

Background: A putative peptide:N-glycosidase (PNGase), distinct from the well characterized PNGase F, was identified in Elizabethkingia meningoseptica.
Results: The candidate protein possesses PNGase activity and a distinct substrate spectrum and domain structure.
Conclusion: The candidate (named PNGase F-II) is a novel PNGase.
Significance: Characterization of PNGase F-II may provide an alternative tool for glycobiology.
Keywords: Bacteria; Crystal Structure; Enzyme; Glycoprotein; N-Linked Glycosylation; Peptide:N-Glycosidase (PNGase), PNGase F, Glycosylation, Deglycosylation, Glycoprotein, Crystal Structure
Abstract
Peptide:N-glycosidase (PNGase) F, the first PNGase identified in prokaryotic cells, catalyzes the removal of intact asparagine-linked oligosaccharide chains from glycoproteins and/or glycopeptides. Since its discovery in 1984, PNGase F has remained as the sole prokaryotic PNGase. Recently, a novel gene encoding a protein with a predicted PNGase domain was identified from a clinical isolate of Elizabethkingia meningoseptica. In this study, the candidate protein was expressed in vitro and was subjected to biochemical and structural analyses. The results revealed that it possesses PNGase activity and has substrate specificity different from that of PNGase F. The crystal structure of the protein was determined at 1.9 Å resolution. Structural comparison with PNGase F revealed a relatively larger glycan-binding groove in the catalytic domain and an additional bowl-like domain with unknown function at the N terminus of the candidate protein. These structural and functional analyses indicated that the candidate protein is a novel prokaryotic N-glycosidase. The protein has been named PNGase F-II.
Introduction
Peptide:N-glycosidase (PNGase5; EC 3.5.1.52), also known as peptide-N4-(N-acetyl-β-d-glucosaminyl) asparagine amidase, catalyzes the cleavage and release of N-linked glycan moieties from glycoproteins and/or glycopeptides. The first PNGase (PNGase A) was identified in almond emulsion in 1977 (1). Subsequently, similar enzymes with deglycosylation activities were identified in other plants and seeds (2–4). A fungal cytoplasmic PNGase was identified in the budding yeast, Saccharomyces cerevisiae (5). In 1991, it was observed that free oligosaccharides accumulate in mature fish eggs and during the early stages of embryogenesis of medaka fish (Oryzias latipes). This led to the discovery of PNGase in animal sources, and a possible role for PNGase-mediated deglycosylation in the embryogenesis of fish was suggested (6). Studies on the tissue distribution and subcellular localization of PNGase in higher eukaryotes illustrated the basal level of PNGase expression in all of the tissues tested and relatively high levels of expression in the testis, liver, and brain (7, 8). Further studies suggested that PNGase is a non-lysosomal soluble protein that may be involved in the regulation of the thermal stability of glycosylated proteins. Chantret et al. (9) reported that down-regulation of the PNGase gene led to 30% inhibition of total free oligosaccharides possessing the di-N-acetylchitobiose moiety in human HepG2 cells. Cytoplasmic studies also indicated that PNGase might be involved in the recycling of misfolded glycoproteins and endoplasmic reticulum-associated protein degradation-mediated events (10, 11).
The first prokaryotic PNGase was isolated from Elizabethkingia meningoseptica (formerly known as Flavobacterium meningosepticum) in 1984 (12). It was named peptide:N-glycosidase F (PNGase F), after the organism in which it was first identified, and has been subjected to extensive structural and functional analyses (13–16). PNGase F hydrolyzes a broad spectrum of asparagine-linked glycoproteins to generate carbohydrate-free peptides and detached full-length oligosaccharides. PNGase F has been used extensively in protein glycosylation/deglycosylation studies. Despite the fact that PNGase F remained the only proximal N-glycosidase identified in bacteria for many years, its biological significance has remained elusive. Recently, several genes encoding proteins with predicted PNGase structures, including candidate PNGase proteins from Deinococcus radiodurans (GenBankTM identifier (GI): 15807985) and Bacteroides fragilis (GI: 3288840), were identified in genome projects. However, to the best of our knowledge, these proteins have not been characterized so far.
In 2012, our group isolated an E. meningoseptica strain (FMS-007) from a T-cell non-Hodgkin's lymphoma patient and obtained its complete genome sequence in one contig (GenBankTM accession number CP006576).6 In addition to the known PNGase F gene, a putative gene encoding a protein with significant structural homology to PNGase F at its C-terminal end was identified by bioinformatics analysis in this E. meningoseptica strain. The protein was expressed, purified, and subjected to biochemical and structural analyses. The results suggest that the candidate protein is a new PNGase identified from Elizabethkingia meningoseptica with a novel domain structure and catalytic specificity. It has been named PNGase F-II or type 2 PNGase F.
EXPERIMENTAL PROCEDURES
Bioinformatics Analysis of the Novel PNGase from E. meningoseptica
The DNA sequence of the putative PNGase gene was obtained from the whole genome sequence of the clinical isolate (FMS-007) of E. meningoseptica. Conserved domains in the putative PNGase were identified using the Basic Local Alignment Search Tool (BLASTp) from the National Center for Biotechnology Information (NCBI) Web site. Protein sequences for the other PNGase used in the multiple-sequence alignment were downloaded from GenBankTM. For proteins with known structures, the .pdb files were obtained from the Protein Data Bank. Multiple sequence alignments were performed using Align X®. A phylogenetic tree was constructed using the neighbor joining method in the Molecular Evolutionary Genetics Analysis (MEGA version 5.2.2) software. The position of the signal peptide on the putative PNGase was predicted using the online program, SignalP (version 4.1).
