

Background: Kaposi sarcoma herpesvirus (KSHV) causes malignancies in patients with AIDS or immunosuppression.
Results: KSHV represses tumor suppressor gene PDLIM2, whose reconstitution suppresses tumorigenesis of KSHV-associated cancer cells.
Conclusion: KSHV suppresses PDLIM2 for tumorigenesis.
Significance: These studies not only reveal an important mechanism underlying KSHV pathogenesis but also provide immediate therapeutic strategies for KSHV-mediated cancers.
Keywords: Cancer, NF-kappa B (NF-KB), STAT3, Tumor Suppressor Gene, Tumor Virus, HIV/AIDS-related Malignancies, KSHV/HHV8, PDLIM2
Abstract
Kaposi sarcoma herpesvirus (KSHV) is the most common cause of malignancies among AIDS patients. However, how KSHV induces tumorigenesis remains largely unknown. Here, we demonstrate that one important mechanism underlying the tumorigenesis of KSHV is through transcriptional repression of the tumor suppressor gene PDZ-LIM domain-containing protein 2 (PDLIM2). PDLIM2 expression is repressed in KSHV-transformed human umbilical vascular endothelial cells as well as in KSHV-associated cancer cell lines and primary tumors. Importantly, PDLIM2 repression is essential for KSHV-induced persistent activation of nuclear factor κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3) and subsequent tumorigenesis and tumor maintenance. Our mechanistic studies indicate that PDLIM2 repression by KSHV involves DNA methylation. Notably, the epigenetic repression of PDLIM2 can be reversed by 5-aza-2-deoxycytidine and vitamin D to suppress KSHV-associated cancer cell growth. These studies not only improve our understanding of KSHV pathogenesis but also provide immediate therapeutic strategies for KSHV-mediated cancers, particularly those associated with AIDS.
Introduction
KSHV,3 also known as human herpesvirus 8 (HHV8), was identified in 1990s as the etiological agent of three malignancies mainly associated with AIDS and immunosuppression: Kaposi sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman disease (1–3). Since then, the molecular biology of this oncovirus has been extensively studied (4–6). However, the molecular mechanisms by which KSHV induces tumorigenesis still remain obscure. Those characterized to date suggest that although its lytic infection contributes in a paracrine manner, the latent infection of KSHV is the direct driving force in tumor formation and maintenance with the expression of a limited set of viral genes capable of hijacking cellular signaling pathways important for host cell proliferation and survival.
Among those host cell signaling pathways hijacked by KSHV, the NF-κB and STAT3 pathways, which have been shown to play a causative role in the formation and therapeutic resistance of several tumor types, are particularly important for the pathogenesis of KSHV. In fact, constitutive activation of NF-κB and STAT3 has been found to be essential for all key steps of KSHV tumorigenesis, from viral latency to cell transformation to tumor formation and maintenance (7–32).
The molecular mechanisms of how KSHV activates NF-κB and STAT3 have been well defined (7–32). However, it remains unknown how the activated NF-κB and STAT3 fail to be turned off in KSHV tumorigenesis. Under normal conditions, activation of NF-κB and STAT3 is usually transient. One essential mechanism for terminating activation of both NF-κB RelA and STAT3 involves polyubiquitination and proteasomal degradation of their nuclear activated forms mediated by PDLIM2 (33–35). Accordingly, the expression of PDLIM2 has been found to be epigenetically repressed in several tumors associated with persistent activation of RelA (36–39). Thus, it is of interest and importance to examine whether and how PDLIM2 is repressed by KSHV and whether PDLIM2 repression by KSHV is involved in the persistent RelA and STAT3 activation and subsequent tumor formation and maintenance. These studies will not only increase our understanding of KSHV pathogenesis but may also form novel therapeutic strategies for KSHV-associated tumors, particularly those related to AIDS.
EXPERIMENTAL PROCEDURES
Expression Vectors and Reagents
Expression vectors encoding Myc-tagged PDLIM2, PDLIM2 PDZ, or LIM domain deletion mutant, NF-κB- or STAT3-driven firefly luciferase reporters, thymidine kinase-driven Renilla luciferase reporter, and antibodies specifically recognizing Myc, PDLIM2, 20 S proteasome, promyelocytic leukemia protein, and SC-35 have been described before (36, 40–42). Lentiviral vector pLL3.7 expressing shRNAs specifically against human PDLIM2, RelA, or STAT3 were generated as described previously (41). Sp1, Hsp90, RelA, and STAT3 antibodies as well as the secondary antibodies were from Santa Cruz Biotechnology (Dallas, TX). Phorbol myristate acetate, sodium butyrate, 5-aza-dC, and 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3) were purchased from Sigma-Aldrich.
