

Background: The function of formyl peptide receptors (FPRs) is incompletely understood.
Results: We report the identification of bacterial signal peptides as potent activators of mammalian FPRs and innate immune responses and define critical features underlying FPR peptide recognition.
Conclusion: These findings identify a molecular signature for FPR activation.
Significance: Our results define a novel mechanism for sensing bacteria.
Keywords: Calcium Imaging, Chemical Biology, G-protein-coupled Receptor (GPCR), Innate Immunity, Molecular Pharmacology, Pathogen-associated Molecular Pattern (PAMP), Pattern Recognition Receptor (PRR), Vomeronasal Organ (VNO)
Abstract
Formyl peptide receptors (FPRs) are G-protein-coupled receptors that function as chemoattractant receptors in innate immune responses. Here we perform systematic structure-function analyses of FPRs from six mammalian species using structurally diverse FPR peptide agonists and identify a common set of conserved agonist properties with typical features of pathogen-associated molecular patterns. Guided by these results, we discover that bacterial signal peptides, normally used to translocate proteins across cytoplasmic membranes, are a vast family of natural FPR agonists. N-terminally formylated signal peptide fragments with variable sequence and length activate human and mouse FPR1 and FPR2 at low nanomolar concentrations, thus establishing FPR1 and FPR2 as sensitive and broad signal peptide receptors. The vomeronasal receptor mFpr-rs1 and its sequence orthologue hFPR3 also react to signal peptides but are much more narrowly tuned in signal peptide recognition. Furthermore, all signal peptides examined here function as potent activators of the innate immune system. They elicit robust, FPR-dependent calcium mobilization in human and mouse leukocytes and trigger a range of classical innate defense mechanisms, such as the production of reactive oxygen species, metalloprotease release, and chemotaxis. Thus, bacterial signal peptides constitute a novel class of immune activators that are likely to contribute to mammalian immune defense against bacteria. This evolutionarily conserved detection mechanism combines structural promiscuity with high specificity and enables discrimination between bacterial and eukaryotic signal sequences. With at least 175,542 predicted sequences, bacterial signal peptides represent the largest and structurally most heterogeneous class of G-protein-coupled receptor agonists currently known for the innate immune system.
Introduction
The initial sensing of infection depends on innate pattern recognition receptors (PRRs)3 that recognize evolutionarily conserved structures of microorganisms known as pathogen-associated molecular patterns (PAMPs). Toll-like receptors represent a prime example of such PRRs (1, 2). Formyl peptide receptors (FPRs) belong to a class of G-protein-coupled receptors (GPCRs) involved in host defense against pathogens in the innate immune system (3–5). FPR function is best known in phagocytic leukocytes (e.g. neutrophils and monocytes) that, in response to microbial chemoattractants, migrate and accumulate at sites of infection, where they release reactive oxygen species (ROS) and other factors to combat invading microorganisms (6–8). Activation of FPRs triggers classical GPCR signaling cascades involving G-protein-dependent phospholipase C stimulation, leading to intracellular Ca2+ mobilization (9–11). Humans are known to express three FPR genes, FPR1, FPR2, and FPR3, but this number varies across mammalian species (12, 13).
FPRs have been proposed to function as PRRs (5, 7, 14, 15), but their pathogen-associated molecular pattern remains unclear. In fact, one of the most puzzling features of the FPR family is its unusually high degree of molecular promiscuity (3, 7, 16). Although FPRs have been named according to their capability to detect formylated peptides (5, 17), these receptors can recognize structurally diverse agonists with no obvious common pattern in amino acid sequence or natural origin (3, 16). Such ligands include N-formylated, C-amidated, and unmodified peptides from bacterial and viral pathogens as well as host-endogenous mitochondrial peptides and several non-peptide agonists, such as resolvin D1 and lipoxin A4 (3, 5, 18). FPRs detect a wide range of structurally diverse pro- and anti-inflammatory ligands associated with important human diseases, such as amyloidosis, Alzheimer disease, HIV, and inflammatory pain (19, 20).
Although FPR expression has been initially described in immune cells, it is becoming increasingly clear that FPRs are also expressed in other cell types and tissues, from the nervous system (13, 21–23) to internal organs, including lung and gut (24, 25), suggesting that FPRs could be generally involved in the sensing and management of the microbiome of the body. An important development has been the finding that a set of FPR-like proteins represents a distinct receptor family in chemosensory neurons of the mouse vomeronasal organ (13, 23) and that some of these neurons recognize formylated peptides (23, 26), suggesting an evolutionary link between recognition mechanisms in immune cells and subsets of sensory neurons of the vomeronasal organ (27–29).
To gain new insight into the function and recognition capabilities of FPRs and to better understand the molecular promiscuity of these receptors, we investigated conserved features in the structure of disparate peptide agonists required for activation of human and mouse FPRs. Through this approach, we generated critical knowledge leading to the discovery of an unsuspected, extremely large family of natural FPR agonists with common structural properties: bacterial signal peptides and their short breakdown products. Signal peptides are N-terminal protein signatures that are required for directing the transfer of bacterial proteins through the plasma membrane, during which they are cleaved off to give rise to the native form of membrane-associated or secreted proteins (30, 31). We observed a remarkably high degree of sensitivity, selectivity, and functional conservation of signal peptide recognition across species and receptor subtypes for FPR1 and FPR2, strongly arguing for an important role of this novel agonist family during evolution of mammalian FPR function. Dynamic measurements in human and mouse innate immune cells demonstrate that all tested signal peptides are recognized by these cells and trigger classical innate immune responses, such as intracellular Ca2+ mobilization, generation of reactive oxygen species, release of metallopeptidase, and chemotactic cell migration. These observations argue that mammalian FPRs may have evolved originally as germ line-encoded pattern recognition receptors that recognize structurally conserved export motifs of bacterial signal sequences as their cognate, pathogen-associated molecular pattern.
EXPERIMENTAL PROCEDURES
Cloning of FPR Genes
All FPR genes were cloned as described previously (32). Sequences of the coding regions are annotated in supplemental Table S1.
Cell Culture and Transient Transfection
HEK293T PEAKrapid cells (ATCC) were cultivated as described previously (32). For immunostaining and intracellular Ca2+ measurements, cells were seeded at 20–30% confluence on poly-d-lysine-coated 96-well μ-clear plates (Greiner). Cells were grown to 50–70% confluence and transfected with equal amounts of receptor and G-protein subunit Gα16 using jetPEI (PeqLab) according to the manufacturer's protocol.
Isolation and Cultivation of Human Monocytes
Peripheral blood leukocytes enriched in leukocyte-reducing system chambers were kindly provided by the Institute of Clinical Hemostaseology and Transfusion Medicine, University of Saarland School of Medicine. Monocytes were isolated by Ficoll (PAA Laboratories) density gradient centrifugation in Leucosep filter columns (Invitrogen) according to the manufacturer's protocol. Following centrifugation, leukocytes were washed with HBSS (Sigma), erythrocytes and thrombocytes were lysed by resuspension in lysis buffer (in 155 mm NH4Cl, 10 mm KHCO3, and 0.13 mm EDTA in H2O, pH 7.3) for 2 min. Cells were harvested by centrifugation, and the pellet was rinsed with HBSS and resuspended in PBS with 0.5% BSA. Subsequently, monocytes were further enriched through an adhesion protocol. Cells were first kept in standard culture flasks containing RPMI 1640 with 10% FCS and 1% penicillin/streptomycin. After 2 h, the medium was exchanged to remove non-adhesive cells. The remaining cell fraction was further incubated overnight in culture medium. On the following day, cells were rinsed with PBS plus 0.5% BSA, scratched from the flask surface, and seeded in Ultra-Low cluster 24-well plates (Corning Inc.) at a density of 1–4 × 106 cells/ml in culture medium. After an additional overnight incubation, monocytes were harvested by gentle trituration and seeded at 30,000 cells/well on uncoated 96-well μ-clear plates (Greiner).
Isolation of Human Granulocytes
10 ml of blood from healthy adult volunteers was collected into S-Monovette Li-Heparin® tubes (SARSTEDT). Subsequently, 17 ml of cold Ca2+/Mg2+-free Dulbecco's PBS (Invitrogen) was added. The cell suspension was carefully overlaid by 13 ml of Ficoll (FicoLIte-H, Linaris) and centrifuged at 560 × g for 25 min at room temperature. The granulocyte-containing phase was transferred to a 50-ml Falcon tube, the volume was adjusted to 25 ml with Dulbecco's PBS, and the cell suspension was mixed with 25 ml of 3% T500-dextran solution (Pharmacosmos A/S). After 15–30 min at room temperature, the supernatant was transferred to a new tube, adjusted to 50 ml with Dulbecco's PBS, and centrifuged (6 min at 339 × g at 4 °C). The supernatant was removed, and the cell-pellet was resuspended by gentle swirling. For erythrocyte lysis, the pellet was gently resuspended in 5 ml of sterile water and immediately mixed with 45 ml of Dulbecco's PBS. After centrifugation (10 min at 339 × g at 4 °C) the supernatant was removed, and the lysis procedure was repeated. The pellet was gently resuspended in RPMI 1640 (Invitrogen) supplemented with 2 mm l-glutamine, 1% penicillin/streptomycin, 1% sodium pyruvate, and 1% autologous human serum.
Isolation of Mouse Leukocytes
Three different mouse strains were used in these experiments: 1) wild type mice (C57BL/6J); 2) mice harboring a global deficiency in Fpr1 (C57BL/6NTac-Fpr1Tm1GaoN6, denoted as Fpr1−/− mice) (Taconic) (12); and 3) their heterozygous littermate controls (Fpr1+/−). To obtain sufficient quantities of cells from these strains, leukocytes were extracted from bone marrow (33). Mice were killed, and femur and tibia from both hind legs were isolated. The distal tips of each bone were cut off, and the bone marrow was obtained by forced rinsing with ice-cold Ca2+- and Mg2+-free HBSS buffer. The cell suspension was filtrated with a 100-μm cell strainer (BD Biosciences) and then centrifuged (300 × g for 8 min at 4 °C). After removal of the supernatant, cells were resuspended in HBSS containing 1 mm Ca2+, 1 mm Mg2+, and 2 μm Fluo4-AM. Approximately 150,000 cells/well were seeded on μ-clear plates (Greiner) and incubated for 45 min at room temperature. Cells were rinsed two times with HBSS before Ca2+ recordings were performed. Staining with nuclear dyes and Ly6G antibody (a neutrophil marker) showed that this leukocyte preparation contains 50% mature neutrophils in wild type, Fpr1+/−, or Fpr1−/− mice. When we isolated mouse neutrophils directly from peripheral blood using Ly6G beads (pluriSelect), the cell number was reduced by 10-fold, but these cells showed comparable Ca2+ responses.
Immunocytochemistry
Cells were seeded in 96-well plates as described above. For cell surface expression studies, cells were fixed for 4 min in methanol-free paraformaldehyde (Polysciences Inc.), 4% in PBS. For immunostaining, cells were blocked with 5% heat-inactivated FCS in PBS for 30 min and incubated overnight with the following monoclonal primary antibodies: anti-human FPR1 mouse IgG2A (R&D Systems, MAB3744, 1 μg/ml); mouse IgG2A isotype control (R&D Systems, MAB0031, 1 μg/ml); anti-human CD14 mouse IgG1,κ (Biolegend, 325601, 0.5 μg/ml); mouse IgG1,κ isotype control (Biolegend, 400101, 0.5 μg/ml). Staining was obtained by 45-min incubation with a polyclonal donkey anti-mouse Alexa Fluor 555 antibody (Invitrogen, 2 μg/ml) and a counterstaining of cell nuclei (Hoechst, 33342, 1 μg/ml). All solutions were diluted in PBS containing 5% heat-inactivated FCS. Images were taken with the BD Pathway Bioimager 855 imaging system (BD Biosciences) and quantified either with BD-image Explorer software (BD Biosciences) or ImageJ version 1.46r (32). Monocytes were treated as described above for HEK cell experiments except that 3% methanol-free paraformaldehyde was used. To correlate Ca2+ responses with receptor expression, we used post hoc antibody staining. Cells were fixed for 5 min by the addition of 2% paraformaldehyde immediately following the experiment to prevent receptor internalization.