Protein Expression and Purification
The target gene was cloned into the pET28a vector (kanr; Novagen, Darmstadt, Germany) and expressed in Escherichia coli BL21(DE3) (Tiangen, Shanghai, China) as follows. The candidate gene without the predicted signal peptide was amplified by PCR using genomic DNA isolated from E. meningoseptica strain FMS-007, and a pair of designed oligonucleotides (5′-atccatggcccagacttatgaaattacttatc-3′ and 5′-acctcgagttcttgccctaagagaacg-3′). The amplified PCR product was ligated between the NcoI and XhoI cloning sites on the pET28a plasmid. The constructed expression vector was transformed into E. coli BL21(DE3), and the transformants were cultured at 37 °C for 12 h in Luria-Bertani medium containing 50 μg/ml kanamycin. Isopropyl 1-thio-β-d-galactopyranoside (1.0 mm) was used to induce the expression of the candidate protein at 28 °C for 12 h.
The expressed protein was purified in sequential steps. The cells were harvested by centrifugation at 2683 × g (Beckman Coulter AllegraTM 25R centrifuge, TA-14-250 rotor) for 20 min, and the pellet was resuspended in a lysis buffer (20 mm Tris-HCl, 500 mm NaCl, 25 mm imidazole, pH 8.0). The suspended cells were treated with lysozyme (1 mg/ml) (Sigma-Aldrich) for 30 min and sonicated (program: 3 s of sonication followed by a pause (5 s) per cycle, 30% power, φ = 10) for 20 min on ice or disrupted using a high pressure homogenizer (JNBIO®, Guangzhou, China). The debris was removed by centrifugation (30 min, 15,455 × g using a TA-14-50 rotor) at 4 °C. The supernatant was purified using a B-PER His6 fusion protein purification kit (Thermo Scientific Pierce) or a HisTrapTM column (GE Healthcare). The loaded column was washed twice with a wash buffer (20 mm Tris, 500 mm NaCl, 25 mm imidazole, pH 8.0), and the candidate protein was eluted in an elution buffer (20 mm Tris, 500 mm NaCl, 300 mm imidazole, pH 8.0). The protein was further purified by gel filtration using a Superdex® 200 16/60 preparation grade column (GE Healthcare) in a gel filtration buffer (10 mm Tris-HCl, 100 mm NaCl, pH 8.0).
For the in vitro biochemical assays, the elution buffer was replaced with 10 mm PBS (pH 7.4) by ultrafiltration through a filter with MWCO of 30,000 (Millipore, Bedford, MA) at 2173 × g (Beckman Coulter AllegtaTM 25R, TA-14-50 rotor) for 30 min at 4 °C. The protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific Pierce) or by measuring the absorption of UV light at A280. The purity of the candidate protein was verified by SDS-PAGE analysis. The amount of purified PNGase F-II obtained from different batches was 70–90 mg/liter of culture. PNGase F-II was stable at −80, −20, and 4 °C in PBS buffer for 1 month. Prior to crystallization, the purified protein was concentrated to 60 mg/ml using an Amicon® Ultra centrifugal device from Millipore. All other chemicals were purchased from Sigma unless indicated otherwise. PNGase F was used as the positive control for all subsequent assays. A similar procedure was adopted to clone, express, and purify PNGase F with a pair of PNGase F-specific oligonucleotides (5′-atccatggatgctccggctgataatacc-3′ and 5′-ccctcgaggtttgtaactatcggagcactaat-3′).
Enzymatic Assay for PNGase Activity
To verify the N-glycosidase activity of the candidate protein, bovine pancreatic ribonuclease B (RNase B) with N-linked high-mannose oligosaccharide (Sigma-Aldrich) was chosen as the standard substrate glycoprotein and subjected to cleavage by PNGase F (New England Biolabs) and the candidate protein. The oligosaccharides cleaved by the two enzymes were analyzed by MALDI-TOF MS (17, 18). RNase B was first dissolved in 50 mm ammonium bicarbonate buffer (pH 8.0) at a concentration of 10 mg/ml. For each reaction, 100 μg of RNase B in ammonium bicarbonate reaction buffer was denatured in a 95 °C water bath for 10 min and then mixed with 90 μl of PBS containing 5 μg of PNGase F and the purified candidate protein in separate vials. The digestion was carried out at 37 °C for 12 h, and the released glycans were collected by ultrafiltration using a filter with a molecular weight cut-off of 3000 (Millipore, Bedford, MA) at 2683 × g for 30 min at 4 °C. One microliter of the filtered solution was deposited on the MALDI plate with 1 μl of saturated 2,5-dihydroxybenzoic acid using the dried droplet method for MS analysis. Profiling of the glycans was performed in the positive ion reflectron mode in an AXIMA MALDI-quadrupole ion trap TOF mass spectrometer (Shimadzu Corp., Kyoto, Japan) using a nitrogen pulsed laser (337 nm) and an acceleration voltage of 20 kV (17, 19). The enzymatic activity was also analyzed by SDS-PAGE.
Substrate Specificity
A panel of representative substrate N-glycoproteins, including RNase B with N-linked high-mannose oligosaccharides, ovalbumin with N-linked hybrid oligosaccharides, human IgG with N-linked complex oligosaccharides (provided by the Chinese Center for Disease Control and Prevention), and horseradish peroxidase (HRP) with α→1,3 core-fucosylated oligosaccharides (which is resistant to PNGase F digestion; Biodee Biotechnology, Beijing, China) were selected for use in this study. Glycoprotein (10 mg/ml) dissolved in ultrapure water (native substrate) or heat-denatured at 100 °C for 10 min (denatured substrate) was assayed. All reactions were conducted in 10 mm PBS buffer (pH 7.4) at 37 °C for 12 h. The treated samples were analyzed by SDS-PAGE.