Cells and Viruses
HUVECs were cultured in VascuLife® VEGF cell culture medium (Lifeline Cell Technology). Human PEL cell lines BCBL-1, BC-1, and BCP-1 were maintained in RPMI 1640 medium supplemented with 10% FBS. BCBL-1 cells were treated with phorbol myristate acetate (20 ng/ml) and sodium butyrate (1 mm) for 3 days to produce infectious KSHV viruses for in vitro transformation of HUVECs.
Quantitative Polymerase Chain Reaction (qPCR) Analysis
Cells were subjected to RNA extraction, RNA reverse transcription, and real-time PCR as described (37–39). The expression levels of PDLIM2 were normalized to that of GAPDH. Primer pairs for GAPDH, PDLIM2, DNMT1, DNMT3a, and DNMT3b were published previously (37). Other primers are: Bcl-xL, forward 5′-GAATGACCACCTAGAGCCTTGG-3′, reverse 5′-TGTTCCCATAGAGTTCCACAAAAG-3′; survivin, forward 5′-TGACGACCCCATAGAGGAACA-3′, reverse 5′-CGCACTTTCTCCGCAGTTTC-3′; and cyclin D1, forward 5′-CCGTCCATGCGGAAGATC-3′, reverse 5′-ATGGCCAGCGGGAAGAC-3′.
Retroviral Transduction and Generation of Stable Transfectant
PEL cell lines stably expressing PDLIM2, PDLIM2 mutants, shRNAs against RelA, STAT3, or PDLIM2 were generated as described before (43).
Soft Agar Assays
Cells suspended in culture medium containing 0.6% SeaPlaque low melting agarose were plated on the top of 1% agarose in culture medium as described before (43). Colonies in soft agar were counted 12 days after plating.
Establishment of Tumors and Ascites in Mice
Four- to six-week-old SCID mice were injected intraperitoneally or subcutaneously with 5 × 106 PEL cells for ascites and/or tumor formation as described previously (44). The protocols were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Histopathology and Immunohistochemistry (IHC) Assays
Formalin-fixed human normal and KS tissues were embedded in paraffin, sectioned, and then subjected to IHC staining as described previously (45).
Immunoblotting (IB) Analysis
Whole cell lysates and nuclear extracts were prepared and used for SDS-PAGE and IB as described previously (46, 47). The purity of cell nuclear fractions was confirmed by the detection of Sp1 (nuclear marker) but no Hsp90 (cytosolic marker) in IB.
Confocal Microscopic Analysis
The indicated PEL stable cell lines were subjected to immunofluorescence staining as described before (36). The subcellular localization of stained proteins was visualized by an Olympus FluoView 1000 confocal microscope (Melville, NY).
Luciferase Gene Reporter Assays
The indicated cells were transfected with NF-κB- or STAT3-driven firefly luciferase reporters together with thymidine kinase-driven Renilla luciferase reporter. At 40 h after transfection, Dual-Luciferase activities were measured as described previously (41).
Bisulfite Genomic DNA Sequencing
As described before (38, 39), genomic DNAs from 5-aza-dC-treated or mock-treated cells were isolated, and aliquots were then treated with sodium bisulfite followed by PCR to amplify the PDLIM2 promoter and DNA sequencing to determine the methylation status of the CpG dinucleotides within the pdlim2 promoter.
Statistical Analysis
Data were reported as mean ± S.D. The Student's t test (two-tailed) was used to assess significance of differences between two groups. p values < 0.05 and 0.01 were considered statistically significant and highly statistically significant, and indicated by * and **, respectively.
RESULTS
PDLIM2 Expression Is Repressed in KSHV-transformed Cells and Primary Tumor Tissues
To investigate whether PDLIM2 is involved in the pathogenesis of KSHV, we initially examined the expression levels of PDLIM2 in human PEL cell lines BCBL-1, BC-1, and BCP-1. In comparison with the virus-free lymphoblastoid B-cell line BJAB, all three PEL cell lines had much lower expression of PDLIM2 RNA (Fig. 1A). Similarly, the RNA expression level of PDLIM2 was significantly suppressed in KSHV-transformed HUVECs, in comparison with normal HUVECs (Fig. 1B). These data suggest that PDLIM2 expression is repressed by KSHV starting from the very early stage of tumorigenesis.
FIGURE 1.