Calcium Imaging
For automated high throughput Ca2+ measurements of cell populations using a fluorescence imaging plate reader system (Molecular Devices), mean Ca2+ changes of cell populations (HEK293T, ∼50,000 cells/well; human monocytes, ∼30,000 cells/well; human granulocytes, 100,000 cells/well; mouse leukocytes, 150,000 cells/well) were recorded essentially as described previously (32). Briefly, HEK cells were loaded 48 h post-transfection with 2 μm Fluo4-AM (Molecular Probes) in C1 solution (130 mm NaCl, 10 mm HEPES, 5 mm KCl, 2 mm CaCl2, 5 mm glucose, pH 7.2) at room temperature for 2 h. Monocytes were loaded in Ringer for 1 h, whereas human and mouse granulocytes were loaded for 45 min in RPMI or HBSS, respectively. For automated high content Ca2+ imaging of human monocytes and HEK cells with single-cell resolution, we used the BD Pathway Bioimager 855 system (BD Biosciences) (32). Cells were loaded with 2 μm Fura2-AM (Molecular Probes) for 1 h at room temperature. Ca2+-dependent fluorescence changes of cells were recorded at 0.5 Hz and quantified either with BD-image Explorer software (BD Bioscience) or ImageJ version 1.46r. Peak values of stimulus-evoked Ca2+ transients were determined and correlated with the fluorescence intensity of post hoc antibody staining for each single cell. A Ca2+ response was defined as an increase of the excitation ratio 340 nm/380 nm that was at least 4 times higher than the mean baseline noise after bath application.
Matrix Metallopeptidase Release
The release of matrix metallopeptidase 9 (MMP-9) from human granulocytes was determined using the Human Total MMP-9 DuoSet (R&D Systems) according to the manufacturer's protocol. 96-well plates were coated overnight with a mouse anti-human MMP-9 (1 μg/ml) antibody. On the following day, the plates were first incubated with 50 μl/well of the test samples for 2 h, followed by a 2-h incubation with a biotinylated goat anti-human antibody (100 ng/ml) and a 20-min incubation with streptavidin-HRP (1:200). For detection, a tetramethylbenzidine-containing substrate solution was added. The reaction was stopped after 20 min, and the optical density was determined with a microplate reader (2030 Multilabel Reader Victor X4, PerkinElmer Life Sciences) at 450 nm excitation and a wavelength correction set to 540 nm. Measurements were always carried out as duplicates. MMP-9 concentrations were determined via concentration standards.
Chemotactic Cell Migration
Granulocyte migration assays were performed in duplicates using 96-well HTS transwell chambers (Corning Inc.) with 3-μm pore size according to the manufacturer's protocol. Directly after isolation, 100,000 cells/well were seeded in the upper chamber in 75 μl of RPMI medium and subsequently maintained for 2 h in a cell culture incubator. Thereafter, the number of transmigrated cells was determined by automated counting using the BD Pathway Bioimager 855 system.
Ligands
Custom made peptides were routinely synthesized by VCPBIO LAB, United Peptides, and Genscript Corp. The sources of commercially available agonists and antagonists are listed in Table 1. Purity of ligands was typically >95%. Agonists were routinely dissolved in C1 buffer or in Ringer as a 0.2–1 mm stock solution. Strongly hydrophobic substances were dissolved as 10 mm stocks in DMSO (99.7%, Sigma). Stock solutions were routinely stored in small aliquots at −20 °C until use. More details on sequence, purity, source, solvents, and storage conditions are given in Table 1. To increase solubility, the highest concentration of the working aliquot was routinely heated for 3 min to 80 °C prior to further dilutions.
TABLE 1.
Source, sequence, and purity of the FPR agonist and antagonists used in this study
Peptide sequences of all essential amino acids are given in one-letter code. Capital letters denote an l-isomer; lowercase letters denote a d-isomer. hcy, homocysteine; orn, ornithine. N- and C-terminal modifications are abbreviated as follows. f, formylated N terminus; CONH2, C terminus with amidation; CHO, aldehyde group; COOCH3, methyl ester group; Ac, acetylation.
| Ligand | Structure | Storage | Source and purity | Solvent |
|---|---|---|---|---|
| Organic compounds | ||||
| AG09/1 | C16H14N4O4S | 4 °C | Sigma, ≥98% | DMSO |
| A14 | C23H20N2O5 (*1/4 H2O) | Ambient | Tocris, >99% | DMSO |
| W-peptide library | ||||
| W-peptide/WKYMVm-CONH2 | WKYMVm-NH2 | −20 °C | VCPBIO, >96.55% | C1 |
| l-W-peptide | WKYMVM-NH2 | −20 °C | Tocris, 99.2% | C1 |
| M-peptide | MMHWAm-NH2 | −20 °C | GenScript, >99.6% | C1 |
| l-M-peptide | MMHWAM-NH2 | −20 °C | GenScript, >96.8% | C1 |
| Rev-W-peptide | f-MVMYKW | −20 °C | GenScript, >97.8% | C1 |
| Rev-M-peptide | f-MAWHMM | −20 °C | VCPBIO, >98.47% | C1 |
| Library peptide 1 | AAAWKYMVm-NH2 | −20 °C | GenScript, >98.8% | C1 |
| Library peptide 2 | AAWKYMVm-NH2 | −20 °C | GenScript, >96.3% | C1 |
| Library peptide 3 | AWKYMVm-NH2 | −20 °C | GenScript, >98% | C1 |
| Library peptide 4 | KYMVm-NH2 | −20 °C | GenScript, >99% | C1 |
| Library peptide 5 | YMVm-NH2 | −20 °C | GenScript, >99% | C1 |
| Library peptide 6 | MVm-NH2 | −20 °C | GenScript, >95% | C1 |
| Library peptide 7 | Vm-NH2 | −20 °C | GenScript, >95% | C1 |
| Library peptide 8 | WKFMVm-NH2 | −20 °C | GenScript, >96.7% | C1 |
| Library peptide 9 | WKKMVm-NH2 | −20 °C | GenScript, >96.8% | C1 |
| Library peptide 10 | WKEMVm-NH2 | −20 °C | GenScript, >97.2% | C1 |
| Library peptide 11 | WKQMVm-NH2 | −20 °C | GenScript, >96.7% | C1 |
| WKYMVm-COOCH3 | WKYMVm-COOCH3 | −20 °C | Thermo Fisher, >99.07% | C1 |
| WKYMVm-CHO | WKYMVm-CHO | −20 °C | American Peptide Co., 86.9% | C1 |
| WKYMVm-COO- | WKYMVm | −20 °C | GenScript, >97% | C1 |
| Library peptide 12 | WKYMVi-NH2 | −20 °C | GenScript, >97.3% | C1 |
| Library peptide 13 | WKYMV[hcy]-NH2 | −20 °C | GenScript, >95.2% | C1 |
| Library peptide 14 | WKYMVf-NH2 | −20 °C | GenScript, >98.6% | C1 |
| Library peptide 15 | WKYMVc-NH2 | −20 °C | GenScript, >95% | C1 |
| Library peptide 16 | WKYMVC-NH2 | −20 °C | GenScript, 97.2% | C1 |
| Library peptide 17 | WKYMV[orn]-NH2 | −20 °C | GenScript, >99.9% | C1 |
| Library peptide 18 | WKYMVk-NH2 | −20 °C | GenScript, >99.7% | C1 |
| Library peptide 19 | WKYMVe-NH2 | −20 °C | GenScript, >97.5% | C1 |
| Bacterial signal peptides | ||||
| Streptococcus-SP1 | f-MGFFIS | −20 °C | VCPBIO, >95.43% | C1 |
| Streptococcus-SP1 non-f | MGFFIS | −20 °C | United Biosystems, >95.49% | C1 |
| Streptococcus-SP1(2–7) | GFFISQ | −20 °C | United Biosystems, >96.13% | C1 |
| Streptococcus-SP1(7–12) | QSKQHY | −20 °C | United Biosystems, >95.23% | C1 |
| Streptococcus-SP1(20–25) | GVCSAL | −20 °C | United Biosystems, >95.23% | C1 |
| Streptococcus-SP1(32–37) | GTRVAA | −20 °C | United Biosystems, >97.09% | C1 |
| Streptococcus-SP1(32–37)-NH2 | GTRVAA-NH2 | −20 °C | United Biosystems, >99,9% | C1 |
| Streptococcus-SP1 full | f-MGFFISQSKQHYGIRKYKVGVCSALIALSILGTRVAA | −20 °C | VCPBIO, >95.18% | Ringer |
| Bacillus-SP2 | f-MKNFKG | −20 °C | VCPBIO, >96.42% | C1 |
| Staphylococcus-SP3 | f-MFIYYCK | −20 °C | VCPBIO, >97.24% | C1 |
| Salmonella-SP4 | f-MAMKKL | −20 °C | VCPBIO, >96.14% | C1 |
| Haemophilus-SP5 | f-MVMKFK | −20 °C | VCPBIO, >95.74% | C1 |
| Psychrosomonas-SP6 | f-MLFYFS | −20 °C | VCPBIO, >95.59% | DMSO |
| Shewanella-SP7 | f-MLFKYS | −20 °C | VCPBIO, >95.18% | C1 |
| Desulfotomaculum-SP8 | f-MLFYLA | −20 °C | VCPBIO, >97.04% | C1 |
| Desulfotomaculum-SP8 full | f-MLFYLALPCTLVIFFASKALYAI | −20 °C | VCPBIO, >95.59% | Ringer |
| Borrelia-SP9 | f-MLKKVY | −20 °C | VCPBIO, >95.24% | C1 |
| Vibrio-SP10 | f-MPKLNR | −20 °C | United Biosystems, >95.32% | Ringer |
| Vibrio-SP11 | f-MVKIIF | −20 °C | United Biosystems, >96.63% | Ringer |
| Staphylococcus-SP12 | f-MNKKLL | −20 °C | United Biosystems, >95.21% | Ringer |
| Clostridium-SP13 | f-MKKNLV | −20 °C | United Biosystems, >95.67% | Ringer |
| Corynebacterium-SP14 | f-MEQQNK | −20 °C | United Biosystems, >95.72% | Ringer |
| Streptomyces-SP15 | f-MVPISI | −20 °C | United Biosystems, >95.85% | Ringer |
| Hydrogenobacter-SP16 | f-MKKFLL | −20 °C | United Biosystems, >95.78% | Ringer |
| Bacillus-SP17 | f-MMKMEG | −20 °C | United Biosystems, >96.38% | Ringer |
| Listeria-SP18 | f-MKKIML | −20 °C | United Biosystems, >95.27% | Ringer |
| Desulfovibrio-SP19 | f-MKFCTA | −20 °C | United Biosystems, >95.44% | Ringer |
| Zymomonas-SP20 | f-MTNKIS | −20 °C | United Biosystems, >95.91% | Ringer |
| Neisseria-SP21 | f-MKTSIR | −20 °C | United Biosystems, >96.04% | Ringer |
| Other peptides and proteins | ||||
| f-MLF | f-MLF | −20 °C | Sigma, ≥97% | C1 |
| CO1 | f-MFINRWLFS | −20 °C | GenScript, >96,9% | DMSO |
| ND1 | f-MFFINTLTL | −20 °C | GenScript, >98,2% | DMSO |
| T20 | Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 | −20 °C | Anaspec, ≥95% | C1 |
| Ac2–26 | Ac-AMVSEFLKQAWFIENEEQEYVQTVK | −20 °C | Tocris, 95% | C1 |
| MMK-1 | LESIFRSLLFRVM | −20 °C | Tocris, 96% | C1 |
| μPAR | Ac-AVTYSRSRYLEC-NH2 | −20 °C | Anaspec, >95% | C1 |
| Humanin | MAPRGFSCLLLLTSEIDLPVKRRA | −20 °C | VCPBIO, 95.46% | C1 |
| f-Humanin | f-MAPRGFSCLLLLTSEIDLPVKRRA | −20 °C | VCPBIO, >96.02% | C1 |
| f-Humanin(1–6) | f-MAPRGF | −20 °C | VCPBIO, >95.69% | C1 |
| Humanin(1–6) | MAPRGF | −20 °C | VCPBIO, >95.11% | C1 |
| Mouse MIP1-α | APYGADTPTACCFSYSRKIPRQFIVDYFETSSLCSQPGVIFLTKRNRQICADSKETWVQEYITDLELNA | −20 °C | Peprotech, >98% | Ringer |
| RANTES | SPYSSDTTPCCFAYIARPLPRAHIKEYFYTSGKCSNPAVVFVTRKNRQVCANPEKKWVREYINSLEMS | −20 °C | Peprotech, >98% | Ringer |
| Antagonists | ||||
| t-Boc2 | N-tert-butoxy-FlFlF | −20 °C | Bachem, 98% | DMSO |
| Cyclosporin H | C62H111N11O12 | −20 °C | Santa Cruz Biotechnology, 95% | DMSO |
Data Analysis
Experiments were routinely performed as duplicates and averaged over at least three independent transfections/donors. Response amplitudes (ΔF/F0) were calculated by dividing maximal fluorescence change after ligand application by baseline fluorescence. Maximal amplitudes were set to 100%, and curves were calculated with GraphPad Prism 5.0 using the equation for sigmoidal dose response with variable slope. S.D. values were calculated as averages from independently obtained EC50 values using the STDEV.P function of Microsoft Excel 2010. Single-cell imaging videos of monocytes and analysis of post hoc immunochemistry were done with ImageJ version 1.46r. All cells responding to Ringer bath application (<5%) were excluded from evaluations. Post hoc correlations of Ca2+ signals and receptor staining were plotted with Origin version 8.6.