Deglycosylation of HRP
Periodic acid-Schiff staining was applied for positive and negative detection of the glycosylated substrate and deglycosylated product, respectively. Denatured HRP was treated with PNGase F and PNGase F-II separately in PBS buffer (pH 7.4) at 37 °C for 12 h and separated by electrophoresis. The gel was cut into two halves; one half was stained using Coomassie Blue for protein detection, and the other half was stained using periodic acid-Schiff reagent, according to the instructions provided in the glycoprotein staining kit manual (Thermo Scientific Pierce). Western blot using anti-HRP polyclonal antibodies (LifeSpan BioSciences) was performed to verify the HRP protein. The digestion of α→1,3 core-fucosylated asparagine oligosaccharide by PNGase F-II was verified by MALDI-TOF-MS.
Crystallization of PNGase F-II
Selenomethionine (Se-Met)-substituted PNGase F-II was obtained by expressing the protein in M9 medium supplemented with 60 mg/liter Se-Met (Sigma-Aldrich). Se-Met PNGase F-II was purified as described above, except that 1 mm DTT was added to the gel filtration buffer. Se-Met PNGase F-II (15 mg/ml) was crystallized using the hanging-drop vapor diffusion method at 16 °C. The optimized crystallization condition contained 10% polyethylene glycol (PEG) 4000, 0.01 m MgCl2, 0.2 m KCl, and 0.05 m sodium cacodylate, pH 6.5. Native PNGase F-II crystals were obtained in a crystallization condition comprising 12% PEG 3350 and 0.1 m sodium malonate, pH 7.0. The crystals were flash-frozen in liquid nitrogen in the mother liquor supplemented with 20% glycerol as a cryoprotectant.
Data Collection and Structure Determination
X-ray diffraction data were collected at the BL17U beamline of the Shanghai Synchrotron Radiation Facility. All diffraction data were indexed, integrated, and scaled using HKL2000 (20). The crystal structure of native PNGase F-II was determined by molecular replacement using the crystal structure of PNGBf (B. fragilis; Protein Data Bank code 3KS7) as the search model. The crystal structure for Se-Met PNGase F-II was determined by molecular replacement using the structure of native PNGase F-II as the search model. All of the structures were displayed using the PyMOL molecular graphics system (version 1.4.1).
Refinement and Model Building
All structural refinement was carried out using the REFMAC5 program of the CCP4 suite (21). Five percent of the data were randomly selected and set aside for free R-factor cross-validation calculations. 2Fo − Fc and Fo − Fc maps were used for manual rebuilding of the peptide chain as well as for the addition of solvent molecules using COOT (22). The refinement was continued until convergence of the free R-factor. The final refinement statistics are listed in Table 1.
TABLE 1.
Crystallographic data collection, processing, structure refinement, and model quality statistics
| Se-Met PNGase F-II | Native PNGase F-II | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.0000 | 1.0000 |
| Space group | C2 | P21 |
| Unit cell parameters (Å) | ||
| a | 161.449 | 81.864 |
| b | 55.431 | 94.165 |
| c | 71.187 | 165.815 |
| α | 90.000 | 90.000 |
| β | 102.231 | 91.403 |
| γ | 90.000 | 90.000 |
| Resolution range (Å)a | 30.00–1.90 (1.97–1.90) | 30.00–2.80 (2.90–2.80) |
| No. of unique observations | 47,585 | 55,844 |
| Completeness (%) | 97.9 (86.1) | 91.7 (82.0) |
| Rsym (%) | 9.9 (42.4) | 13.6 (42.0) |
| I/σI | 15.56 (3.02) | 9.87 (2.28) |
| Redundancy | 4.0 (2.8) | 2.8 (2.0) |
| Refinement | ||
| Resolution (Å) | 29.97–1.90 | 29.50–2.81 |
| Rwork | 0.165 | 0.199 |
| Rfree | 0.203 | 0.257 |
| No. of reflections | 45,187 | 52,973 |
| Model quality | ||
| Estimated coordinate error (Å) | 0.13 | 0.44 |
| Root mean square deviation bonds (Å) | 0.010 | 0.009 |
| Root mean square deviation angles (degrees) | 1.386 | 1.156 |
a Values in parentheses are for the highest resolution shell.
b Rwork = Σ‖Fo| − |Fc‖/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
c Rfree = Σ‖Fo| − |Fc‖/Σ|Fo| for 5% of the data not used at any stage of structural refinement.
Preparation of PNGase F-IIΔNBL
To further define the function of the catalytic domain of PNGase F-II, a similar procedure was applied for the cloning, expression, and purification of PNGase F-IIΔNBL (residues 184–537, without the NBL domain). The mutant gene was amplified by PCR using the following primer pair (5′-atccatgggaaagagcagatttatcactattcctg-3′ and 5′-acctcgagctattcttgccctaagagaacg-3′). The amplified product was inserted between the NcoI and XhoI cloning sites on the pET28a plasmid. The constructed expression vector was transformed into E. coli BL21(DE3), and the transformants were cultured at 37 °C for 12 h in Luria-Bertani medium containing 50 μg/ml kanamycin. The enzymatic activity of the mutant protein PNGase F-II-ΔNBL was subjected to the same assays applied for PNGase F-II.
RESULTS
A Novel Candidate PNGase Gene (pngF-II) Was Identified in the Whole Genome Sequence of E. meningoseptica
FMS-007 is a clinical strain of E. meningoseptica that was isolated from the sputum collected from a T-cell non-Hodgkin's lymphoma patient; its whole genome sequence (comprising 3,938,967 base pairs in one contig) was obtained in 2012. Bioinformatics analysis of the genome led to the identification of an ORF with a mature protein of 537 amino acids plus a signal peptide of 30 amino acids. The protein contained a domain with unknown function at the N terminus (residues 1–170) followed by a PNGase F domain (PNGF domain; residues 184–537). The protein was named PNGase F-II. The pngF-II gene (FMSX7GL000700) was localized in the Scaffold1:749867–751570 (−) of the genome sequence. Phylogenetic analysis of the PNGF domain with other PNGase (both published and predicted) in GenBankTM revealed that the proteins could be divided into two separate clades, with PNGase F in one clade and the newly predicted PNGase in the other (Fig. 1C). Clade-specific conserved amino acids were also identified from the alignment (Fig. 1B). These results indicated that the candidate protein might have PNGase activity distinct from that reported for well characterized PNGase F protein.