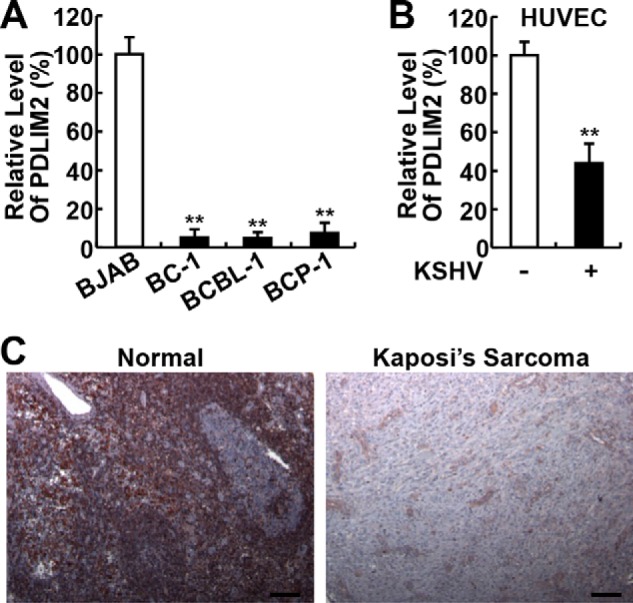
PDLIM2 expression is repressed in KSHV-transformed cells and KS primary tissues. A, qPCR analyses showing decreased PDLIM2 RNA levels in KSHV-associated PEL cell lines. B, qPCR analyses showing decreased PDLIM2 RNA levels in KSHV-transformed HUVECs. C, IHC analyses showing decreased PDLIM2 protein levels in KS primary tissues. Scale bar: 100 μm. Data are mean ± S.D. **, p < 0.01.
To validate the clinic significance of this finding, we examined the expression of PDLIM2 in human primary tissues of KS associated with KSHV. In line with its ubiquitous expression in murine tissues/cells (48), PDLIM2 was readily detected in various cell types of KSHV-negative normal tissues (Fig. 1C, left panel). However, little PDLIM2 was detected in primary KS cells (Fig. 1C, right panel). These data indicated that PDLIM2 repression is relevant to KSHV-associated tumors both pathogenically and clinically.
PDLIM2 Reconstitution Blocks HUVEC Transformation by KSHV and Suppresses the Tumorigenicity of KSHV-associated PEL Cells Both in Vitro and in Vivo
To determine the significance of PDLIM2 down-regulation in the pathogenesis of KSHV, we first examined whether PDLIM2 reconstitution prevents KSHV-mediated transformation of HUVECs. To do so, we generated HUVECs stably expressing exogenous PDLIM2 or an empty vector (Fig. 2A, upper panels). Expression of exogenous PDLIM2 had no obvious effect on the morphology of HUVECs (Fig. 2A, middle panel). In agreement with previous studies (49), infection of KSHV resulted in dramatic induction of spindle-shaped morphology, a hallmark of KS, in HUVECs expressing empty vector (Fig. 2A, bottom panel, left). However, no significant morphology change was observed in HUVECs expressing PDLIM2 after the same infection (Fig. 2A, bottom panel, right). These data suggest that PDLIM2 suppresses KSHV-mediated cell transformation.
FIGURE 2.

PDLIM2 reconstitution blocks KSHV-mediated transformation of HUVECs and suppresses the growth and tumorigenicity of KSHV-associated PEL cells both in vitro and in vivo. A, upper panels, IB assays showing exogenous PDLIM2 expression in HUVEC stable cell lines. Lower panels, morphology assays indicating that PDLIM2 reconstitution suppresses KSHV-mediated transformation of HUVECs. Scale bar: 100 μm. B, upper panels, IB assays showing reconstituted PDLIM2 in PEL cell lines. Lower panels, cell counting assays showing decreased growth rate of PEL cells reconstituted with PDLIM2. C, soft agar assays showing decreased anchorage-independent growth of PEL cells reconstituted with PDLIM2. D, SCID mouse xenograft assays showing decreased tumor formation ability of PEL cells reconstituted with PDLIM2. E, SCID mouse xenograft assays showing decreased ascites and tumor formation of PEL cells reconstituted with PDLIM2. Data are mean ± S.D. *, p < 0.05; **, p < 0.01.
Next, we examined whether PDLIM2 reconstitution can reverse the malignant phenotype of PEL cells. When compared with their vector-expressing control cells, BCBL-1 and BC-1 cells stably expressing PDLIM2 showed a slight, but statistically significant, decrease in growth rate even when 10% FBS was provided in their culture medium (Fig. 2B). The difference was much more dramatic when the supplemented FBS was decreased to 5%. More importantly, the PDLIM2 stable cell lines formed far fewer colonies in soft agar (Fig. 2C). These data clearly suggest that PDLIM2 re-expression suppresses the tumorigenicity of KSHV-associated PEL cells in vitro.
To confirm the in vitro studies in vivo, we subcutaneously inoculated the vector control and PDLIM2-expressing PEL cell lines into the opposite flanks of SCID mice. As shown in Fig. 2D, both vector control and PDLIM2-expressing PEL cell lines developed tumors at the inoculation sites in mice. However, the tumors formed by the PDLIM2-expressing cancer cell lines were significantly smaller than those formed by the vector control cell lines. To validate these studies in a different in vivo model of human PEL, the PEL stable cell lines were intraperitoneally injected into SCID mice. In agreement with previous studies (44), SCID mice injected with the vector control PEL cell lines developed malignant ascites and effusion lymphomas in various organs (Fig. 2E). However, the severity of both ascites and lymphomas in mice injected with the PDLIM2 stable PEL cell lines was significantly lower. These data together indicate that PDLIM2 down-regulation is one important mechanism underlying both the tumorigenesis and the tumor phenotype maintenance of KSHV-associated tumors.