Estimating the Size of the Ligand Family
To provide an estimate of the size of the signal peptide ligand family, we tested a set of 12 randomly selected signal sequences. All peptides were chosen independently of their amino acid sequence among all 1,168 experimentally confirmed signal peptides present in the database (see the Signal Peptide Database Website). We found that all 12 hexapeptides were activators of FPRs. We then calculated the probability to obtain this result by chance using the basic urn model. In this approach, we randomly drew 12 peptides and assumed different percentages of FPR activators among the total number of peptides in the library. We calculated the probability for a range from 100 to 70% of FPR activators using the formula, x12 = p (where x denotes the percentages of FPR agonists and p denotes the probability that this ratio is obtained by chance). If we assume that 90, 80, or 70% of the sequences are agonists, the respective p values for an x of 90, 80, or 70% are 0.9012 = 0.28, 0.8012 = 0.07, and 0.7012 = 0.01. Thus, the respective probability to obtain our result by chance is 28, 7, or 1%, respectively. Calculation of the 95% confidence interval (0.7812 = 0.05) leads to the prediction that 78% of the peptides in the database should be FPR activators with a confidence level of 95%.
Fluorescence-based Hydrogen Peroxide Measurement
Monocyte-dependent hydrogen peroxide (H2O2) production was measured using H2O2 reactive Amplex®UltraRed (Molecular Probes) that forms the fluorescent dye Resorufin during the reaction. Signals were recorded at 535 nm excitation and 590 nm emission in a Tecan GENios Pro plate reader with bottom reading settings using black 96-well plates (Greiner). All measurements were performed as duplicates with ∼30,000 monocytes per well in Ringer supplied with 50 μm Amplex®UltraRed, 0.1 unit/ml horseradish peroxidase and 10 units/ml superoxide dismutase. H2O2 concentrations were calculated from relative fluorescent units 10 min after application using calibration curves.
Peptide Modeling
The secondary structure of the peptides was modeled with the molecular graphics software package Accelrys Discovery Studio version 1.6 and Visualizer version 3.5 installed on a PC. For comparison, selected structures were also modeled with QUANTA (Release 4.1, MSI) running on a Silicon Graphics Indy work station. For the modified groups (e.g. formylation and amidation) the atom types were adequately adjusted. Each structure was minimized in energy by the implemented Dreiding or charmm23 force field using the “steepest descent” algorithm until a threshold was achieved. Selected structures were further processed by molecular dynamics calculations (QUANTA) to monitor structural flexibility (34). The minimal energy structures of the various peptides were superimposed by fit of C-a pairs (formylated or amidated peptides) or by tethers on backbone atoms (formylated and amidated cases) for minimal root mean square superposition. For comparison of formylated and amidated peptides, rotamers of the side chains were selected to achieve close similarity of side chain orientation. These structures were again minimized in energy. From the NMR structures of f-MLF (Protein Data Bank entry 1Q7O), one conformational state was selected and minimized in energy after editing atom types. This structure was overlaid on the first α-turn of a reference peptide. The side chain orientations were adjusted by rotation around freely rotatable bonds. The methionine (FME1) was rotated by 184°, and for MTY3, the phenyl group was rotated around N-CA by 64° and around CA-CB by 58°. After minimization, both structures showed nearly identical energies (51.19 kcal/mol versus 51.52 kcal/mol). The adjusted and minimized structure was used for final overlay to the reference peptide. To visualize the space-filling of the relevant side groups, a transparent soft solvent surface was rendered for each peptide. Chemical structures of ligands were drawn with Accelrys Draw version 4.1 (Accelrys).
Electron Paramagnetic Resonance Spectrometry
These experiments were performed with a Bruker spectrometer (ESP300e) equipped with a standard 4102ST cavity, which holds the capillary support quartz glass finger. The glass finger temperature was controlled by a BioIII-TGC device (Noxygen) and set to 37 °C for work with cells. The modulation amplitude was set to 0.1 millitesla, and the microwave power was set to 20 milliwatts as a standard condition for all experiments. Spectra were recorded with scan times of 60 s and stored consecutively to monitor the kinetic behavior of the signal. All experiments were performed in Ringer's buffer at 37 °C with an oxygen concentration of about 200 μm. The activity of monocytes was initially tested with phorbol ester (1 μm). The redox-activated cyclic hydroxylamine spin trap 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH) was added last in 300 μm concentration for all experiments to monitor superoxide production. 1 × 105 to 2.5 × 105 monocytes/experiment were stimulated with the designated peptides and immediately transferred into a 50-μl glass capillary in the capillary holder. The reduction of CMH by superoxide leads to formation of a three-line EPR signal typical for the nitroxide-centered radical. The temporal evolution of the CMH-radical signals was evaluated with the in-house program Medeia, which automatically determines the double integral of one or several lines of the radical visible in the time series with a resolution of 1 s. In the absence of line width changes and saturation effects, the information can be translated to radical concentration by comparing it with the stable nitroxide radical TEMPOL at 100 μm. The output data were further processed with Origin version 8.5 for evaluation and presentation. In control experiments, each component of the assays and CMH were tested for unwanted radical production. The superoxide radical was identified as the reacting species by suppression of the CMH signal by superoxide dismutase (100 units/ml) as a scavenger.
RESULTS
Structurally Dissimilar Peptide Agonists Imply a Conserved FPR Recognition Mechanism
To investigate molecular mechanisms underlying FPR ligand recognition and activation, we first used heterologous expression of 13 FPRs from six mammalian species in combination with high-throughput measurements of intracellular Ca2+ mobilization and examined ligand-induced activation (32). This strategy enabled us to systematically investigate the effects of a large number of combinations between potential ligands and receptors.
Initial experiments revealed that two peptides exhibiting highly divergent chemical structures, MMHWAm (M-peptide) (35) and WKYMVm (W-peptide) (36) (Fig. 1A), that carry an amidated d-methionine at their C terminus and their stereoisomers with an amidated l-methionine caused surprisingly similar activation of mFpr-rs1, one of the FPRs expressed in the mouse vomeronasal organ, with respect to concentration, stereoselective preference, and the relative potency of d- versus l-isoforms (Fig. 1, B–D). Recognition capabilities toward M- and W-peptide seemed to be highly conserved between different receptor subtypes and different species. Both peptides activated FPR1 from mouse, human, rat, dog, rabbit, and hamster (Fig. 1E). Conserved activation patterns extended also to human and mouse FPR2 as well as rFpr-rs2a, rFpr-rs2b, and rFpr-rs2c from rats (Fig. 1E). Thus, recognition capabilities of distinct FPRs from different species for these structurally divergent synthetic peptides were closely similar, resembling that of the best known natural ligand f-MLF, whereas the response profile to another synthetic peptide agonist, MMK-1, showed very little correlation (Fig. 1E). Further support for a strong degree of functional conservation came from concentration-response measurements of W- and M-peptide using human and mouse FPR1 and FPR2 (Fig. 1, F and G).
FIGURE 1.

Activation by disparate peptides suggests a conserved FPR recognition mechanism. A, chemical structures of W- and M-peptide. Amino acid sequences are given in one-letter code. Both peptides contain a C-terminal d-methionine with an amidation (m-NH2). In the l-isoform of both peptides the d-methionine is replaced by an amidated l-methionine. B, representative single cell imaging traces demonstrating relative cytosolic Ca2+ changes of HEK293T cells transiently transfected with mFpr-rs1. Red traces, cells responding to M-peptide and W-peptide; gray traces, cells not responding to both stimuli; black trace, mean response. x scale, 1 min; y scale, 10% increase in A340/380 ratio. Cells were extensively rinsed for 3 min with 20-fold bath exchange prior to the next stimulus application. Similar results were obtained in three independent experiments. C, percentage of M-peptide-responding cells that also responded to a second stimulus. Data are representative of three experiments. Numbers above each bar show the total amount of cells responding to a given stimulus. Note that 97% of the M-peptide-responding cells also reacted to W-peptide, whereas responses to typical agonists for immune FPRs were below 3%. Error bars, S.D. D, mean concentration-response curves of mFprs-rs1 activation by W-peptide (green) or M-peptide (red). Error bars, S.D.; experimental numbers are given in parentheses. E, Ca2+ peak responses representing activation of FPRs from six different species upon stimulation with M-peptide, W-peptide, f-MLF, and MMK-1 (each at 10 μm). Similar results were obtained in three independent experiments. F, concentration-response curves of human (green) and mouse (black) FPR1 and FPR2 activation by W- and M-peptide. Curves denote mean responses from 3–9 independent experiments. Error bars, S.D. G, concentration-response curves of hFPR2 activation by W-peptide (green) and M-peptide (red).
Thus, despite major structural differences between the M- and W-peptide, they activate some FPRs in a strikingly similar manner, suggesting shared structural properties that could not be deduced from their amino acid sequences. This high level of conservation across different species is unexpected because there should be no evolutionary pressure in mammals toward maintaining sensitive detection of these synthetic molecules. Hence, we reasoned that these findings could reflect the existence of a conserved recognition mechanism for a family of natural FPR agonists exhibiting mutual structural features.
A Conserved Secondary Structure of FPR Peptide Agonists
To define the conserved structural features essential for agonist activity, we next used a combination of experimental and molecular modeling approaches. We previously identified a core agonist motif for mFpr-rs1 activation (32). To explore whether a similar agonist motif is recognized by immune FPRs, we performed a detailed comparison of all five human and mouse immune FPRs with mFpr-rs1 by analyzing EC50 values derived from 343 distinct concentration-response experiments to stimulation with W-peptide and 22 W-peptide derivatives (Fig. 2A).
FIGURE 2.

Key residues in the sequence of W-peptide required for FPR activation. A, EC50 values (in nm, with S.D.) obtained from concentration-response curves of hFPR1, hFPR2, hFPR3, mFpr1, mFpr2, and mFpr-rs1 for different W-peptide derivatives. The number of experiments (n) is given in parentheses. The color code indicates the loss in receptor sensitivity relative to the W-peptide response (gray): light blue, 6–15-fold reduced activity; green, 25–600-fold reduced activity; dark blue, ≥1,000-fold reduced activity. B, key residues in W-peptide that are important for activation of human and mouse FPRs. The analysis of dose-response experiments using 22 W-peptide derivatives identified residues at C3, C4, and C6 as crucial for agonist-receptor interaction. The importance of residues for receptor activation is indicated by color code and letter size. C3, C4, and C6 denote the distance of the positions from the N-terminal residue of W-peptide. Peptide sequences of all essential amino acids are given in one-letter code. Capital letters denote an l-isomer; lowercase letters denote a d-isomer. hcy, homocysteine; orn, ornithine. N- and C-terminal modifications are abbreviated as follows. f, formylated N terminus; CONH2, C terminus with amidation; CHO, aldehyde group; COOCH3, methyl ester group.