FIGURE 1.

Sequence alignment of the candidate protein (putative PNGase) with PNGase listed in GenBankTM. A, schematic diagram of the domain architecture of the predicted PNGase from E. meningoseptica (FMS-007). B, multiple-sequence alignment of the catalytic domain of the candidate protein with PNGase from the following species: E. meningoseptica (PNGEm; GI: 157833537), Salmo salar (PNGSs; GI: 223648018), Danio rerio (PNGDre; GI: 33417217), D. radiodurans (PNGDra; GI: 15807985), B. fragilis, (PNGBf; GI: 60680363), and Bacillus massiliensis (PNGBm; GI: 492737344). C, unrooted phylogenetic tree of PNGase from different species.
The pngF-II Gene Encoding a Protein with PNGase Activity
The profile of the released glycans from RNase B (Fig. 2A) was compared with that obtained for the PNGase F control (Fig. 2B). The result generated by PNGase F was consistent with that reported in previous studies (23). The glycans released by the candidate protein were identical to the ones produced by PNGase F (Fig. 2). This result indicated that the putative PNGase has activity similar to that of PNGase F; they both hydrolyzed the glycan moiety in the innermost N-acetylglucosamine (GlcNAc) from denatured RNase B. Therefore, the candidate protein was named PNGase F-II.
FIGURE 2.

MALDI-TOF mass spectra of the glycans released from denatured RNase B. A, spectra of the glycans released by the candidate PNGase. B, spectra of the glycans released by PNGase F.
Substrate Specificity of the Candidate PNGase
To define the substrate specificity of PNGase F-II, three representative glycoproteins with N-linked glycans (RNase B (high mannose type), ovalbumin (hybrid type), and IgG (complex type)) were digested with PNGase F-II and PNGase F. Substrate digestion was verified by the downward shift of the substrate glycoprotein band observed on SDS-PAGE analysis (Fig. 3). The results revealed that PNGase F-II could release N-linked glycans from denatured but not native RNase B and ovalbumin (Fig. 3, A1 and A2), whereas PNGase F could cleave both denatured and native substrates (Fig. 3, B1 and B2). Both enzymes were more active toward native IgG than toward denatured IgG (Fig. 3, A3 and B3).
FIGURE 3.
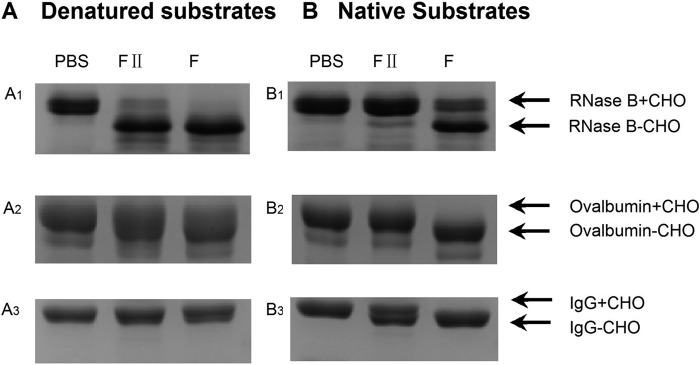
SDS-PAGE analysis of N-glycoproteins treated with PNGase F-II. A, comparison of the deglycosylation activities of PNGase F-II (F-II) and PNGase F (F) toward denatured substrates (glycosylated and deglycosylated forms of the substrates are indicated by +CHO and −CHO, respectively). B, comparison of the deglycosylation of native substrates by PNGase F-II and PNGase F.
Only PNGase F-II Could Release α→1,3 Core-fucosylated Glycans from HRP
To further distinguish the activities of PNGase F-II and PNGase F, plant N-glycoprotein HRP was used as the substrate in the deglycosylation assay. HRP contains α→1,3 core-fucosylated glycans and is known to be resistant to PNGase F digestion (24). When heat-denatured HRP was treated with PNGase F-II and PNGase F, a band down-shifted to an approximate size of 35 kDa was detected only for the PNGase F-II-treated sample (Fig. 4A). Western blot analysis indicated that anti-HRP antibodies recognized the down-shifted band. To confirm that the down-shifted band was a deglycosylated product of PNGase F-II digestion, the proteins on the gel were subjected to glycoprotein staining. The 35 kDa band was negative for glycan staining, whereas the intact HRP band stained positive (Fig. 4A). The glycans released from HRP on PNGase F-II digestion were analyzed by MALDI-TOF MS; the resultant profile was consistent with the reported N-glycan profile for HRP (25). Because PNGase F does not show activity toward proteins with α→1,3 core fucosylation, the digestion of HRP by PNGase F-II indicated that PNGase F-II might have a function distinct from that of PNGase F.
FIGURE 4.

Deglycosylation of HRP by PNGase F-II. A, deglycosylation of HRP by PNGase F-II. PNGase F-II-digested HRP was subjected to Coomassie Blue staining (left), glycoprotein staining (middle), and Western blot analysis with polyclonal anti-HRP antibodies (right). PBS, only substrate; FII, PNGase F-II and substrate. Glycosylated and deglycosylated forms of HRP are indicated by +CHO and −CHO, respectively. B, MALDI-TOF mass spectra of glycans released from HRP.