PDLIM2 Reconstitution Inhibits Oncogenic NF-κB RelA and STAT3 Activation to Suppress KSHV Tumorigenesis
To define the molecular mechanism by which PDLIM2 down-regulation contributes to the pathogenesis of KSHV, we focused on RelA and STAT3, two downstream targets of PDLIM2. As expected, a high nuclear expression of RelA and STAT3 was detected in the BCBL-1-vector and BC-1-vector cells (Fig. 3, A and B). However, the nuclear expression of RelA and STAT3 was inhibited in the BCBL-1-PDLIM2 and BC-1-PDLIM2 cells. The decrease of nuclear RelA and STAT3 was due to proteasomal degradation because treatment of the proteasome inhibitor MG132 led to increase of RelA and STAT3 at the nuclear matrix but not the promyelocytic leukemia or SC-35 bodies of those PDLIM2 stable cell lines (Fig. 3A). Consistently, the transcription activities and downstream target genes of NF-κB and STAT3, such as Bcl-xL, survivin, and cyclin D1, were also suppressed in those PDLIM2 stable cell lines (Fig. 3, C and D). It is noteworthy that Bcl-xL, survivin, and cyclin D1 are known drivers of cell survival and proliferation.
FIGURE 3.

PDLIM2 reconstitution inhibits oncogenic NF-κB RelA and STAT3 activation to suppress KSHV tumorigenesis. A, confocal microscopic assays showing decreased RelA and STAT3 nuclear expression in BCBL-1 cells reconstituted with PDLIM2, which can be reversed by proteasome inhibition. B, IB assays showing decreased RelA and STAT3 nuclear expression in the PEL cells reconstituted with PDLIM2. Lanes 1 and 3 represent PEL cells stably expressing an empty vector, while lanes 2 and 4 represent PEL cells stably expressing PDLIM2. C, gene reporter assays showing decreased transcriptional activity of NF-κB in PEL cells reconstituted with PDLIM2. D, qPCR analysis showing PDLIM2 down-regulation of Bcl-xL, survivin, and cyclin D1 in BCBL-1 cells. E, generation of BCBL-1 cells stably expressing PDLIM2 mutants defective in RelA and STAT3 suppression. The expression levels of these PDLIM2 mutants as well as the nuclear expression levels of RelA and STAT3 in the stable cell lines were examined by IB assay. Lanes 1–4 represent BCBL-1 cells stably expressing empty vector, PDLIM2 WT, ΔPDZ, and ΔLIM mutants, respectively. F, cell counting assays showing that PDLIM2 mutants defective in RelA and STAT3 termination lose the ability to suppress the growth of BCBL-1 cells in culture. G, soft agar assays showing that PDLIM2 mutants defective in RelA and STAT3 termination lose the ability to suppress the anchorage-independent growth of BCBL-1 cells. H, cell counting assays showing that knockdown of RelA or STAT3 inhibits the growth of BCBL-1 cells. Data are mean ± S.D. *, p < 0.05; **, p < 0.01.
To validate whether PDLIM2 suppresses KSHV tumorigenesis via termination of RelA and STAT3 activation, we utilized the PDLIM2 PDZ or LIM domain deletion mutants. In line with previous studies (33, 34, 38–40), the two mutants failed to prevent the nuclear expression of RelA and STAT3 in PEL cells (Fig. 3E). Importantly, the two mutants also failed to suppress the growth and colony formation of PEL cells (Fig. 3, F and G). In further support of this, knockdown of RelA or STAT3 resulted in significant growth inhibition of PEL cells (Fig. 3H). Altogether, our studies suggest that KSHV somehow represses PDLIM2 expression, leading to persistent RelA and STAT3 activation and subsequent tumorigenesis and tumor phenotype maintenance.