For hFPR1, hFPR2, mFpr1, and mFpr2, the results revealed remarkably potent activation by almost all tested peptides (Fig. 2A). By contrast, hFPR3 and mFpr-rs1 reacted to fewer peptides and, if they reacted, responses were less sensitive (Fig. 2A). Despite these differences, through systematic variation of the W-peptide, we arrived at the conclusion that all six receptors detect similar agonist key positions (Fig. 2B). Both human and mouse FPR1 and FPR2 tolerate N-terminal elongations by three amino acids and deletions by two residues without a strong loss of agonist potency (Fig. 2A). Deletion of the third and fourth N-terminal residue drastically reduced the agonist potency for both FPRs. This suggests that these amino acids are especially important for receptor-ligand interactions. Furthermore, d-methionine at C6, methionine at C4, tyrosine at C3, and the C-terminal amidation (CONH2) all critically influence agonist potency. Nonetheless, a considerable amount of structural variation is tolerated at the C3 and C4 positions (Fig. 2A). However, larger residues are preferred, suggesting that these amino acids preferentially contribute to van der Waals interactions. By contrast, little variation at C6 is permitted. This implies that methionine at C6 is a key element in the agonist structure. The C-terminal amide group (CONH2) represents another key structure because its removal results in a strong loss of agonist potency (Fig. 3A). Surprisingly, replacement of this CONH2 group by methyl ester (COOCH3) or aldehyde (CHO) did not drastically change the agonist potency (Fig. 3B). This argues against a direct interaction of the amine (NH2) with the receptor and favors a model in which NH2 influences the charge properties of the adjacent carbonyl group (C=O). A C=O group with similar donor/acceptor properties is also contained in the methyl ester and aldehyde derivative (Fig. 3, C and F). In the drastically less potent COO− peptide, the C=O donor/acceptor properties are clearly altered through the negative charge of this group (Fig. 3D).
FIGURE 3.

A conserved secondary structure of FPR peptide agonists. A, mean concentration-response curves of hFPR1 (red) and mFpr1 (black) activation by W-peptide and a derivative lacking C-terminal amidation. Experimental numbers are given in parentheses. Error bars, S.D. B, mean concentration-response curves to derivatives in which the amidation at the C-terminal methionine was replaced by a methyl ester or an aldehyde. C, chemical structure of methionine carrying a C-terminal amide or methylester. D, structure of methionine with a C-terminal COOH group without modifications. E, comparison between the predicted three-dimensional structure of M- (red) and W-peptide (green). Both peptides have similarities in their spatial orientation, shape, and chemical properties that cannot be easily predicted from their primary sequences. The residues at C3, C4, and C6 form a predominantly hydrophobic trilobular structure, indicated by a gray dotted line. F, structural similarities between methionine carrying a C-terminal aldehyde group (blue circle) and N-terminally formylated methionine (red circle). G, comparison between the predicted three-dimensional structure of W-peptide (green) and a formylated derivative with reversed amino acid sequence (rev-W-peptide, blue). The C-terminal methionine of W-peptide is aligned with the N-terminal methionine of rev-W-peptide. Despite large variations in linear amino acid chain key residues, both peptides show a similar spatial orientation, forming a characteristic trilobular structure indicated by the dotted line. H, comparison of the effects of W-peptide (green) and rev-W-peptide (blue) on the activation of mFpr1 (top curves) and hFPR1 (bottom curves). I, comparison between the three-dimensional structure of rev-W-peptide based on our modeling (blue) and an experimentally determined NMR structure for f-MLF (left, cyan); the N terminus of the mitochondrial protein NADH reductase subunit 1 (ND1, middle, pink), a potent FPR agonist; and the N terminus of the neuroprotective mitochondrial protein humanin (right, yellow), a less potent FPR agonist. Note that the C3 lobe in humanin appears considerably smaller. J, representative Ca2+ responses of mouse and human immune FPRs to the formylated N terminus of the mitochondrial protein NADH reductase subunit 1 (ND1). y scale, 100,000 relative fluorescent units/square; x scale, 3 min/square). Similar results were obtained in four independent experiments. K, representative Ca2+ responses of human and immune FPRs to the formylated (left) or unformylated (right) N terminus of humanin. y scale, 50,000 relative fluorescent units/square; x scale, 3 min/square. Similar results were obtained in three independent experiments.
We next compared M- and W-peptide at the key residues at C6, C4, and C3. Both peptides have an amidated methionine (m-CONH2) at C6, further supporting our hypothesis that this residue is the central element. M-peptide exhibits tryptophan instead of Met at C4 and histidine instead of tyrosine at C3. Despite considerable structural variations, all of these residues have a large molecular surface that permits van der Waals interactions. This indicates that the residues at C3 and C4, together with Met at C6, may form a spatially conserved three-dimensional structure instead of a linear motif. We therefore employed three-dimensional modeling to investigate the spatial orientation of key residues between W- and M-peptide. Assuming that these peptides form an α-turn, the most frequent conformation occurring in similar peptides (37), alignment of both peptides revealed that the residues at C4 and C3 are indeed closely overlapping (Fig. 3E). Tyr at C3 of W-peptide has a similar spatial orientation and molecular surface as His at C3 of M-peptide. At C4, the non-polar residues Trp and Met are in close proximity. Together, the three key residues at C6, C4, and C3 form a predominantly hydrophobic tripartite structure that is oriented symmetrically around a carbonyl group in a clawlike fashion. Interestingly, an N-terminally formylated Met resembles a C-terminal met with an aldehyde substitution (Fig. 3F).
The symmetrical organization of this motif (Fig. 3E), together with our finding that C-terminal CHO and CONH2 probably act through the C=O group, suggest a mechanism in which an amidated peptide first interacts with the receptor binding pocket via its C-terminal Met, whereas a formylated peptide binds first through its N terminus. Comparison of the model structure of W-peptide with a de novo peptide in which the amino acid sequence is reversed (f-MVMYKW or rev-W-peptide) demonstrates that the three N-terminal residues at C1, C3, and C4 of this peptide form a similar hydrophobic tripartite structure as that formed by C6, C4, and C3 at the C terminus of W-peptide (Fig. 3G), predicting that rev-W-peptide should function as a potent FPR activator. When we tested this, we found indeed nearly identical EC50 values for this peptide on hFPR1 and mFpr1 (Fig. 3H). With an EC50 of 0.19 ± 0.13 (n = 3) for hFPR1 and 0.09 ± 0.12 (n = 6) for mFpr1, the reversed form of M-peptide (rev-M-peptide or f-MAWHMM) was even ∼100-fold more potent than M-peptide that had EC50 values of 205 ± 25 (n = 3) for hFPR1 and 11 ± 6.9 (n = 5) for mFpr1. Together, these results demonstrate that the position of certain functional groups and their spatial arrangement are more important for FPR-ligand interactions than their linear amino acid sequences. These results offer a plausible mechanistic explanation for our finding that FPRs can be activated with equal sensitivity by sequence-divergent peptides that are either N-terminally formylated or C-terminally amidated.
Importantly, the structural constraints deduced from our analysis of synthetic peptides apply also to natural FPR ligands. We compared the NMR structure of f-MLF (38) (PDB entry 1Q70) with the model of rev-W-peptide and found clear similarities in shape and spatial orientation (Fig. 3I). Three-dimensional modeling using two known mitochondrial FPR agonists, humanin (39) and ND1 (40), revealed a tripartite clawlike motif at the N terminus of ND1 and humanin that showed clear structural similarities to our synthetic peptides (Fig. 3I). Subsequent functional measurements indeed confirmed that peptide fragments containing the N terminus of humanin and ND1 activate human and mouse FPRs (Fig. 3, J and K), and, as expected, the addition of an N-terminal formyl group substantially improved the potency of humanin (Fig. 3K). Hence, we hypothesized that these results define a common structural basis, or molecular signature, for FPR activation that could be used by FPRs to detect natural molecules associated with the presence of pathogens.
Bacterial Signal Peptides Constitute a Novel Family of FPR Agonists
Guided by the results illustrated in Figs. 1–3, we proposed the existence of an as yet undiscovered family of natural FPR ligands that are most likely released by bacteria and had a significant impact on shaping FPR structure and function during mammalian evolution. Our strategy to identify such ligands was based on the following assumptions. 1) They are likely to be short peptides with variable amino acid sequence and a minimal length of three amino acids. 2) Their N terminus should consist of a formylated methionine, or their C terminus should terminate with an amidated methionine. 3) The second and/or third residue next to this methionine should comprise amino acids that preferentially allow for van der Waals interactions. 4) These ligands should possess a well defined secondary structure, probably containing an α-turn, which resembles the trilobular motif identified above. 5) The agonist structure should be highly conserved across many different pathogens, and such pathogens should affect a wide range of mammalian species, including humans and mice.
These predictions enabled us to identify classical bacterial signal peptides, normally used to target newly synthesized proteins to the membrane transport machinery, as novel natural agonists of human and mouse FPRs (Figs. 4 and 5 and Tables 2 and 3). Such signal peptides are variable in their amino acid sequence but have a conserved secondary structure that is largely α-helical (41). They contain three typical domains: a 3–6-amino acid-long N-terminal region (n) starting with a methionine; an α-helical hydrophobic h-region (h); and a c-region (c) containing a conserved signal peptidase (SPase) recognition motif (Fig. 4A) (30, 41).
FIGURE 4.

Bacterial signal peptides activate hFPR1 and hFPR2. A, signal peptides dependent on the Sec protein translocation pathway have a typical motif permitting a high degree of sequence flexibility in combination with a conserved three-dimensional topology. Bacterial signal peptides consist of an n-region (blue) with an N-terminal formylated methionine, an α-helical hydrophobic h-region (green), and a c-region (red) containing a conserved signal peptidase (SPase) recognition motif. B, comparison of the calculated van der Waals surface between the first six amino acids of a signal peptide (purple) and the de novo agonist rev-W-peptide (blue). C, the N-terminal hexapeptide fragment from the signal peptide of Streptococcus suis hyaluronidase elicits robust dose-dependent Ca2+ transients in HEK cells transfected with human and mouse FPR1 or FPR2. y scale, 100,000 relative fluorescent units/square; x scale, 3 min/square. D, specificity and sensitivity of hFPR1 (circles, solid lines) and hFPR2 (squares, dotted lines) to structurally diverse N-terminal signal peptide fragments as observed through mean concentration-response curves. Numbers of experiments are given in parentheses. Error bars, S.D. E, mean concentration-response curves of hFPR1 (full circles) and hFPR2 (full squares, dotted lines) activation by three signal peptides starting with an f-MLF motif. F, mean concentration-response curves of hFPR1 activation by N-terminal hexapeptide fragment (blue squares) versus the full-length signal peptide of Streptococcus-SP1 comprising 36 residues (gray circles). G, mean Ca2+ peak responses (n = 3) of different human and mouse FPRs to stimulation with the indicated bacterial signal peptide fragments from the n- (blue), h- (green), or c- (red) region of Streptococcus-SP1 (full-length sequence indicated in the figure). Error bars, S.D. H, mean concentration-response curves of hFPR1 and mFpr1 to the formylated (blue circles) and non-formylated peptide (gray squares) comprising the first six N-terminal residues of Streptococcus-SP1.
FIGURE 5.