Crystal Structure of PNGase F-II
PNGase F-II, crystallized in two different space groups, C2 (Fig. 5, A and B) and P21 (Fig. 5, C and D). Native PNGase F-II crystallized in the P21 space group with four molecules in the asymmetric unit; the crystals diffracted x-rays to a resolution of 2.8 Å. Se-Met PNGase F-II crystallized in the C2 space group with one molecule in the asymmetric unit; the crystals diffracted x-rays to a resolution of 1.9 Å. The crystal structures of both forms were solved by molecular replacement. Because the structures of both forms were identical, the Se-Met PNGase F-II structure was used as the reference structure for all structural analyses.
FIGURE 5.

The crystal structure of PNGase F-II. A, structure of PNGase F-II in the C121 space group. The two domains are marked with different colors. The N-terminal bowl-like domain is colored green, whereas the PNGF domain is colored violet. The hexahistidine tag at the C terminus was bound with a metal ion (blue sphere). B, the four molecules in the asymmetric unit of the P21 space group. Chain A is colored green, chain B is cyan, chain C is light magenta, and chain D is yellow. C, angle shift observed in the different chains. The five chains were superimposed on each other. Chain A is colored green, chain B is cyan, chain C is magenta, chain D is yellow, and Se-Met PNGase F-II is red. The angles are represented by black lines.
The structure of PNGase F-II contained two spatially independent domains, an N-terminal bowl-like (NBL) domain with unknown function (residues 1–170; Fig. 5A) and a core catalytic domain. The bridge between the NBL domain and the PNGF domain was a “linker” α-helix. In the P21 space group (native PNGase F-II), there were four molecules in the asymmetric unit: chains A, B, C, and D (Fig. 5C). These four chains and the single chain in the C2 asymmetric unit (Se-Met PNGase F-II) shared a common structure with minor differences. Structural alignment of the five different chains revealed that the “linker” α-helix might function to provide flexibility (Fig. 5D) between the two domains. Chains B and C of the P21 space group had the same structure, whereas chains A, D, and Se-Met PNGase F-II showed minor variations. When the PNGF domains of chains A, D, and Se-Met PNGase-II were aligned with that of chain B/C as the reference molecule, the NBL domains of chains A, D, and Se-Met PNGase-II were shifted by 9.2, 11.9, and 17.6°, respectively, from the “linker” α helix. Thus, the “linker” α-helix may play a role in coordinating the location of the NBL domain with respect to the PNGF domain. The hexahistidine tag and the bound Zn2+ at the C terminus were derived from the pET28a expression vector.
PNGase F-II Contains a Larger N-Glycan-binding Groove
Like all of the other PNGases, the core catalytic domain of PNGase F-II consists of two eight-stranded antiparallel β sheets, PNGF-N (β1 and β4–10) and PNGF-C (β11–15 and β17–19) (Figs. 1B and 5A). These two eight-stranded antiparallel β sandwiches lie side by side with parallel principal axes. Unlike PNGase F, which contains three pairs of disulfide bonds, only one disulfide bond was detected in PNGase F-II (between Cys460 and Cys488), and these two residues were conserved in the PNGase family (Fig. 1B).
The substrate-binding region of the core catalytic domain was identified by superimposing its crystal structure on the structure of PNGase F with or without a glycan product (Fig. 6, A and B; EmPNG (Protein Data Bank code 1PNF), crystal structure complexed with the product N,N′-diacetylchitobiose; EmPNG (Protein Data Bank code 1PNG), crystal structure without the product). As in PNGase F, an N-glycan-binding groove located between the two eight-stranded antiparallel β sandwiches with a negatively charged surface was identified (Fig. 6F). Previous studies indicated that the residues Asp60, Glu118, and Glu206 in EmPNG are essential for catalytic activity, with Asp60 as the primary catalytic residue (26). Two of these three residues were identified at corresponding positions in PNGase F-II (Asp245 and Glu435). Residue Glu118 in EmPNG was found to be replaced by residue Gly350 in PNGase F-II at the corresponding position. However, the carboxylic group of Glu389 in PNGase F-II was found in the same position as that of Glu118 in PNGase F (Fig. 6, C–E). These results suggest that PNGase F-II and PNGase F may have similar glycan-binding grooves and catalytic sites. Notably, the side chain of Trp191 in EmPNG is extended toward the outside of the groove (Fig. 6D), whereas the side chain of His427 in PNGase F-II is positioned on the inner surface of the groove (Fig. 6E). This provides more room for proximal GlcNAc inside the glycan-binding groove of PNGF-II (Fig. 6F). This may allow PNGF-II to bind, cleave, and release glycans from substrates with core α→1,3 fucosylation.
FIGURE 6.

Substrate-binding region and active site of PNGase F-II. A, superimposition of the PNGF domain of PNGase F-II and EmPNG (Protein Data Bank code 1PNG). The PNGF domain of PNGF-II is colored violet, and EmPNG is colored cyan. B, superimposition of the PNGF domain of PNGase F-II and EmPNG with product (N,N′-diacetylchitobiose) (Protein Data Bank code 1PNF). The PNGF domain of PNGF-II is colored violet, and EmPNG is colored aquamarine. The three insertions in PNGF-II are colored red. C, superimposition of the glycan-binding sites of PNGase F and PNGase F-II. D and E, glycan-binding sites of PNGase F and F-II, respectively. F, electrostatic surface potential diagrams of the glycan-binding grooves. The grooves are marked by yellow lines. G, PNGase F-II with an N,N′-diacetylchitobiose molecule modeled in the glycan-binding groove. The putative peptide-binding cleft is marked by green lines.
Based on the configuration of the proximal GlcNAc inside the glycan-binding groove, the peptide-binding site and the core mannose-binding site could be located on the right and left sides of the groove, respectively (Fig. 6G). Together, these three sites form a substrate-binding region for glycoproteins. Interestingly, three insertions surrounding the substrate-binding region were identified only in the PNGase F-II structure (Figs. 1B and 6B). Insertion I is the largest (residues 259–293), whereas insertions II (residues 312–324) and III (residues 469–478) are on the predicted peptide-binding side of the substrate-binding region (Fig. 6G). These two extended insertions may function as spatial barriers to prevent the binding of native proteins, providing a possible explanation for the preference of PNGase F-II for denatured substrates (Fig. 3).