PDLIM2 Repression Involves Its Promoter Methylation and Can Be Reversed by 5-aza-dC and Vitamin D to Inhibit Growth of KSHV-associated Tumor Cells
Given the role of PDLIM2 in KSHV pathogenesis and its potential application in the therapy of KSHV-associated tumors, it is of interest and importance to determine the molecular mechanism by which KSHV represses PDLIM2 expression. Our previous studies showed that PDLIM2 is epigenetically repressed in several cancer types (37–39). Thus, we examined the methylation status of the pdlim2 promoter in PEL cells. We found that the pdlim2 promoter was indeed hyper-methylated in PEL cells, which could be reversed by the demethylation drug 5-aza-dC (Fig. 4A). Moreover, the treatment of 5-aza-dC resulted in significant recovery of PDLIM2 expression in the PEL cells (Fig. 4B). Interestingly, 5-aza-dC had a dose-dependent effect on PDLIM2 recovery at a concentration lower than 0.5 μm. These data are consistent with the fact that 5-aza-dC induces expression of methylated genes at low doses and mainly exerts its cytotoxicity through inducing DNA damage at high doses. To determine how KSHV induces PDLIM2 promoter hyper-methylation for its repression, we examined the expression of all three DNA methyltransferases in PEL cells. We found that DNMT3a, but not DNMT1 or DNMT3b, was dramatically induced in BCBL-1 and BC-1 cells when compared with the virus-free BJAB cells (Fig. 4C). These data suggest that KSHV represses PDLIM2 expression through inducing DNMT3a expression and pdlim2 promoter methylation.
FIGURE 4.

PDLIM2 repression by KSHV involves its promoter methylation and can be reversed by 5-aza-2-dC and vitamin D to inhibit growth of KSHV-associated tumor cells. A, bisulfite genomic DNA sequencing showing epigenetic repression of PDLIM2 in BCBL-1 cells. Each circle represents a CpG site, and the ratio of the filled area in each circle represents the percentile of methylation in the CpG site. The position of each CpG nucleotide relative to the PDLIM2 transcription initiation site (+1) is indicated at the side. 5-aza-dC treatment: 0.2 μm for 4 days. DMSO, dimethyl sulfoxide. B, qPCR analyses showing dose- and time-dependent re-induction of PDLIM2 by 5-aza-dC in BCBL-1 cells. 5-aza-dC treatment: left, 4 days; right, 0.2 μm. C, qPCR analyses showing increased RNA level of DNMT3a, but not DNMT1 or DNMT3b in PEL cells. D, qPCR analyses showing dose- and time-dependent re-induction of PDLIM2 by vitamin D in BCBL-1 cells. 1,25(OH)2D3 treatment: left, 4 days; right, 0.1 μm. E, qPCR analyses showing cooperation of 5-aza-dC and vitamin D in PDLIM2 re-induction in BCBL-1 cells. F, gene reporter assays showing inhibition of transcriptional activity of NF-κB and STAT3 by 5-aza-dC and vitamin D in BCBL-1 cells. G, cell counting assays showing synergistic growth inhibition of BCBL-1 cells by 5-aza-dC and vitamin D. H, cell counting assays showing that growth inhibition of BCBL-1 cells by 5-aza-dC and/or vitamin D (3-day treatment) can be blocked by PDLIM2 knockdown. For E–H, 5-aza-dC, 0.05 μm; 1,25(OH)2D3, 0.1 μm. Data are mean ± S.D. *, p < 0.05; **, p < 0.01.
A recent study showed that vitamin D can induce PDLIM2 re-expression in human breast cancer cells through both DNA methylation-dependent and DNA methylation-independent mechanisms (50). As expected, 1,25(OH)2D3, the biologically active form of vitamin D3, induced PDLIM2 re-expression in PEL cells (Fig. 4D). Interestingly, 5-aza-dC and 1,25(OH)2D3 could synergize in inducing PDLIM2 re-expression in PEL cells (Fig. 4E). Of note, re-induction of PDLIM2 by 5-aza-dC and 1,25(OH)2D3 was associated with decreased transcriptional activity of NF-κB and STAT3, as well as growth inhibition of PEL cells (Fig. 4, F and G).
To determine whether PDLIM2 re-induction is involved in the antitumor activity of 5-aza-dC and 1,25(OH)2D3, we generated PEL cells stably expressing PDLIM2 shRNA, in which PDLIM2 re-induction by 5-aza-dC and 1,25(OH)2D3 could be sufficiently blocked. Remarkably, the growth inhibition of PEL cells by 5-aza-dC and 1,25(OH)2D3 was significantly inhibited by PDLIM2 knockdown (Fig. 4H). These data not only provide the mechanistic insights into the KSHV-mediated repression of PDLIM2, but also suggest an immediate strategy to target PDLIM2 for the prevention and treatment of KSHV-associated tumors.
DISCUSSION
Twenty years ago, KSHV was demonstrated as the causative agent of three major AIDS-associated tumors. However, little progress has been made in targeting the virus for the treatment of these incurable tumors (51). Recent studies suggest that persistent activation of NF-κB and STAT3 plays critical roles in both KSHV-associated tumors and HIV-associated AIDS, therefore providing ideal therapeutic targets. However, clinical trials show that it is impractical to block NF-κB activation for cancer therapy using classical NF-κB inhibitors because of the physiological importance of NF-κB in humans (52). It also seems unfeasible to block virus-specific activation of NF-κB for cancer therapy because NF-κB activation by KSHV involves multiple mechanisms and multiple viral genes (17–26). A similar situation also applies to STAT3 (16, 27–32, 45). To meet these challenges, we have been focusing our studies on the termination mechanisms of NF-κB and STAT3 activation in KSHV-associated tumors because the fundamental difference between physiological and pathogenic activation of NF-κB and STAT3 is that the former is rapidly turned off and therefore transient, whereas the latter cannot be turned off and is therefore persistent.
