souda K, Sakaguchi T, Nakamura H, and Suda T. Does hepatic oxidative stress enhance activation of nuclear factor-E2-related factor in patients with nonalcoholic steatohepatitis? Antioxid Redox Signal 20: 538–543, 2014 [DOI] [PubMed] [Google Scholar]
- 53.Thannickal VJ. and Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem 270: 30334–30338, 1995 [DOI] [PubMed] [Google Scholar]
- 54.Tilg H. The role of cytokines in non-alcoholic fatty liver disease. Dig Dis 28: 179–185, 2010 [DOI] [PubMed] [Google Scholar]
- 55.Tschopp J. and Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10: 210–215, 2010 [DOI] [PubMed] [Google Scholar]
- 56.Ulman EA, Ifft KH, Kari FW, and Visek WJ. Fatty liver of growing rats fed excess lysine and its prevention by adenine or allopurinol. Lipids 16: 393–396, 1981 [DOI] [PubMed] [Google Scholar]
- 57.Valeri F, Boess F, Wolf A, Goldlin C, and Boelsterli UA. Fructose and tagatose protect against oxidative cell injury by iron chelation. Free Radic Biol Med 22: 257–268, 1997 [DOI] [PubMed] [Google Scholar]
- 58.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, and Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17: 179–188, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vos MB. and Lavine JE. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 57: 2525–2531, 2013 [DOI] [PubMed] [Google Scholar]
- 60.Zhang HF, Shi LJ, Song GY, Cai ZG, Wang C, and An RJ. Protective effects of matrine against progression of high-fructose diet-induced steatohepatitis by enhancing antioxidant and anti-inflammatory defences involving Nrf2 translocation. Food Chem Toxicol 55: 70–77, 2013 [DOI] [PubMed] [Google Scholar]
- 61.Zhao C, Gillette DD, Li X, Zhang Z, and Wen H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J Biol Chem 289: 17020–17029, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou R, Tardivel A, Thorens B, Choi I, and Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11: 136–140, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Zhou R, Yazdi AS, Menu P, and Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011 [DOI] [PubMed] [Google Scholar]
- 64.Zou X, Yan C, Shi Y, Cao K, Xu J, Wang X, Chen C, Luo C, Li Y, Gao J, Pang W, Zhao J, Zhao F, Li H, Zheng A, Sun W, Long J, Szeto IM, Zhao Y, Dong Z, Zhang P, Wang J, Lu W, Zhang Y, Liu J, and Feng Z. Mitochondrial dysfunction in obesity-associated nonalcoholic fatty liver disease: the protective effects of pomegranate with its active component punicalagin. Antioxid Redox Signal 21: 1557–1570, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.