The N-terminal Domain of PNGase F-II Is Not Required for Its Deglycosylation Activity
A DALI search revealed that the NBL domain has a unique structure without any significant homologs. This domain has a bowl-like structure comprising 10 β-sheets and two α helices (Fig. 7A). The pocket of this bowl is ∼27 Å in length and 24 Å in width. A divalent ion was found outside the bowl in coordination with residues Glu51 and Asp158 (Fig. 7A). The electrostatic surface potential diagram showed that the bottom of the bowl is relatively hydrophobic, whereas one side of the bowl is positively charged and the other is negatively charged (Fig. 7B).
FIGURE 7.

NBL domain of PNGase F-II. A, the NBL domain of PNGF-II. The domain is represented in rainbow colors. The N terminus, C terminus, and divalent ion are colored blue, red, and orange, respectively. B, electrostatic surface potential diagram of the NBL domain. C, SDS-PAGE analysis of RNase B treated with PNGase F-IIΔNBL. PBS, only RNase B; FII, PNGase F-II and RNase B; FIIΔN, PNGase F-IIΔNBL and RNase B; F, PNGase F and RNase B. Glycosylated and deglycosylated forms of RNase B are indicated by +CHO and −CHO, respectively.
To further define the functional domain of PNGase F-II, a deletion mutant (PNGase F-IIΔNBL, comprising residues 184–537 and without the NBL domain) was constructed, expressed, purified, and subjected to functional assays. The enzymatic activities of PNGase F-IIΔNBL and PNGase F-II were compared. PNGase F-IIΔNBL could release N-linked glycan moieties from heat-denatured RNase B (Fig. 7C) and HRP (data not shown). These results indicated that the C-terminal PNGF domain of PNGase F-II is sufficient for its PNGase activity and that the NBL domain is probably involved in the other functions, such as substrate selectivity, protein stability, and interaction with other molecules. Unlike PNGase F-II, the purified shorter protein (PNGase F-IIΔNBL) precipitated easily at 4 °C (data not shown).
DISCUSSION
PNGase (EC 3.5.1.52) is a critical enzyme implicated in many biological processes. It catalyzes the cleavage of the full-length glycan from the proximal GlcNAc residue on N-glycoproteins and N-glycopeptides. Although PNGase isolated from different species may have different biological functions, they all act on substrates composed of two biologically important but structurally different polymers: a peptide with a signature sequence of Asn-X-Thr/Ser and a polysaccharide with a core oligosaccharide of di-N,N′-acetylchitobiosyl plus three mannose residues. Altmann et al. (27) showed that the size of the carbohydrate moiety in the substrate has little influence on the enzymatic activities of N-glycosidase and that the hydrolysis rates of PNGase enzymes are primarily determined by the length of the peptide. Studies on the substrate requirements of PNGase F showed that Asn-linked GlcNAc occupied the most critical position for substrate binding and that PNGase F requires at least the di-N,N′-acetylchitobiosyl core unit and a tripeptide for its activity (28).
In vitro biochemical studies demonstrated that both PNGase F-II and PNGase F showed activity toward substrates with high mannose (RNase B), hybrid N-glycans (ovalbumin), and complex N-glycans (IgG) of bi-, tri-, and tetra-antennary oligosaccharides (12, 15), but only PNGase F-II showed activity toward horseradish peroxidase, a plant N-glycoprotein with fucose linked α→1,3 to Asn-GlcNAc (25). The resistance of HRP to PNGase F digestion observed in this study is consistent with a previous report (24). These observations indicate that the substrate specificity of the novel PNGase characterized in this study differs from that of the well established PNGase F and that this PNGase might serve as an alternative for PNGase F and complement its function. In this study, deglycosylation assays were first conducted with denatured substrates. In order to explore the potential biological function of the candidate PNGase, the assays were also conducted with non-denatured substrates. Our results indicated that both enzymes showed activity toward denatured RNase B, denatured ovalbumin, and native IgG (IgG, hepatitis B immune globin, and globins; data not shown). PNGase F had relatively low activity toward native RNase B and ovalbumin, whereas PNGase F-II showed no activity. The biological significance of these observations is yet to be elucidated.
A previous mutagenesis study of the active sites suggested that Asp60, Glu118, and Glu206 were essential amino acids for PNGase F activity (26). Identical residues were found at two of the corresponding sites in PNGase F-II (AspF60/F-II245 and GluF206/F-II435). Although GluF118 in PNGase F is replaced by GlyF-II350 in PNGase F-II, the Glu389 residue in the active site of PNGase F-II might compensate for this substitution.
On a core-fucosylated GlcNAc molecule, the α1–3- and α1–6-linked fucose moieties are located on different sides of the central line defined by the β1–4 bond formed between two GlcNAc molecules. An earlier study has shown that α1–6-linked fucose is located on the solvent side of the glycan binding site (26), indicating that α1–3-linked fucose is probably sitting on the interface between the substrate and PNGase F-II.