Our studies presented in this study indicate that epigenetic repression of PDLIM2 is one important mechanism underlying the persistent activation of NF-κB and STAT3 in KSHV tumorigenesis. Given the specific role of PDLIM2 in repressing pathogenic, but not physiologic, activation of NF-κB and STAT3, PDLIM2-based therapies will alleviate NF-κB- and STAT3-mediated tumorigenesis and chemoradioresistance while keeping the physiological functions of NF-κB and STAT3 largely intact in patients. Indeed, the expression of PDLIM2 can be restored by 5-aza-dC and vitamin D to suppress oncogenic NF-κB and STAT3 activation and tumorigenicity of PEL cells. Of note, 5-aza-dC and vitamin D have already been used in humans and show very low toxicity. Thus, our studies not only provide mechanistic insights into the regulation and actions of the newly defined tumor suppressor PDLIM2, the oncogenic activation of NF-κB and STAT3, the pathogenesis of KSHV-associated tumors, and the antitumor activity of 5-aza-dC and vitamin D, but also suggest the clinical feasibility of PDLIM2-based therapies for KSHV-associated tumors and other tumors mediated by deregulated PDLIM2/NF-κB/STAT3.
Acknowledgments
We thank Drs. Yuan Chang, Patrick Moore, and Gutian Xiao for various research reagents as well as for critical reading and helpful suggestions. We also thank Dr. Leizhen Wei for technique assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant P30CA047904-24S through the NCI (to Z. Q.). This work was also supported by a grant from the Competitive Medical Research Fund (CMRF).
- KSHV
- Kaposi sarcoma herpesvirus
- KS
- Kaposi sarcoma
- PDLIM2
- PDZ-LIM domain-containing protein 2
- NF-κB
- nuclear factor κB
- PEL
- primary effusion lymphoma
- shRNA
- short hairpin RNA
- HUVEC
- human umbilical vascular endothelial cell
- 5-aza-dC
- 5-aza-2-deoxycytidine
- 1,25(OH)2D3
- 1α,25-dihydroxyvitamin D3
- IB
- immunoblotting
- IHC
- immunohistochemistry
- qPCR
- quantitative polymerase chain reaction.
REFERENCES
- 1. Chang Y., Cesarman E., Pessin M. S., Lee F., Culpepper J., Knowles D. M., Moore P. S. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266, 1865–1869 [DOI] [PubMed] [Google Scholar]
- 2. Cesarman E., Chang Y., Moore P. S., Said J. W., Knowles D. M. (1995) Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191 [DOI] [PubMed] [Google Scholar]
- 3. Soulier J., Grollet L., Oksenhendler E., Cacoub P., Cazals-Hatem D., Babinet P., d'Agay M. F., Clauvel J. P., Raphael M., Degos L., et al. (1995) Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86, 1276–1280 [PubMed] [Google Scholar]
- 4. Cai Q., Verma S. C., Lu J., Robertson E. S. (2010) Molecular biology of Kaposi's sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 78, 87–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wen K. W., Damania B. (2010) Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett. 289, 140–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cousins E., Nicholas J. (2014) Molecular biology of human herpesvirus 8: novel functions and virus-host interactions implicated in viral pathogenesis and replication. Recent Results Cancer Res. 193, 227–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiao G., Rabson A. B., Young W., Qing G., Qu Z. (2006) Alternative pathways of NF-κB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 17, 281–293 [DOI] [PubMed] [Google Scholar]
- 8. Qu Z., Xiao G. (2011) Human T-cell lymphotropic virus: a model of NF-κB-associated tumorigenesis. Viruses 3, 714–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiao G., Fu J. (2011) NF-κB and cancer: a paradigm of Yin-Yang. Am. J. Cancer Res. 1, 192–221 [PMC free article] [PubMed] [Google Scholar]
- 10. de Oliveira D. E., Ballon G., Cesarman E. (2010) NF-κB signaling modulation by EBV and KSHV. Trends Microbiol. 18, 248–257 [DOI] [PubMed] [Google Scholar]
- 11. Keller S. A., Schattner E. J., Cesarman E. (2000) Inhibition of NF-κB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 96, 2537–2542 [PubMed] [Google Scholar]
- 12. Brown H. J., Song M. J., Deng H., Wu T. T., Cheng G., Sun R. (2003) NF-κB inhibits γherpesvirus lytic replication. J. Virol. 77, 8532–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keller S. A., Hernandez-Hopkins D., Vider J., Ponomarev V., Hyjek E., Schattner E. J., Cesarman E. (2006) NF-κB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood 107, 3295–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He Z., Zhao J., Zhang J., Jung J. U., Feng P. (2014) NF-κB activation coordinated by IKKβ and IKKϵ enables latent infection of Kaposi's sarcoma-associated herpesvirus. J. Virol. 88, 444–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aoki Y., Feldman G. M., Tosato G. (2003) Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 101, 1535–1542 [DOI] [PubMed] [Google Scholar]