Conserved signal peptide recognition by mFpr1 and mFpr2, the effects of randomly chosen signal peptide fragments, and signal peptide recognition by hFPR3 and mFpr-rs1. A, mean concentration-response curves of mFpr1 (gray circles) and mFpr2 (gray squares) activation by several signal peptides. Corresponding curves for hFPR1 and hFPR2 are plotted in Figs. 4D and 3E. Numbers of experiments are given in parentheses. B, comparison of mean Ca2+ peak responses (n = 4) of hFPR1 and hFPR2 (red) with mFpr1 and mFpr2 (gray) to the formylated N-termini of 12 randomly chosen bacterial signal peptides (each at 30 μm except for Corynebacterium-SP14, which was used at 100 μm). Mock control (black) indicates empty vector transfection. Error bars, S.D. C, mean Ca2+ peak responses (n = 3) of hFPR3 and mFpr-rs1 to 21 bacterial signal peptides (each at 30 μm). Error bars, S.D. D, mean concentration-response curves of mFpr-rs1 activation by two distinct signal peptides as indicated.
TABLE 2.
Bacterial signal peptides as FPR agonists
Listed is a set of nine bacterial signal peptides that we initially identified, their sources, sequences, accession numbers (UniProt database), and mean EC50 values (in nm) with respect to the activation of heterologously expressed hFPR1, mFpr1, hFPR2, mFpr2, hFPR3, and mFpr-rs1. EC50 values were obtained by measuring full concentration-response curves. Number of experiments (n) is given in parentheses. n.d., no data; n.r., no response with the highest tested concentration (30 μm).
| Signal peptide | Source | Species | Symptoms | Monocytes | hFPR1 | mFpr1 | hFPR2 | mFpr2 | hFPR3 | mFpr-rs1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Streptococcus-SP1 | Hyaluronidase (Q8VLQ8) | Streptococcus suis | Meningitis, sepsis, skin lesions | 6.4 ± 7.7 (13) | 1.8 ± 1.6 (4) | 2.2 ± 0.09 (3) | 1343 ± 614 (4) | 1013 ± 84 (3) | n.r (4) | n.r (3) |
| Streptococcus-SP1-full | Hyaluronidase (Q8VLQ8) | Streptococcus suis | Meningitis, sepsis, skin lesions | 24 ± 17 (4) | 30 ± 29 (2) | n.d. | 54 ± 35 (2) | n.d. | n.d. | n.d. |
| Bacillus-SP2 | Hemolysin (Q45105) | Bacillus cereus | Food poisoning | n.d. | 12850 (1) | 1981 ± 732 (3) | 568 (1) | 78 ± 34 (3) | n.r (1) | n.r (3) |
| Staphylococcus-SP3 | Cell wall hydrolase LytN (Q6GHI8) | Staphylococcus aureus | Abscess, cellulitis, pneumonia | 164 ± 134 (3) | 61 ± 31 (3) | 26 ± 15 (3) | 293 ± 143 (3) | 437 ± 155 (3) | n.r (3) | n.r (3) |
| Salmonella-SP4 | Membrane protein YaeT (Q8Z9A3) | Salmonella typhimurium | Enteric fever | n.d. | 11520 ± 1888 (2) | 6862 ± 3985 (3) | 31 ± 16 (2) | 417 ± 318 (4) | n.r (2) | n.r (4) |
| Haemophilus-SP5 | Tail-specific protease (P45306) | Haemophilus influenzae | Fever, meningitis, bronchitis | 232 ± 97 (5) | 705 ± 238 (2) | 100 ± 48 (3) | 8 ± 2.0 (2) | 62 ± 60 (3) | n.r (2) | n.r (3) |
| Psychromonas-SP6 | Phosphatetransferase (A1ST24) | Psychromonas ingrahamii | n.d. | 0.19 ± 0.14 (3) | 0.15 ± 0.06 (4) | 0.07 ± 0.03 (4) | 38 ± 34 (4) | 93 ± 58 (4) | >10000 (4) | 284 ± 79 (4) |
| Shewanella-SP7 | Siderophore receptor (A6WUI7) | Shewanella baltica | n.d. | 64 ± 32 (3) | 7.7 ± 4.5 (3) | 2.4 ± 3.1 (3) | 6.7 ± 4.0 (3) | 680 ± 920 (3) | n.r (3) | n.r (3) |
| Desulfotomaculum-SP8 | Membrane protein (A4J7E6) | Desulfotomaculum reducens | n.d. | 0.09 ± 0.04 (7) | 0.25 ± 0.17 (4) | 0.17 ± 0.26 (4) | 34 ± 25 (4) | 144 ± 226 (4) | >10000 (4) | 269 ± 245 (3) |
| Desulfotomaculum-SP8-full | Membrane protein (A4J7E6) | Desulfotomaculum reducens | n.d. | 735 ± 202 (4) | 1090 ± 9 (2) | n.d. | 1512 ± 249 (2) | n.d. | n.d. | n.d. |
| Borrelia-SP9 | Basic membrane protein D (Q44743) | Borrelia burgdorferi | Lyme borreliosis | n.d. | 11379 ± 7930 (3) | 5806 ± 3150 (4) | 957 ± 640 (3) | 1067 ± 584 (3) | n.r (4) | n.r (3) |
TABLE 3.
Summary of FPR response profiles for 21 bacterial signal peptides identified in this study
Listed are sequences, sources, pathogenicity, frequency of database hits for residues 1–6 or 1–4, and the response profiles of hFPR1, hFPR2, hFPR3, mFpr1, mFpr2, and mFpr-rs1 for 21 N-terminal peptide fragments. Database hits refer to the number of identical sequence entries in the signal peptide database for the first four or six amino acids of a given signal sequence. Accession numbers refer to the UniProt database. Quantification of receptor sensitivity: Full large circles, activation threshold ≤100 nm; full small circles, activation threshold ≤1000 nm; open small circles, activation threshold ≤30,000 nm; minus sign, no response at 30 μm. n.d., no data.
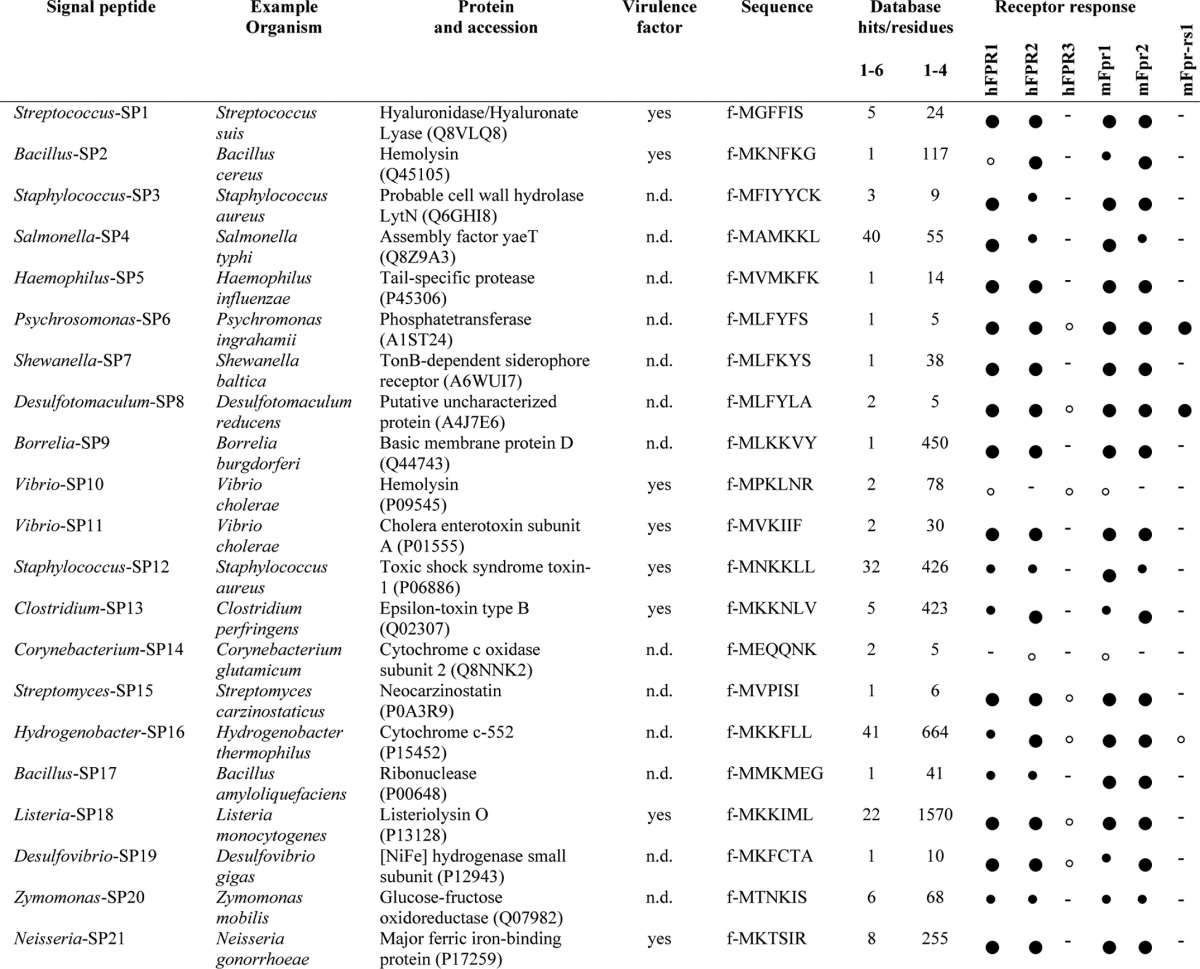
Through database analyses (see the Signal Peptide Database Website), we first identified N-terminal signal peptide fragments on the basis of their similarity with our core agonist motif (Fig. 4B) and initially tested nine hexapeptides (SP1–SP9) derived from distinct bacterial strains (Table 2). All nine peptides turned out to function as potent activators of both hFPR1 and hFPR2 (Fig. 4 (C–E) and Table 2). We examined the sensitivity and selectivity of hFPR1 and hFPR2 for these peptides by analyzing full concentration-response curves (Fig. 4 (D and E) and Table 2). Streptococcus-SP1 potently activated hFPR1 with an EC50 value of 1.8 ± 1.6 nm, whereas the EC50 value of hFPR2 was 1,343 ± 614 nm. A similar preference for hFPR1 was observed by Staphylococcus-SP3, with EC50 values of 61 ± 31 nm for hFRP1 and 293 ± 143 nm for hFPR2. By contrast, Bacillus-SP2, Salmonella-SP4, and Haemophilus-SP5 all preferentially activated hFPR2, with EC50 values of 568, 31, and 8 nm, respectively, whereas they activated hFPR1 at ∼100-fold higher concentrations. Thus, at low nanomolar concentrations, these signal peptides are selectively recognized by either of the two receptors, whereas at higher concentrations, they activate both receptors.
We found that the sequence of the classical FPR agonist f-MLF is contained in signal peptides of at least 305 distinct bacterial strains, including the highly pathogenic Yersinia pestis, Clostridium botulinum, and Bacillus cereus. These signal peptides could constitute a potential natural source for f-MLF and its derivatives. We therefore tested three signal peptides (SP6–SP8) that contain an f-MLF at their N terminus: f-MLFYLS from Psychromonas ingrahamii, f-MLFKYS from Shewanella baltica, and f-MLFYLA from Desulfotomaculum reducens (Fig. 4E and Table 2). For hFPR1 and hFPR2, EC50 values were 0.15 ± 0.06 and 38 ± 34 nm for Psychromonas-SP6, 7.7 ± 4.5 and 6.7 ± 4.0 nm for Shewanella-SP7, and 0.25 ± 0.17 nm and 34 ± 25 nm for Desulfotomaculum-SP8, respectively. Thus, two of these tested peptides are among the most potent natural FPR agonists identified thus far.