We noticed that, in PNGase F-II, His427 is located on the inner surface of the groove, whereas in PNGase F, Trp191 is on the outer surface of the groove, providing a barrier in the space between the substrate and the enzyme. To address the role of His427 in PNGase F-II, site-directed mutants H427W, H427A, and H427K were produced, and their substrate specificities were analyzed (data not shown). Three mutants retained enzymatic activities toward RNase B, but lost the activity toward HRP, indicating that His427 of PNGase F-II was responsible for its unique substrate specificity toward HRP. A similar approach was used to examine the significance of Trp119 in the inner surface of the groove in the crystal structure of PNGase F-II; three mutants (W119H, W119A, and W119Y) were active toward RNase B but not toward HRP. This result indicated that a single residue change was not enough to create room for the binding of HRP in PNGase F. These structural features explain why PNGase F showed activity toward α→1,6-fucosylated substrates only, whereas PNGase F-II showed activity toward α→1,3-fucosylated glycoproteins as well. A substrate-binding region was identified in the catalytic domain (Fig. 6H), with a glycan-binding groove at the center and two predicted binding sites on either side. Three insertions in the PNGase F-II surround the substrate-binding region and provide additional space barriers to prevent the binding of native glycoproteins. Therefore, PNGase F-II has low activity toward most native glycoproteins.
Compared with PNGase F, PNGase F-II had an additional domain with a bowl-like structure at its N terminus. A deletion assay conducted in this study suggested that this domain is not required for PNGase activity. Additional domains involved in non-catalytic functions, such as protein-protein interactions, have been reported for eukaryotic PNGases. Mouse PNGase has N- and C-terminal domains in addition to its central catalytic domain. The N-terminal domain is known to be involved in protein-protein interaction (29), whereas the C-terminal domain may function as a mannose-binding module (30). Allen et al. reported that the N-terminal PUB domain of human PNGase consists of five α helices that pack into a short three-stranded antiparallel β sheet. This domain may assist the binding of the p97 protein to the D1 ATPase domain, the D2 ATPase domains, or both, thereby coordinating the ATP hydrolysis-dependent substrate-extracting activity of p97 with PNGase-catalyzed deglycosylation (31). Although the extra non-catalytic domain is probably a common feature of some PNGase proteins, elucidating the structures and functions of these domains merits further studies.
In conclusion, we identified a new PNGase from E. meningoseptica that has structure and substrate specificity different from that of PNGase F. It contains a bowl-like domain of unknown function at its N terminus. The identification of this isozyme may help us to understand the biological role of bacterial PNGase. In addition, to the best of our knowledge, this is the first study to demonstrate the presence of two PNGase proteins in a bacterium. The co-existence of two isozymes in E. meningoseptica indicates that PNGase activity is critical for this species. Because protein N-glycosylation is a rare event in bacteria, we speculate that PNGase activity may provide a niche in host-bacterium interactions.
Acknowledgments
We thank Prof. Dangsheng Li and Prof. Yumei Wen for critical reading of the manuscript. We thank the beamline scientists at the Shanghai Synchrotron Radiation Facility for help with data collection.
This work was supported in part by Fudan University Grant EZF101507. A part of this work was performed under the National Science and Technology Major Project of China (2012ZX10003008-010). This work was also supported in part by Ministry of Science and Technology of China Grants 2012CB910502 and 2011CB966304.
The atomic coordinates and structure factors (codes 4R4X and 4R4Z) have been deposited in the Protein Data Bank (http://wwpdb.org/).
G. Sun and L. Chen, submitted for publication.
- PNGase
- peptide:N-glycosidase
- PNGF domain
- PNGase F domain
- NBL
- N-terminal bowl-like
- GlcNAc
- N-acetylglucosamine
- Se-Met
- selenomethionine
- contig
- group of overlapping clones.
REFERENCES
- 1. Takahashi N. (1977) Demonstration of a new amidase acting on glycopeptides. Biochem. Biophys. Res. Commun. 76, 1194–1201 [DOI] [PubMed] [Google Scholar]
- 2. Berger S., Menudier A., Julien R., Karamanos Y. (1995) Do de-N-glycosylation enzymes have an important role in plant cells? Biochimie 77, 751–760 [DOI] [PubMed] [Google Scholar]
- 3. Plummer T. H., Jr., Phelan A. W., Tarentino A. L. (1987) Detection and quantification of peptide-N4-(N-acetyl-β-glucosaminyl)asparagine amidases. Eur. J. Biochem. 163, 167–173 [DOI] [PubMed] [Google Scholar]
- 4. Sugiyama K., Ishihara H., Tejima S., Takahashi N. (1983) Demonstration of a new glycopeptidase, from jack-bean meal, acting on aspartylglucosylamine linkages. Biochem. Biophys. Res. Commun. 112, 155–160 [DOI] [PubMed] [Google Scholar]
- 5. Chiba Y., Suzuki M., Yoshida S., Yoshida A., Ikenaga H., Takeuchi M., Jigami Y., Ichishima E. (1998) Production of human compatible high mannose-type (Man5GlcNAc2) sugar chains in Saccharomyces cerevisiae. J. Biol. Chem. 273, 26298–26304 [DOI] [PubMed] [Google Scholar]
- 6. Seko A., Kitajima K., Inoue Y., Inoue S. (1991) Peptide:N-glycosidase activity found in the early embryos of Oryzias latipes (Medaka fish): the first demonstration of the occurrence of peptide:N-glycosidase in animal cells and its implication for the presence of a de-N-glycosylation system in living organisms. J. Biol. Chem. 266, 22110–22114 [PubMed] [Google Scholar]