- 16. Burger M., Hartmann T., Burger J. A., Schraufstatter I. (2005) KSHV-GPCR and CXCR2 transforming capacity and angiogenic responses are mediated through a JAK2-STAT3-dependent pathway. Oncogene 24, 2067–2075 [DOI] [PubMed] [Google Scholar]
- 17. Pati S., Cavrois M., Guo H. G., Foulke J. S., Jr., Kim J., Feldman R. A., Reitz M. (2001) Activation of NF-κB by the human herpesvirus 8 chemokine receptor ORF74: evidence for a paracrine model of Kaposi's sarcoma pathogenesis. J. Virol. 75, 8660–8673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwarz M., Murphy P. M. (2001) Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-κB and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J. Immunol. 167, 505–513 [DOI] [PubMed] [Google Scholar]
- 19. Brinkmann M. M., Glenn M., Rainbow L., Kieser A., Henke-Gendo C., Schulz T. F. (2003) Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 77, 9346–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Konrad A., Wies E., Thurau M., Marquardt G., Naschberger E., Hentschel S., Jochmann R., Schulz T. F., Erfle H., Brors B., Lausen B., Neipel F., Stürzl M. (2009) A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-κB activation. J. Virol. 83, 2563–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun Q., Zachariah S., Chaudhary P. M. (2003) The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-κB activation. J. Biol. Chem. 278, 52437–52445 [DOI] [PubMed] [Google Scholar]
- 22. Chugh P., Matta H., Schamus S., Zachariah S., Kumar A., Richardson J. A., Smith A. L., Chaudhary P. M. (2005) Constitutive NF-κB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 102, 12885–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin D., Galisteo R., Ji Y., Montaner S., Gutkind J. S. (2008) An NF-κB gene expression signature contributes to Kaposi's sarcoma virus vGPCR-induced direct and paracrine neoplasia. Oncogene 27, 1844–1852 [DOI] [PubMed] [Google Scholar]
- 24. Matta H., Mazzacurati L., Schamus S., Yang T., Sun Q., Chaudhary P. M. (2007) Kaposi's sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IκB kinase complex to selectively activate NF-κB without JNK activation. J. Biol. Chem. 282, 24858–24865 [DOI] [PubMed] [Google Scholar]
- 25. Bottero V., Kerur N., Sadagopan S., Patel K., Sharma-Walia N., Chandran B. (2011) Phosphorylation and polyubiquitination of transforming growth factor β-activated kinase 1 are necessary for activation of NF-κB by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J. Virol. 85, 1980–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lei X., Bai Z., Ye F., Xie J., Kim C. G., Huang Y., Gao S. J. (2010) Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nat. Cell Biol. 12, 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cousins E., Nicholas J. (2013) Role of human herpesvirus 8 interleukin-6-activated gp130 signal transducer in primary effusion lymphoma cell growth and viability. J. Virol. 87, 10816–10827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Punjabi A. S., Carroll P. A., Chen L., Lagunoff M. (2007) Persistent activation of STAT3 by latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 81, 2449–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Molden J., Chang Y., You Y., Moore P. S., Goldsmith M. A. (1997) A Kaposi's sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J. Biol. Chem. 272, 19625–19631 [DOI] [PubMed] [Google Scholar]
- 30. Gwack Y., Hwang S., Lim C., Won Y. S., Lee C. H., Choe J. (2002) Kaposi's sarcoma-associated herpesvirus open reading frame 50 stimulates the transcriptional activity of STAT3. J. Biol. Chem. 277, 6438–6442 [DOI] [PubMed] [Google Scholar]
- 31. Muromoto R., Okabe K., Fujimuro M., Sugiyama K., Yokosawa H., Seya T., Matsuda T. (2006) Physical and functional interactions between STAT3 and Kaposi's sarcoma-associated herpesvirus-encoded LANA. FEBS Lett. 580, 93–98 [DOI] [PubMed] [Google Scholar]
- 32. Suthaus J., Stuhlmann-Laeisz C., Tompkins V. S., Rosean T. R., Klapper W., Tosato G., Janz S., Scheller J., Rose-John S. (2012) HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood 119, 5173–5181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tanaka T., Grusby M. J., Kaisho T. (2007) PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 8, 584–591 [DOI] [PubMed] [Google Scholar]