To determine in more detail signal peptide key features crucial for FPR activation, we next analyzed signal peptides of different lengths, fragments from different signal peptide domains, and the effects of formylation (Fig. 4, F–H). We compared concentration-response curves of hFPR1 to the full-length, 36- and 23-amino acid-containing signal peptides of Streptococcus-SP1 and Desulfotomaculum-SP8 with their corresponding N-terminal hexapeptide fragments (Fig. 4F and Table 2). In the case of Streptococcus-SP1, the hexapeptide and the full-length signal peptide activated hFPR1 at comparable concentrations. For Desulfotomaculum-SP8, the small N-terminal fragment was considerably more potent than the full-length signal peptides. However, both full-length peptides could activate hFPR1 at nanomolar concentrations (Table 2), demonstrating that cleavage products of variable lengths can be recognized. To test whether peptides derived from different domains of a given signal peptide are detected by FPRs, we compared activation of several FPRs by Streptococcus-SP1 with that of fragments from the n-, h-, and c-regions of the corresponding full-length signal peptide. Whereas N-terminal Streptococcus-SP1 hexapeptide robustly activated hFPR1 and hFPR2 at nanomolar concentrations, none of the tested peptides from other domains were potent agonists (Fig. 4G). Moreover, in order to function as efficient agonists, signal peptides require a formylated N-terminal methionine. Complete removal of this residue abolished the response of hFPR1 and hFPR2 (Fig. 4G), whereas removal of the N-terminal formylation alone was sufficient to reduce the activation of hFPR1 by 770-fold (Fig. 4H). Interestingly, this latter result enables FPR discrimination between bacterial and host-endogenous signal peptides; whereas bacteria use an N-terminally formylated methionine to initiate protein biosynthesis, this methionine in eukaryotic cells remains normally unformylated (42).
Signal Peptide Recognition Is Evolutionarily Conserved between Human and Mouse FPRs
Our findings obtained with human FPRs extend also to mouse Fprs. Comparison of activation profiles of hFPR1 and hFPR2 with mFpr1 and mFpr2 revealed several striking similarities (Figs. 4 (C–H) and 5A and Table 2). Streptococcus-SP1 hexapeptide derived from the n-domain of the signal peptide robustly activated mFpr1 and mFpr2 at nanomolar concentrations (Fig. 4A), whereas both receptors failed to recognize peptides from other domains (Fig. 4G). Removal of the N-terminal methionine led to a greater than 1,000-fold increase in the activation threshold of mFpr1 and completely abolished the response of mFpr2 (Fig. 4G). Removal of the N-terminal formylation alone was sufficient to reduce the activation of mFpr1 by 280-fold (Fig. 4H). Both mouse receptors showed agonist selectivity at nanomolar concentrations but could be activated by all signal peptides when tested at higher concentrations (Fig. 5A and Table 2). If a signal peptide preferentially activated hFPR1, it was usually also preferred by mFpr1. Similarly, peptides that reacted more potently on hFPR2 preferentially activated mFpr2 (Table 2). Furthermore, responses to Streptococcus-SP1, Psychromonas-SP6, and Desulfotomaculum-SP8 were all nearly identical for both receptors between both species (Figs. 4 (C–E) and 5A and Table 2). These observations strongly argue for an evolutionarily conserved recognition mechanism of this agonist family by human and mouse FPR1 and FPR2.
The Size of the Ligand Family
Given that all nine signal peptides that we initially tested are activators of human and mouse FPR1 and FPR2 (Table 2), we asked how many of the currently annotated 175,542 bacterial signal peptides are potentially recognized by these two receptors. Database searches using the first four amino acids of SP1–SP9 (containing the key motif) revealed 55 identical hits for the hexapeptides and 717 hits for the core sequence comprising the first four amino acids (Table 3). These results imply that the number of bacterial signal peptides functioning as FPR activators could be very large. All peptides that we tested thus far were selected on the basis of structural considerations. To provide a more unbiased estimate of the size of the ligand family, we next used an additional set of 12 signal sequences (SP10–SP21) that were randomly chosen from the 1,168 experimentally confirmed signal peptides present in the database (Table 3 and Fig. 5). For this peptide selection, database searches revealed another 123 identical hits for the hexapeptide and 3,576 hits for their core motifs (Table 3). Remarkably, all 12 hexapeptides turned out to function as activators of human and mouse FPR1 and FPR2 although with differential activation thresholds and selectivity for each receptor (Fig. 5B and Table 3). These results allowed us to estimate the size of the ligand family for FPR1 and FPR2. Probability calculations predict with a confidence level of 95% that these two receptors should be capable of detecting at least 78% of the 175,542 sequences currently annotated in the signal peptide database (see “Experimental Procedures”).
Strongly Restricted Signal Peptide Recognition by hFPR3 and mFpr-rs1
Having established that human and mouse FPR1 and FPR2 are both capable of recognizing a wide range of bacterial signal peptides, we asked whether this applies also to hFPR3 and mFpr-rs1. These receptors did not recognize all tested signal peptides, but we identified seven signal peptides that activated hFPR3 (SP6, SP8, SP10, SP15, SP16, SP18, and SP19). Three of these peptides also activated mFpr-rs1 (SP6, SP8, and SP16) (Fig. 5 (C and D) and Tables 2 and 3). hFPR3 responded at concentrations of ≥1 μm; mFpr-rs1 was potently activated by Psychromonas-SP6 and Desulfotomaculum-SP8 with EC50 values of 284 ± 79 and 269 ± 245 nm, respectively (Fig. 5D and Table 3). These results indicate that hFPR3 and mFpr-rs1 could also function as signal peptide receptors, but, compared with FPR1 and FPR2, their recognition capabilities seem to be far more restricted because they detect only a limited number of the peptides tested thus far.
Signal Peptides Trigger Innate Defense Mechanisms in Phagocytic Leukocytes
To further validate the biological significance of our findings, we investigated the recognition capabilities of bacterial signal peptides by native human and mouse immune cells. We first examined primary human monocytes that naturally express FPR1 and FPR2 (3) and performed single-cell Ca2+ imaging experiments. Exposure to Streptococcus-SP1 produced robust intracellular Ca2+ mobilization in ∼89% of monocytes (Fig. 6A). Consistent with our results from the in vitro assay, monocytes recognize a wide range of signal peptides (Fig. 6B), and short hexapeptides are more potent than the corresponding full-length signal peptides (Fig. 6C and Table 2).
FIGURE 6.

Human immune cells recognize bacterial signal peptides. A, overlay of single-cell Ca2+ responses obtained from 186 human leukocytes present in the visual field of a representative experiment (n = 3). Of these, 167 cells responded to Streptococcus-SP1 (70 nm). B, average Ca2+ peak responses of human monocytes stimulated with a 10 μm concentration of the indicated peptides. Number of experiments (each from a different human donor) is shown in parentheses. Error bars, S.D. C, concentration-response curves of human monocytes to the N-terminal hexapeptide and the full-length signal peptide of Streptococcus-SP1 comprising 36 residues. D, inhibition of signal peptide-induced Ca2+ responses by FPR antagonists. 100 nm Streptococcus-SP1 was applied either alone or in combination with the hFPR1 inhibitors CsH (1 μm) or t-Boc2 (10 μm). E, left, representative images showing immunostainings of hFPR1 (green) and the corresponding mock staining using an isotype KLH antibody as negative control. Similar results were obtained in three independent experiments. Red, cell nuclei; scale bar, 50 μm. Right, percentage of cells expressing hFPR1 or the monocyte marker hCD14 (n = 3). As isotype controls, we used a KLH antibody for hFPR1 and a MOPC-21 antibody for hCD14. F, left, representative correlation between intensity of hFPR1 staining in individual cells and peak amplitude of Ca2+ responses to Streptococcus-SP1. Similar results were obtained in three independent experiments. The black line indicates a best curve fit of the data points. Right, quantitative comparison of monocyte Ca2+ responses to Streptococcus-SP1 and post hoc hFPR1 immunostaining. G, concentration-response curves of human monocytes (red) and heterologously expressed hFPR1 (black) to the indicated signal peptides. Curves denote average responses ± S.D. H, concentration-response curves of human granulocytes (green) and heterologously expressed hFPR1 (black) to the indicated signal peptide. Error bars, S.D. I, mean Ca2+ peak responses of leukocytes from wild type, Fpr1+/−, and Fpr1−/− mice to stimulation with Streptococcus-SP1 (1 μm) or control buffer (mock). Stimulation with the FPR agonist f-MLF (1 μm) and the macrophage inflammatory protein 1α (MIP-1α, 10 μm) that activates a chemokine receptor served as positive controls. Bars, mean responses ± S.D. from two independent experiments. J, representative electron paramagnetic resonance spectrometry measurements of O2⨪ production by human monocytes upon stimulation with 10 μm Streptococcus-SP1, 10 μm f-MLF, or control buffer (mock). Similar results were obtained in three independent experiments. K, Amplex measurements of H2O2 production by human monocytes stimulated with four different signal peptides (each at 10 μm except for Borrelia-SP9, which was used at 100 μm) and rev-W-peptide. W-peptide and f-MLF (each at 10 μm) were used as positive controls. Bath application served as mock control. L, comparison of concentration dependence between Ca2+ signals of human monocytes (red, left) or granulocytes (green, right) and their respective H2O2 production (blue) ± S.D. upon stimulation with Streptococcus-SP1. M, release of MMP-9 by human neutrophils. Left, MMP-9 release ± S.D. upon stimulation with Streptococcus-SP1 (1 μm), Hydrogenobacter-SP16 (1 μm). Stimulation with lipopolysaccharide (LPS 100 ng/ml) served as positive control, and culture medium served as negative control. Right, stimulus-response curves for MMP-9 release (dark blue) and Ca2+ mobilization (green) in human granulocytes following stimulation with Streptococcus-SP1. N, chemotactic response of human granulocytes to stimulation with signal peptides. Left, representative images showing accumulation of human neutrophils following exposure to Streptococcus-SP1 (100 nm). Culture medium served as negative control; scale bar, 100 μm. Similar results were obtained in three independent experiments. Right, number of migrated granulocytes ± S.D. after stimulation with Streptococcus-SP1 (10 nm), Salmonella-SP4 (100 nm), or Hydrogenobacter-SP16 (10 nm). Numbers denote the total amount of cells counted.
Monocyte Ca2+ responses to Streptococcus-SP1 and f-MLF were inhibited by the established FPR antagonists t-Boc2 and cyclosporine H (Fig. 6D). Streptococcus-SP1 and f-MLF strongly cross-desensitized each other, whereas RANTES, a CCR1 (C-C chemokine receptor 1) receptor agonist (43), did not affect subsequent Ca2+ responses to Streptococcus-SP1 and f-MLF in monocytes (not shown). We next combined Ca2+ imaging with post hoc immunocytochemistry using antibodies specific for hFPR1 (Fig. 6 (E and F) and supplemental Video 1) and found that the vast majority of all cells expressing hFPR1 (92 ± 10%) responded to Streptococcus-SP1 (Fig. 6F). There was a clear correlation between receptor staining intensity and strength of the Ca2+ signal (Fig. 6F). We compared concentration-response curves for six signal peptides in human monocytes with curves obtained from the in vitro experiments and found them to be remarkably similar (Fig. 6 (G and L) and Table 2). Equivalent Ca2+ responses to bacterial signal peptides were also observed in human granulocytes (Fig. 6H), another immune cell type known to naturally express FPR1 and FPR2 (5).
To directly demonstrate that FPR1 is required for signal peptide recognition by immune cells, we also performed some mouse experiments comparing Ca2+ responses in leukocytes of wild type, Fpr1−/−, and Fpr1+/− mice (Fig. 6I). These results demonstrate that Streptococcus-SP1 can be recognized by mouse leukocytes and that this response, along with the response to f-MLF, is selectively abolished in Fpr1−/− cells, whereas the response to MIP1-α, a chemokine receptor agonist (44), remains normal (Fig. 6I). Together, our results indicate that bacterial signal peptides are recognized by native immune cells and that this recognition process depends on FPR function.
Because the physiological function of monocytes is host defense, we tested whether bacterial signal peptides are capable of activating known classical innate defense mechanisms. To accomplish this, we first examined whether signal peptides trigger the production of superoxide radicals (O2⨪) and other reactive oxygen species that are produced by leukocytes during immune responses (5). In human monocytes, we monitored the kinetics of short lived free O2⨪ production in response to Streptococcus-SP1 using electron paramagnetic resonance (EPR) spectroscopy (45). Streptococcus-SP1 induced dose-dependent production of O2⨪ with a kinetic response very similar to that of f-MLF (Fig. 6J). For a larger selection of stimuli, we also used a 96-well Amplex-based ROS assay that monitors fluorescence changes induced by H2O2 (45). All four tested signal peptides caused robust H2O2 production (Fig. 6K). Direct comparison of concentration-response curves for Ca2+ mobilization and H2O2 production obtained with Streptococcus-SP1 and f-MLF stimulation revealed a ∼10-fold shift between H2O2 and Ca2+ responses in monocytes and granulocytes (Fig. 6L), consistent with previous studies of f-MLF-mediated ROS formation (46). Further tests with human granulocytes revealed that signal peptides also elicit other classical innate defense reactions, such as the release of MMP-9 as well as chemotactic cell migration (Fig. 6, M and N). Hence, these results demonstrate that bacterial signal peptides are potent molecular triggers of classical innate defense mechanisms against pathogen invasion.