- 7. Kitajima K., Suzuki T., Kouchi Z., Inoue S., Inoue Y. (1995) Identification and distribution of peptide:N-glycanase (PNGase) in mouse organs. Arch. Biochem. Biophys. 319, 393–401 [DOI] [PubMed] [Google Scholar]
- 8. Suzuki T., Kwofie M. A., Lennarz W. J. (2003) Ngly1, a mouse gene encoding a deglycosylating enzyme implicated in proteasomal degradation: expression, genomic organization, and chromosomal mapping. Biochem. Biophys. Res. Commun. 304, 326–332 [DOI] [PubMed] [Google Scholar]
- 9. Chantret I., Fasseu M., Zaoui K., Le Bizec C., Yayé H. S., Dupré T., Moore S. E. (2010) Identification of roles for peptide:N-glycanase and endo-β-N-acetylglucosaminidase (Engase1p) during protein N-glycosylation in human HepG2 cells. PLoS One 5, e11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki T. (2007) Cytoplasmic peptide:N-glycanase and catabolic pathway for free N-glycans in the cytosol. Semin. Cell Dev. Biol. 18, 762–769 [DOI] [PubMed] [Google Scholar]
- 11. Suzuki T., Seko A., Kitajima K., Inoue Y., Inoue S. (1993) Identification of peptide:N-glycanase activity in mammalian-derived cultured cells. Biochem. Biophys. Res. Commun. 194, 1124–1130 [DOI] [PubMed] [Google Scholar]
- 12. Plummer T. H., Jr., Elder J. H., Alexander S., Phelan A. W., Tarentino A. L. (1984) Demonstration of peptide:N-glycosidase F activity in endo-β-N-acetylglucosaminidase F preparations. J. Biol. Chem. 259, 10700–10704 [PubMed] [Google Scholar]
- 13. Plummer T. H., Jr., Tarentino A. L. (1991) Purification of the oligosaccharide-cleaving enzymes of Flavobacterium meningosepticum. Glycobiology 1, 257–263 [DOI] [PubMed] [Google Scholar]
- 14. Kuhn P., Tarentino A. L., Plummer T. H., Jr., Van Roey P. (1994) Crystal structure of peptide-N4-(N-acetyl-β-d-glucosaminyl) asparagine amidase F at 2.2-Å resolution. Biochemistry 33, 11699–11706 [DOI] [PubMed] [Google Scholar]
- 15. Tarentino A. L., Gómez C. M., Plummer T. H., Jr. (1985) Deglycosylation of asparagine-linked glycans by peptide:N-glycosidase F. Biochemistry 24, 4665–4671 [DOI] [PubMed] [Google Scholar]
- 16. Norris G. E., Stillman T. J., Anderson B. F., Baker E. N. (1994) The three-dimensional structure of PNGase F, a glycosylasparaginase from Flavobacterium meningosepticum. Structure 2, 1049–1059 [DOI] [PubMed] [Google Scholar]
- 17. Zhang W., Wang H., Tang H., Yang P. (2011) Endoglycosidase-mediated incorporation of 18O into glycans for relative glycan quantitation. Anal. Chem. 83, 4975–4981 [DOI] [PubMed] [Google Scholar]
- 18. Küster B., Wheeler S. F., Hunter A. P., Dwek R. A., Harvey D. J. (1997) Sequencing of N-linked oligosaccharides directly from protein gels: in-gel deglycosylation followed by matrix-assisted laser desorption/ionization mass spectrometry and normal-phase high-performance liquid chromatography. Anal. Biochem. 250, 82–101 [DOI] [PubMed] [Google Scholar]
- 19. Cai Y., Zhang Y., Yang P., Lu H. (2013) Improved analysis of oligosaccharides for matrix-assisted laser desorption/ionization time-of-flight mass spectrometry using aminopyrazine as a derivatization reagent and a co-matrix. Analyst 138, 6270–6276 [DOI] [PubMed] [Google Scholar]
- 20. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 21. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 22. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Hoek A. N., Wiener M. C., Verbavatz J. M., Brown D., Lipniunas P. H., Townsend R. R., Verkman A. S. (1995) Purification and structure-function analysis of native, PNGase F-treated, and endo-β-galactosidase-treated CHIP28 water channels. Biochemistry 34, 2212–2219 [DOI] [PubMed] [Google Scholar]
- 24. Tretter V., Altmann F., März L. (1991) Peptide-N4-(N-acetyl-β-glucosaminyl) asparagine amidase F cannot release glycans with fucose attached α1→3 to the asparagine-linked N-acetylglucosamine residue. Eur. J. Biochem. 199, 647–652 [DOI] [PubMed] [Google Scholar]
- 25. Yang B. Y., Gray J. S., Montgomery R. (1996) The glycans of horseradish peroxidase. Carbohydr. Res. 287, 203–212 [DOI] [PubMed] [Google Scholar]
- 26. Kuhn P., Guan C., Cui T., Tarentino A. L., Plummer T. H., Jr., Van Roey P. (1995) Active site and oligosaccharide recognition residues of peptide-N4-(N-acetyl-β-d-glucosaminyl)asparagine amidase F. J. Biol. Chem. 270, 29493–29497 [DOI] [PubMed] [Google Scholar]
- 27. Altmann F., Schweiszer S., Weber C. (1995) Kinetic comparison of peptide: N-glycosidases F and A reveals several differences in substrate specificity. Glycoconj. J. 12, 84–93 [DOI] [PubMed] [Google Scholar]
- 28. Fan J. Q., Lee Y. C. (1997) Detailed studies on substrate structure requirements of glycoamidases A and F. J. Biol. Chem. 272, 27058–27064 [DOI] [PubMed] [Google Scholar]
- 29. Zhao G., Zhou X., Wang L., Li G., Kisker C., Lennarz W. J., Schindelin H. (2006) Structure of the mouse peptide N-glycanase-HR23 complex suggests co-evolution of the endoplasmic reticulum-associated degradation and DNA repair pathways. J. Biol. Chem. 281, 13751–13761 [DOI] [PubMed] [Google Scholar]
- 30. Zhou X., Zhao G., Truglio J. J., Wang L., Li G., Lennarz W. J., Schindelin H. (2006) Structural and biochemical studies of the C-terminal domain of mouse peptide-N-glycanase identify it as a mannose-binding module. Proc. Natl. Acad. Sci. U.S.A. 103, 17214–17219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Allen M. D., Buchberger A., Bycroft M. (2006) The PUB domain functions as a p97 binding module in human peptide N-glycanase. J. Biol. Chem. 281, 25502–25508 [DOI] [PubMed] [Google Scholar]