- 34. Tanaka T., Yamamoto Y., Muromoto R., Ikeda O., Sekine Y., Grusby M. J., Kaisho T., Matsuda T. (2011) PDLIM2 inhibits T helper 17 cell development and granulomatous inflammation through degradation of STAT3. Sci. Signal. 4, ra85. [DOI] [PubMed] [Google Scholar]
- 35. Qu Z., Fu J., Ma H., Zhou J., Jin M., Mapara M. Y., Grusby M. J., Xiao G. (2012) PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease. Cell Biosci. 2, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan P., Fu J., Qu Z., Li S., Tanaka T., Grusby M. J., Xiao G. (2009) PDLIM2 suppresses HTLV-I Tax-mediated tumorigenesis by targeting Tax into the nuclear matrix for proteasomal degradation. Blood 113, 4370–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yan P., Qu Z., Ishikawa C., Mori N., Xiao G. (2009) Human T-cell leukemia virus type I-mediated repression of PDZ-LIM domain-containing protein 2 involves DNA methylation but independent of the viral oncoprotein Tax. Neoplasia. 11, 1036–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qu Z., Fu J., Yan P., Hu J., Cheng S. Y., Xiao G. (2010) Epigenetic repression of PDLIM2: implications for the biology and treatment of breast cancer. J. Biol. Chem. 285, 11786–11792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qu Z., Yan P., Fu J., Jiang J., Grusby M. J., Smithgall T. E., Xiao G. (2010) DNA methylation-dependent repression of PDLIM2 in colon cancer and its role as a potential therapeutic target. Cancer Res. 70, 1766–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fu J., Yan P., Li S., Qu Z., Xiao G. (2010) Molecular determinants of PDLIM2 in suppressing HTLV-I Tax-mediated tumorigenesis. Oncogene 29, 6499–6507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fu J., Qu Z., Yan P., Ishikawa C., Aqeilan R. I., Rabson A. B., Xiao G. (2011) The tumor suppressor gene WWOX links the canonical and noncanonical NF-κB pathways in HTLV-I Tax-mediated tumorigenesis. Blood 117, 1652–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qing G., Qu Z., Xiao G. (2005) Regulation of NF-κB2 p100 processing by its cis-acting domain. J. Biol. Chem. 280, 18–27 [DOI] [PubMed] [Google Scholar]
- 43. Qing G., Qu Z., Xiao G. (2007) Endoproteolytic processing of C-terminally truncated NF-κB2 precursors at κB-containing promoters. Proc. Natl. Acad. Sci. U.S.A. 104, 5324–5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boshoff C., Gao S. J., Healy L. E., Matthews S., Thomas A. J., Coignet L., Warnke R. A., Strauchen J. A., Matutes E., Kamel O. W., Moore P. S., Weiss R. A., Chang Y. (1998) Establishing a KSHV+ cell line (BCP-1) from peripheral blood and characterizing its growth in Nod/SCID mice. Blood 91, 1671–1679 [PubMed] [Google Scholar]
- 45. Zhou J., Qu Z., Yan S., Sun F., Whitsett J. A., Shapiro S. D., Xiao G. (2014) Differential roles of STAT3 in the initiation and growth of lung cancer. Oncogene 10.1038/onc.2014.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qu Z., Qing G., Rabson A., Xiao G. (2004) Tax deregulation of NF-κB2 p100 processing involves both β-TrCP-dependent and -independent mechanisms. J. Biol. Chem. 279, 44563–44572 [DOI] [PubMed] [Google Scholar]
- 47. Qu Z., Sun D., Young W. (2011) Lithium promotes neural precursor cell proliferation: evidence for the involvement of the non-canonical GSK-3β-NF-AT signaling. Cell Biosci. 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanaka T., Soriano M. A., Grusby M. J. (2005) SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity 22, 729–736 [DOI] [PubMed] [Google Scholar]
- 49. Matta H., Surabhi R. M., Zhao J., Punj V., Sun Q., Schamus S., Mazzacurati L., Chaudhary P. M. (2007) Induction of spindle cell morphology in human vascular endothelial cells by human herpesvirus 8-encoded viral FLICE inhibitory protein K13. Oncogene 26, 1656–1660 [DOI] [PubMed] [Google Scholar]
- 50. Vanoirbeek E., Eelen G., Verlinden L., Carmeliet G., Mathieu C., Bouillon R., O'Connor R., Xiao G., Verstuyf A. (2014) PDLIM2 expression is driven by vitamin D and is involved in the pro-adhesion, and anti-migration and -invasion activity of vitamin D. Oncogene 33, 1904–1911 [DOI] [PubMed] [Google Scholar]
- 51. Casper C. (2008) New approaches to the treatment of human herpesvirus 8-associated disease. Rev. Med. Virol. 18, 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robe P. A., Martin D. H., Nguyen-Khac M. T., Artesi M., Deprez M., Albert A., Vanbelle S., Califice S., Bredel M., Bours V. (2009) Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 9, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]