DISCUSSION
FPRs are a unique family of GPCRs with an exceptionally broad and promiscuous agonist spectrum showing no obvious common pattern in primary structure or natural origin (3, 7, 8). Because of this molecular promiscuity, there is little understanding of what the biological targets of these receptors are and why FPRs detect a range of structurally dissimilar molecules. Here, we report the ability of FPRs to recognize bacterial signal peptides, a vast family of novel natural FPR agonists that differ extensively in their primary structure but exhibit a well conserved secondary structure. Detailed structural variation analysis of peptide agonists defined the key features that enable both promiscuity and high specificity of FPR molecular detection. Finally, we argue that these results identify a common structural basis or molecular signature for FPR activation that is used to detect natural pathogens. These results provide new insight into the function of these receptors and their molecular promiscuity and make specific inferences about their biological roles and evolution.
Our results indicate that bacterial signal peptides provide an exceptionally large pool of sequence-divergent FPR agonists that all contain a conserved secondary structure. The f-MLF motif alone exists at the N terminus of several hundred signal peptides, whereas the core agonist motif of all 21 signal peptides used in this study occurs at the N terminus of a total of 4,293 peptides (Table 3). Moreover, our results suggest that FPR1 and FPR2 together may recognize far more than 100,000 distinct signal peptides. In fact, there is currently no reason for ruling out that FPR1 and FPR2 could detect the entire set of bacterial signal peptides. As such, bacterial signal peptides represent the largest family of natural FPR agonists and one of the largest families of GPCR ligands known to date. They are also one of the most complex classes of natural activators of the innate immune system discovered thus far.
Not only do our findings demonstrate that bacterial signal peptides are recognized by human and mouse FPR1 and FPR2 in vitro; they also provide extensive evidence that this recognition process occurs in primary leukocytes of the human and mouse innate immune systems. These cells are equipped with native FPRs, and we provide clear evidence that bacterial signal peptides are detected in a sensitive manner by monocytes and granulocytes and that they trigger a range of classical innate immune defense mechanisms, including chemotactic migration, ROS production, and matrix metalloproteinase release. Pharmacologic and genetic manipulation indicates that this recognition process is indeed FPR-dependent.
Our experiments establish that FPR1 and FPR2 function as broad signal peptide receptors. A goal of future experiments will be to determine whether this finding applies to other FPR subtypes as well. The vomeronasal receptor mFpr-rs1 and its sequence orthologue hFPR3 seem to be much more narrowly tuned in the detection of signal peptides. Compared with FPR1 and FPR2, these receptors reacted to a relatively small subset of signal peptides and required higher concentrations. Possibly, hFPR3 and mFpr-rs1 focus on the detection of a specific set of bacterial pathogens, whereas FPR1 and FPR2 function as more general sensors of bacteria.
FPRs were originally discovered as receptors for formylated peptides, such as f-MLF, present in the supernatant of bacterial cultures (6, 47). However, the precise source and release pathway of such peptides has never been identified (5). Our finding that the sequences of these ligands are contained within bacterial signal peptides and that these signal peptides and their N-terminal breakdown products function as potent activators of FPRs identifies a likely natural source for these molecules. Signal peptides are cleaved off the maturing protein by extracellular proteases during membrane targeting and subsequently digested into short fragments (30). Although the fate of bacterial signal peptides after their cleavage is currently not well understood (30), recent mass spectrometry studies of bacterial secretomes found complete signal peptides as well as N-terminal fragments in the extracellular medium of bacteria cultures, demonstrating that these molecules indeed can be secreted by bacteria (48–50). One possibility of how this happens is through lysis, either through autolysis during bacterial growth (51) or through immune cell-mediated lysis during an infection (52). Both mechanisms are well established and, therefore, represent likely pathways by which signal peptides can be made available for the recognition by FPRs. Determination of whether other more specific release mechanisms exist for bacterial signal peptides remains a goal of future research. Compared with eukaryotes, bacteria employ highly complex secretion mechanisms, and their secretory pathways are less well examined (53, 54).
The ability of FPRs to recognize thousands of peptides with distinct amino acid sequences yet maintaining selectivity requires a compromise between broad specificity and high affinity. Our results suggest a remarkable solution to this problem. We provide evidence for an FPR detection mechanism that seems to focus on the recognition of a conserved three-dimensional motif rather than the linear peptide sequences. Several critical features of this mechanism can now be defined. For example, our data predict that FPR peptide agonists possess a well defined secondary structure, probably containing an α-turn. Such agonists can vary considerably in their length, but a minimal size of three amino acids is required. The first (or last) amino acid residue of a given peptide represents a key element for agonist potency because this residue has the most stringent spatial and chemical limitations. The N-terminal residue should consist of a formylated methionine, or, alternatively, the C-terminal residue should be an amidated methionine. Rather than site-specific hydrogen donor/acceptor or ionic bonds, flexible shape-oriented van der Waals interactions of most other amino acid side chains determine agonist potency. Especially the second and/or third residue next to this methionine should comprise amino acids that preferentially permit van der Waals interactions, although a certain amount of polarity can be tolerated. We define a core motif that comprises three key residues forming a hydrophobic, tripartite clawlike structure with an α-turn oriented around a carbonyl group. The symmetrical organization of this agonist motif, together with the observation that a similar carbonyl group exists in both the N-terminal formyl and the C-terminal amide group (Fig. 3F), provides an explanation for the finding that FPRs recognize in very similar ways both N-terminally formylated and C-terminally amidated peptides. We predict that a C-terminally amidated peptide interacts first with the receptor binding pocket via its C-terminal methionine, whereas an N-terminally formylated peptide binds first through its N terminus. Experimental evidence for the validity of this mechanism is provided by our demonstration that C-terminally amidated peptides are equally potent FPR agonists as corresponding peptides in which the amino acid sequence has been reversed and now comprises an N-terminal formylation.
The fact that this mechanistic concept enabled us to identify bacterial signal peptides as a novel class of FPR agonists provides direct support for its usefulness. Bacterial signal peptides have structural features that fit particularly well to our model predictions, including a highly variable primary structure that contains a largely hydrophobic α-helical domain close to a conserved N terminus starting with a formylated methionine (41). Importantly, key features derived from our analyses can also be found in other previously identified FPR agonists, such as mitochondrial peptides, suggesting that these results could be of general significance. N-terminally formylated mitochondrial peptides from membrane proteins, such as ND1, ND4, ND6, and CO1, are well known to function as FPR agonists (9). Mitochondria are of ancient bacterial origin (55), and we predict that these mitochondrial peptides served originally as signal peptides enabling protein translocation through the mitochondrial membrane. Consistent with this idea, the ND1 N terminus reveals clear structural similarities to our agonist model (Fig. 3I). Further structural comparisons demonstrate that other known FPR ligands, such as humanin and f-MLF, also display striking similarities with our agonist model (Fig. 3I). Independent support for this concept comes from a recent study using non-peptide agonists to identify a potential binding motif in FPR2 (56). Moreover, other FPR agonists, such as phenol-soluble modulin peptide toxins (15), amyloid-β(1–42) (8), or μPAR (57), may also fit into this scheme. For example, a recent study demonstrated that the signal peptide of the Staphylococcus aureus quorum-sensing signal, AgrD, shares clear structural and functional similarities with the phenol-soluble modulin family (50). The fact that critical agonist features as described here extend to a number of distinct FPR agonists suggests that our results should prove useful in future developments, such as the identification of the FPR ligand binding site and the discovery of subtype-selective FPR antagonists.
The mammalian innate immune system as the first line of host defense has evolved multiple strategies to detect pathogen-associated molecules to subsequently eliminate infective pathogens in the body. A major challenge for the innate immune system is the recognition of a multitude of rapidly adapting microorganisms via a limited number of germ line-encoded PRRs. Therefore, PRRs focus on the recognition of highly conserved microbial components, so-called PAMPs, which are difficult to alter because they are essential for the survival of an invading pathogen. Bacterial signal peptides show a number of specific molecular properties that are typical hallmarks of PAMPs. First, export processes initiated by signal peptides represent an evolutionarily conserved mechanism that is essential for bacterial survival (30). Second, bacteria use an N-terminally formylated methionine to initiate translation, whereas this methionine in eukaryotic cells is unformylated (42). Because the formylation is critical for FPR recognition, this difference provides an elegant solution to enable discrimination between host-endogenous and microbial signal peptides. Third, our studies using human and mouse leukocytes provide clear evidence that signal peptides are indeed capable of triggering classical innate immune responses mediated by FPRs. Fourth, we observed a remarkably high degree of conservation in signal peptide recognition between human and mouse FPR1 and FPR2, thus arguing for an important role of bacterial signal peptides during evolution of FPR function. Consistent with this concept, human and mouse FPR1 and FPR2 both show relatively broad tuning and respond to a large number of signal peptides, and these receptors display striking similarities in their sensitivity and selectivity toward structurally divergent peptides. In summary, the recognition capabilities that mammalian FPRs have evolved are perfect for sensing the molecular properties of bacterial signal peptides, which combine an exceptionally high sequence variability with a conserved secondary structure. Hence, we propose that mammalian FPRs may have evolved originally as germ line-encoded PRRs that recognize structurally conserved export motifs of bacterial signal sequences as their cognate, pathogen-associated molecular pattern.
Both FPRs and the Toll-like receptors focus on the recognition of invading bacteria. However, these receptors trigger distinct signal transduction cascades. It is tempting to speculate that there could be an interaction between both systems. A critical feature of FPRs is their ability to detect not only molecular patterns associated with pathogens, such as bacterial signal peptides, but also host-endogenous mitochondrial peptides. This specific property enables the FPRs to function not only as PAMP receptors but also as sensors of danger-associated molecular patterns (known as DAMPs), for example in the case of tissue damage. This aspect could be useful in the general management of the microbiome of the body, consistent with the large distribution of FPRs in multiple tissues and organs. Taken together, our findings provide an essential foundation for understanding the function of FPRs, with far reaching consequences for their biological roles.
Supplementary Material
Acknowledgments
We thank S. Plant, C. Hässig, and R. Bender-Omlor for expert technical and G. Mörschbächer for editorial assistance. R. Zimmermann, M. Pyrski, M. Bischoff, and T. Boehm provided critical advice on the manuscript. We thank H. Eichler for providing human blood samples and S. Saul for technical advice regarding monocyte culture.
This work was supported by Deutsche Forschungsgemeinschaft Grants INST 256/271–1 FUGG (to F. Z. and B. B), Sonderforschungsbereich 894/A1 (to M. H.) and A17 (to F. Z.), Sonderforschungsbereich 1027/A2 and C4 (to M. H. and I. B.), and Schwerpunktprogramm 1392 (to F. Z.). This work was also supported by the University of Saarland through HOMFORexcellent Grants 2011 (to B. B.) and 2013 (to I. B.) and a graduate scholarship (GradUS) (to T. S.).

This article contains supplemental Table S1 and Video 1.
- PRR
- pattern recognition receptor
- FPR
- formyl peptide receptor
- GPCR
- G-protein-coupled receptor
- f-MLF
- N-formylmethionine-leucine-phenylalanine
- PAMP
- pathogen-associated molecular pattern
- ROS
- reactive oxygen species
- ND
- NADH-reductase subunit
- RANTES
- regulated on activation, normal T-cell expressed and secreted
- μPAR
- urokinase receptor
- HBSS
- Hanks' buffered saline solution
- HEK
- human embryonic kidney
- CMH
- 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine
- MMP
- matrix metallopeptidase.
REFERENCES
- 1. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 2. Takeuchi O., Akira S. (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820 [DOI] [PubMed] [Google Scholar]
- 3. Migeotte I., Communi D., Parmentier M. (2006) Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 17, 501–519 [DOI] [PubMed] [Google Scholar]
- 4. Liu M., Chen K., Yoshimura T., Liu Y., Gong W., Wang A., Gao J. L., Murphy P. M., Wang J. M. (2012) Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ye R. D., Boulay F., Wang J. M., Dahlgren C., Gerard C., Parmentier M., Serhan C. N., Murphy P. M. (2009) International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 61, 119–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schiffmann E., Corcoran B. A., Wahl S. M. (1975) N-Formylmethionyl peptides as chemoattractants for leucocytes. Proc. Natl. Acad. Sci. U.S.A. 72, 1059–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fu H., Karlsson J., Bylund J., Movitz C., Karlsson A., Dahlgren C. (2006) Ligand recognition and activation of formyl peptide receptors in neutrophils. J. Leukoc. Biol. 79, 247–256 [DOI] [PubMed] [Google Scholar]
- 8. Le Y., Gong W., Tiffany H. L., Tumanov A., Nedospasov S., Shen W., Dunlop N. M., Gao J. L., Murphy P. M., Oppenheim J. J., Wang J. M. (2001) Amyloid b42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 21, RC123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rabiet M. J., Huet E., Boulay F. (2005) Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur. J. Immunol. 35, 2486–2495 [DOI] [PubMed] [Google Scholar]
- 10. Selvatici R., Falzarano S., Mollica A., Spisani S. (2006) Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur. J. Pharmacol. 534, 1–11 [DOI] [PubMed] [Google Scholar]
- 11. Panaro M. A., Acquafredda A., Sisto M., Lisi S., Maffione A. B., Mitolo V. (2006) Biological role of the N-formyl peptide receptors. Immunopharmacol. Immunotoxicol. 28, 103–127 [DOI] [PubMed] [Google Scholar]
- 12. Gao J. L., Lee E. J., Murphy P. M. (1999) Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J. Exp. Med. 189, 657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liberles S. D., Horowitz L. F., Kuang D., Contos J. J., Wilson K. L., Siltberg-Liberles J., Liberles D. A., Buck L. B. (2009) Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc. Natl. Acad. Sci. U.S.A. 106, 9842–9847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kretschmer D., Nikola N., Dürr M., Otto M., Peschel A. (2012) The virulence regulator Agr controls the staphylococcal capacity to activate human neutrophils via the formyl peptide receptor 2. J. Innate Immun. 4, 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kretschmer D., Gleske A. K., Rautenberg M., Wang R., Köberle M., Bohn E., Schöneberg T., Rabiet M. J., Boulay F., Klebanoff S. J., van Kessel K. A., van Strijp J. A., Otto M., Peschel A. (2010) Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 7, 463–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Y., Murphy P. M., Wang J. M. (2002) Formyl-peptide receptors revisited. Trends Immunol. 23, 541–548 [DOI] [PubMed] [Google Scholar]
- 17. Boulay F., Tardif M., Brouchon L., Vignais P. (1990) Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem. Biophys. Res. Commun. 168, 1103–1109 [DOI] [PubMed] [Google Scholar]
- 18. Bäck M., Powell W. S., Dahlén S. E., Drazen J. M., Evans J. F., Serhan C. N., Shimizu T., Yokomizo T., Rovati G. E. (2014) Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br. J. Pharmacol. 171, 3551–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y., Ye D. (2013) Molecular biology for formyl peptide receptors in human diseases. J. Mol. Med. 91, 781–789 [DOI] [PubMed] [Google Scholar]
- 20. Mollica A., Stefanucci A., Costante R., Pinnen F. (2012) Role of formyl peptide receptors (FPR) in abnormal inflammation responses involved in neurodegenerative diseases. Antiinflamm. Antiallergy Agents Med. Chem. 11, 20–36 [DOI] [PubMed] [Google Scholar]
- 21. Lacy M., Jones J., Whittemore S. R., Haviland D. L., Wetsel R. A., Barnum S. R. (1995) Expression of the receptors for the C5a anaphylatoxin, interleukin-8 and FMLP by human astrocytes and microglia. J. Neuroimmunol. 61, 71–78 [DOI] [PubMed] [Google Scholar]
- 22. Chiu I. M., Heesters B. A., Ghasemlou N., Von Hehn C. A., Zhao F., Tran J., Wainger B., Strominger A., Muralidharan S., Horswill A. R., Bubeck Wardenburg J., Hwang S. W., Carroll M. C., Woolf C. J. (2013) Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rivière S., Challet L., Fluegge D., Spehr M., Rodriguez I. (2009) Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature 459, 574–577 [DOI] [PubMed] [Google Scholar]
- 24. Shao G., Julian M. W., Bao S., McCullers M. K., Lai J. P., Knoell D. L., Crouser E. D. (2011) Formyl peptide receptor ligands promote wound closure in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 44, 264–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wentworth C. C., Alam A., Jones R. M., Nusrat A., Neish A. S. (2011) Enteric commensal bacteria induce extracellular signal-regulated kinase pathway signaling via formyl peptide receptor-dependent redox modulation of dual specific phosphatase 3. J. Biol. Chem. 286, 38448–38455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chamero P., Katsoulidou V., Hendrix P., Bufe B., Roberts R., Matsunami H., Abramowitz J., Birnbaumer L., Zufall F., Leinders-Zufall T. (2011) G protein Gαo is essential for vomeronasal function and aggressive behavior in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 12898–12903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chamero P., Leinders-Zufall T., Zufall F. (2012) From genes to social communication: molecular sensing by the vomeronasal organ. Trends Neurosci. 35, 597–606 [DOI] [PubMed] [Google Scholar]
- 28. Leinders-Zufall T., Ishii T., Mombaerts P., Zufall F., Boehm T. (2009) Structural requirements for the activation of vomeronasal sensory neurons by MHC peptides. Nat. Neurosci. 12, 1551–1558 [DOI] [PubMed] [Google Scholar]
- 29. Leinders-Zufall T., Ishii T., Chamero P., Hendrix P., Oboti L., Schmid A., Kircher S., Pyrski M., Akiyoshi S., Khan M., Vaes E., Zufall F., Mombaerts P. (2014) A family of nonclassical class I MHC genes contributes to ultrasensitive chemodetection by mouse vomeronasal sensory neurons. J. Neurosci. 34, 5121–5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dalbey R. E., Wang P., van Dijl J. M. (2012) Membrane proteases in the bacterial protein secretion and quality control pathway. Microbiol. Mol. Biol. Rev. 76, 311–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hegde R. S., Bernstein H. D. (2006) The surprising complexity of signal sequences. Trends Biochem. Sci. 31, 563–571 [DOI] [PubMed] [Google Scholar]
- 32. Bufe B., Schumann T., Zufall F. (2012) Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J. Biol. Chem. 287, 33644–33655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boxio R., Bossenmeyer-Pourié C., Steinckwich N., Dournon C., Nüsse O. (2004) Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol. 75, 604–611 [DOI] [PubMed] [Google Scholar]
- 34. Brooks B. R., Bruccoleri R. R., Olafson B. D., States D. J., Swaminathan S., Karplus M. (1983) CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 10.1002/jcc.540040211 [DOI] [Google Scholar]
- 35. Bae G. H., Lee H. Y., Jung Y. S., Shim J. W., Kim S. D., Baek S. H., Kwon J. Y., Park J. S., Bae Y. S. (2012) Identification of novel peptides that stimulate human neutrophils. Exp. Mol. Med. 44, 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baek S. H., Seo J. K., Chae C. B., Suh P. G., Ryu S. H. (1996) Identification of the peptides that stimulate the phosphoinositide hydrolysis in lymphocyte cell lines from peptide libraries. J. Biol. Chem. 271, 8170–8175 [DOI] [PubMed] [Google Scholar]
- 37. Ruiz-Gomez G., Tyndall J. D., Pfeiffer B., Abbenante G., Fairlie D. P. (2010) Update 1 of: Over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem. Rev. 10.1021/cr900344w [DOI] [PubMed] [Google Scholar]
- 38. Rienstra C. M., Tucker-Kellogg L., Jaroniec C. P., Hohwy M., Reif B., McMahon M. T., Tidor B., Lozano-Pérez T., Griffin R. G. (2002) De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 99, 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ying G., Iribarren P., Zhou Y., Gong W., Zhang N., Yu Z. X., Le Y., Cui Y., Wang J. M. (2004) Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 172, 7078–7085 [DOI] [PubMed] [Google Scholar]
- 40. Shawar S. M., Rich R. R., Becker E. L. (1995) Peptides from the amino-terminus of mouse mitochondrially encoded NADH dehydrogenase subunit 1 are potent chemoattractants. Biochem. Biophys. Res. Commun. 211, 812–818 [DOI] [PubMed] [Google Scholar]
- 41. von Heijne G. (1985) Signal sequences. The limits of variation. J. Mol. Biol. 184, 99–105 [DOI] [PubMed] [Google Scholar]
- 42. Benelli D., Londei P. (2009) Begin at the beginning: evolution of translational initiation. Res. Microbiol. 160, 493–501 [DOI] [PubMed] [Google Scholar]
- 43. Neote K., DiGregorio D., Mak J. Y., Horuk R., Schall T. J. (1993) Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell 72, 415–425 [DOI] [PubMed] [Google Scholar]
- 44. Gear A. R., Camerini D. (2003) Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation 10, 335–350 [DOI] [PubMed] [Google Scholar]
- 45. Bogeski I., Kappl R., Kummerow C., Gulaboski R., Hoth M., Niemeyer B. A. (2011) Redox regulation of calcium ion channels: chemical and physiological aspects. Cell Calcium 50, 407–423 [DOI] [PubMed] [Google Scholar]
- 46. Kemmerich B., Pennington J. E. (1988) Different calcium and oxidative metabolic responses in human blood monocytes during exposure to various agonists. J. Leukoc. Biol. 43, 125–132 [DOI] [PubMed] [Google Scholar]
- 47. Marasco W. A., Phan S. H., Krutzsch H., Showell H. J., Feltner D. E., Nairn R., Becker E. L., Ward P. A. (1984) Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J. Biol. Chem. 259, 5430–5439 [PubMed] [Google Scholar]
- 48. de Souza G. A., Leversen N. A., Målen H., Wiker H. G. (2011) Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J. Proteomics 75, 502–510 [DOI] [PubMed] [Google Scholar]
- 49. Ravipaty S., Reilly J. P. (2010) Comprehensive characterization of methicillin-resistant Staphylococcus aureus subsp. aureus COL secretome by two-dimensional liquid chromatography and mass spectrometry. Mol. Cell Proteomics 9, 1898–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwartz K., Sekedat M. D., Syed A. K., O'Hara B., Payne D. E., Lamb A., Boles B. R. (2014) The AgrD N-terminal leader peptide of Staphylococcus aureus has cytolytic and amyloidogenic properties. Infect. Immun. 82, 3837–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yarmolinsky M. B. (1995) Programmed cell death in bacterial populations. Science 267, 836–837 [DOI] [PubMed] [Google Scholar]
- 52. Martner A., Dahlgren C., Paton J. C., Wold A. E. (2008) Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect. Immun. 76, 4079–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pugsley A. P. (1993) The complete general secretory pathway in Gram-negative bacteria. Microbiol. Rev. 57, 50–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tseng T. T., Tyler B. M., Setubal J. C. (2009) Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol. 9, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sagan L. (1967) On the origin of mitosing cells. J. Theor. Biol. 14, 255–274 [DOI] [PubMed] [Google Scholar]
- 56. Schepetkin I. A., Kirpotina L. N., Khlebnikov A. I., Leopoldo M., Lucente E., Lacivita E., De Giorgio P., Quinn M. T. (2013) 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: molecular modeling of chiral recognition by FPR2. Biochem. Pharmacol. 85, 404–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Resnati M., Pallavicini I., Wang J. M., Oppenheim J., Serhan C. N., Romano M., Blasi F. (2002) The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. U.S.A. 99, 1359–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.